

Summary
Although gut microbiome composition is well defined, the mechanisms underlying community assembly remain poorly understood. Bacteroidales possess three genetic architectures (GA1–3) of the type VI secretion system (T6SS), an effector delivery pathway that mediates interbacterial competition. Here we define the distribution and role of GA1–3 in the human gut using metagenomic analysis. We find that adult microbiomes harbor limited effector and cognate immunity genes, suggesting selection for compatibility at the species (GA1, GA2) and strain (GA3) levels. Bacteroides fragilis GA3 is known to mediate potent inter-strain competition, and we observe GA3 enrichment among strains colonizing infant microbiomes, suggesting competition early in life. Additionally, GA3 is associated with increased Bacteroides abundance, indicating that this system confers an advantage in Bacteroides-rich ecosystems. Collectively, these analyses uncover the prevalence of T6SS-dependent competition and reveal its potential role in shaping human gut microbial composition.
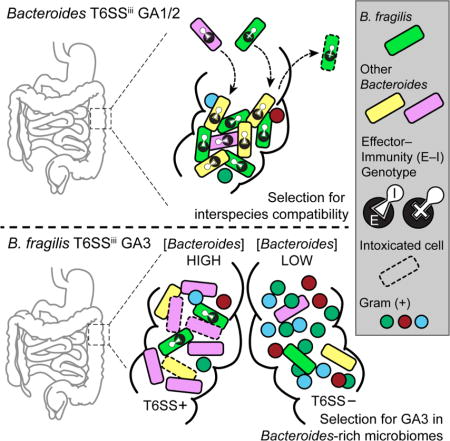
Graphical abstract
The T6SS is an effector delivery system that mediates interbacterial competition. Using metagenomic analyses, Verster et al. investigate the prevalence and role of the T6SS in the human gut microbiome. They demonstrate that the T6SS mediates interactions between Bacteroides strains in the infant microbiome and between species in Bacteroides-rich environments.
Introduction
Bacterial communities are of fundamental importance to natural ecosystems (Prosser et al., 2007). While cooperative interactions between the species comprising such communities can occur (Rakoff-Nahoum et al., 2016), it is clear that bacteria in these settings experience pervasive competition from surrounding cells (Coyte et al., 2015; Hibbing et al., 2010; Levy and Borenstein, 2013). Indeed, the genomes of bacteria encode a wealth of dedicated interbacterial antagonism pathways (Zhang et al., 2012). Some of these function through the production of diffusible small molecules (Riley and Wertz, 2002), whereas others utilize proteinaceous toxins. A prevalent pathway mediating the transfer of toxic proteins between bacteria is the type VI secretion system (T6SS) (Hood et al., 2010). This system has been most thoroughly studied in Proteobacteria, though it is found in several phyla of Gram-negative bacteria, and while it can, in some cases, target eukaryotic cells, it has been primarily investigated as an interbacterial system.
The T6S apparatus transfers toxic effector proteins from donor to recipient bacterial cells by a mechanism dependent upon cell contact (Russell et al., 2014a). Characterized interbacterial effector proteins are thus far without exception enzymes that target conserved, essential features of the bacterial cell, such as peptidoglycan, phospholipids, and nucleic acids. This feature of effector proteins, taken together with the fact that T6SS targeting does not appear to be dependent on a specific receptor, confers broad activity against Gram-negative cells. Indiscriminate effector transfer also extends to kin cells; therefore, cells with the T6SS produce immunity proteins that inactivate cognate toxins through active site occlusion (Benz and Meinhart, 2014).
Given its wide phylogenetic distribution and its capacity to target diverse recipient cells, the T6SS is likely to play an important role in the assembly and composition of bacterial communities. Indeed, there are recent reports consistent with the pathway mediating bacterial interaction in environmental communities. For instance, T6S genes were found to be enriched and under positive selection in the barley rhizosphere (Bulgarelli et al., 2015), and T6S phospholipase effectors were detected in metagenomes from diverse sources (Egan et al., 2015). To date, however, systematic studies of the impact of T6S on microbial community assembly are lacking.
The human gut microbiome is a dense ecosystem whose composition is paramount to its function (Walter and Ley, 2011). Factors such as diet, immune status, and host genetics have each been implicated in shaping the gut community, yet the contribution of direct interbacterial competition to the structure of this community remains poorly understood. Recently, a T6SS-like pathway was detected in Bacteriodetes, the most abundant Gram-negative phylum in the human gut (Coyne et al., 2014; Russell et al., 2014b). Additional work demonstrated that T6S contributes to the fitness of Bacteroides fragilis in competition with other bacteria in vitro and in gnotobiotic mice (Chatzidaki-Livanis et al., 2016; Hecht et al., 2016; Russell et al., 2014b; Wexler et al., 2016). These and other data show that the mammalian GI tract is physically conducive to T6SS-dependent interbacterial antagonism, suggesting a potential impact of this pathway on the composition of the human gut microbiome (Anderson et al., 2017; Sana et al., 2016). Here, we sought to define the distribution of the Bacteroidales T6SS and to explore its function in the human gut microbiome through the analysis of several publicly available metagenomic datasets. These datasets allow us to study the outcome of natural community dynamics in the gut microbiome, and we reasoned that their analysis could therefore provide unique insight into the physiologic role of T6SS-dependent competition in this ecosystem. Our findings reveal the prevalence of this pathway in intact human gut microbial communities, highlight striking and non-random patterns in its distribution across samples, and suggest an active role for the T6SS in intra- and inter-species bacterial interactions in the gut.
Results
Detection of T6SS E–I pairs in the human gut microbiome
We first set out to characterize the prevalence and distribution of T6SS genes in the gut microbiomes of healthy adult individuals. Based on their organization and content, Bacteroidales T6SS gene clusters can be divided into three distinct subtypes, termed genetic architecture 1–3 (GA1–3) (Coyne et al., 2016). Each T6S subtype possesses one or more cassettes at stereotyped positions that contain variable genes predicted or demonstrated to encode effector–immunity (E–I) pairs (Chatzidaki-Livanis et al., 2016; Coyne et al., 2016; Russell et al., 2014b; Wexler et al., 2016). As T6SS-based antagonism is determined by the effector and immunity genes of donor and recipient cells, respectively, the identification of E–I pairs provides information regarding the potential for interbacterial interactions mediated by this system. Furthermore, since these cassettes are variable within, but appear unique among the T6S subtypes, estimation of the abundance of these genes within metagenomes can serve as a proxy for the presence and distribution of GA1–3.
To define the E–I repertoire associated with GA1–3, we searched within T6-associated variable cassettes from Bacteroidales reference genomes and from preassembled metagenome contigs from the Human Microbiome Project (HMP) for genes with hallmarks of known T6SS effector and immunity factors (Human Microbiome Project, 2012). These included fusion to modular adaptor domains, reduced GC content, bicistronic arrangement, and similarity to protein families defined by their association with characterized E–I pairs (see Experimental Procedures for a complete description of annotation criteria; Figures 1A and S1). In total, we identified 12 GA1, 19 GA2, and 14 GA3 putative E–I pairs. As expected, genes with significant homology to GA3 pairs were identified only in B. fragilis reference genomes, whereas GA1 and GA2 pairs were detected throughout the order. Importantly, we did not identify GA1–3 E–I genes outside of Bacteroidales.
Figure 1. Bacteroides T6SS E–I genes are abundant in human gut microbiome samples.
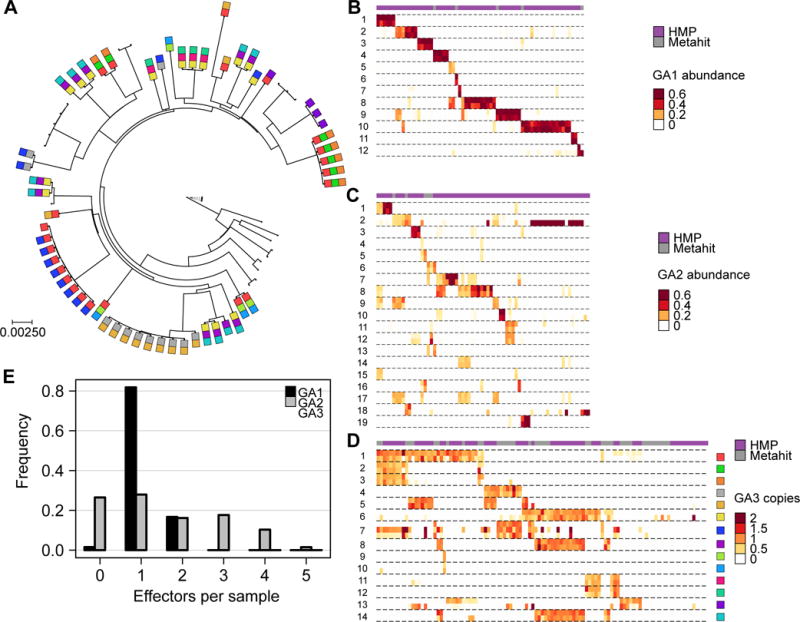
(A) A maximum likelihood phylogeny of B. fragilis reference strains constructed from concatenated marker genes. Phylogenetic distance is measured as substitutions per site on the marker genes. GA3 effector genes are represented as colored squares (using the same color coding as in panel D). (B–D) Each heatmap illustrates the abundance of E–I genes for one of the T6SS subsystems. Each row corresponds to a different E–I pair (effector, top; immunity, bottom). Columns represent the samples analyzed (HMP, purple; MetaHIT, gray). For GA1 and GA2, only samples in which at least 100 reads mapped to the E–I genes of a given subsystem are included, and abundance is measured as fraction of the total abundance of E–I genes in a given sample. For GA3, only samples in which B. fragilis is present are included and E–I abundance is normalized by the abundance of B. fragilis-specific marker genes, hence measuring the average number of copies per B. fragilis genome. (E) Histograms showing the number of effector genes detected (at >10% of the most abundant effector gene) in each sample.
To estimate the abundance of these E–I pairs in gut microbiomes, we obtained metagenomic datasets derived from healthy donor samples of the HMP (Human Microbiome Project, 2012) and MetaHIT (Qin et al., 2010) studies. We next mapped the reads from each sample to our catalog of E–I genes (using 97% sequence identity threshold). Our results indicate that T6S is prevalent in the human gut microbiome; of the 246 samples analyzed, we detected E–I genes in 155 (63%). Moreover, each E–I pair in our list was detected in at least one microbiome sample, with an average of 9.5 occurrences. Importantly, the abundance of GA1–3 effector genes across samples correlates with that of subtype-specific T6SS structural genes with only very few samples containing structural genes and no effector genes, suggesting that our catalog of E–I pairs is comprehensive and approximates the full diversity of such genes in nature (Figure 2A).
Figure 2. Differential associations between T6SS and Bacteroides spp.
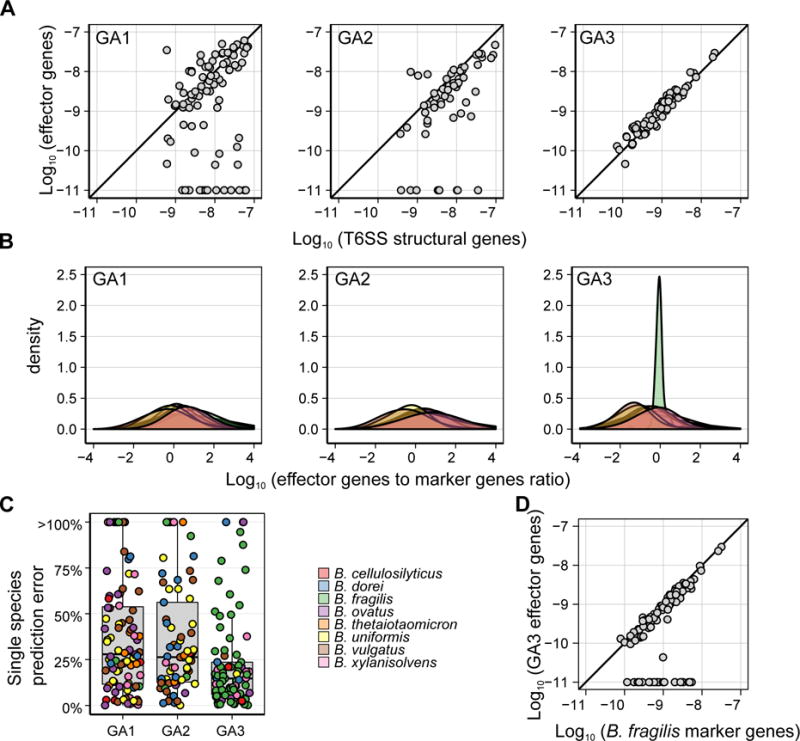
(A) Scatter plot of the average abundance of detected effector genes vs. the average abundance of T6SS structural genes for different subtypes. We have restricted our analysis to samples with at least 25 reads mapping to a given subtype. The strong correlation observed, and the very few samples in which structural genes but no effector genes can be found, testify to the completeness of our E–I pairs catalog. (B) Density plots showing the distribution across samples of the ratio between the average abundance of detected effector genes from each T6SS subsystem and the average abundance of species-specific marker genes for different Bacteroides spp. Only samples in which at least 100 reads mapped to the E–I genes of a given subsystem and only species for which at least 5 genomes were available (and therefore marker genes can be robustly inferred) are included. (C) A boxplot showing the minimal relative error in effector abundance assuming that the T6SS is encoded by a single species. The relative error is defined as the relative difference between the average abundance of detectable effector genes in a sample and the abundance of species with the closest abundance. The color of each point represents the species for which the minimal relative error was obtained. (D) Scatter plot of the average abundance of detected GA3 effector genes vs. the average abundance of B. fragilis-specific marker genes. Only samples in which B. fragilis is present are included. As in (A) each abundance was increased by 10−11. See also Figures S2 and S3.
T6S E–I pairs display low diversity within human gut microbiome samples
The systematic characterization of E–I abundance in metagenomic samples provided a unique opportunity to examine the distribution of the genes associated with each genetic architecture across healthy gut microbiomes. We first focused on GA1 and GA2, which utilize unique complements of effectors, but share the ability to undergo conjugative transfer between species belonging to the order Bacteroidales (Coyne et al., 2016). Surprisingly, we found that the complement of GA1- and GA2-associated E–I genes in a typical microbiome is small, with only a few pairs per sample, comparable to the number of pairs usually detected in a single genome (Figure 1B,C, E). Moreover, in many cases, the same complement of E–I genes was detected in multiple samples. Henceforth, we refer to these combinations as E–I genotypes. This pattern suggests that either each sample is dominated by a single strain that harbors the observed E–I genotype or that there exists selective pressure for compatible E–I genes across multiple strains or species in a sample.
To further explore these possibilities, we focused our attention on the most prominent members of the genus Bacteroides. Other genera in the order Bacteroidales are less abundant constituents of the microbiome and based on reference genomes do not often harbor GA1 or GA2. We identified a set of species-specific single-copy marker genes for each Bacteroides species and estimated their abundance in each sample. Next we compared marker gene abundance to that of GA1 and GA2 E–I genes across samples (see Experimental Procedures). We found that the abundance of these E–I genes was not consistent with that of an individual species (Figures 2B–C). These findings suggest that multiple species co-existing in a microbiome typically encode a single GA1 and/or GA2 E–I genotype, potentially due to selective pressure for maintenance of E–I compatibility.
We next examined GA3 E–I genes, and found, as in GA1 and GA2, that each sample harbors only a small set of E–I pairs (Figure 1D–E). Moreover, observed GA3 E–I genotypes matched those detected in reference genomes (Figure S2A–B), and appeared randomly distributed between the American (HMP) and European (MetaHIT) datasets (Figure 1D). However, in contrast to GA1 and GA2, we found a strong correlation (R = 0.94) between the abundance of GA3 effector and immunity genes and that of a single species, B. fragilis (Figures 2B–D). This finding shows that restriction of GA3 to B. fragilis observed in sequenced reference genomes holds across naturally occurring communities (Coyne et al., 2016).
We hypothesized that the pattern of GA3 E–I genotypes we observed could be explained by the dominance of a single B. fragilis strain within each individual microbiome. Indeed, prior studies suggest that B. fragilis exhibits relatively low diversity within individuals (Yassour et al., 2016). To confirm that this pattern is also observed in HMP and MetaHIT samples, we first measured nucleotide diversity in species-specific markers of Bacteroides spp. We found that within an individual, B. fragilis possesses the lowest average SNP diversity of well represented members of the Bacteroides genus (Figure S2C). We then used a previously developed method for inferring the most likely set of strains in metagenomic samples based on nucleotide variants, combined with a phylogenetic analysis of these inferred strains to determine the number of different monophyletic groups of strains present in each sample (see Experimental Procedures). We found that B. fragilis inferred strains in every HMP and MetaHIT sample formed a single monophyletic group, indicating that extant B. fragilis strains in each sample are likely derived from a single colonization event or outcompeted other strains to obtain dominance. Moreover, the set of E–I pairs detected in each metagenomic sample generally matched the set of E–I pairs found in the reference strains closest in the phylogenetic tree to the inferred strains, especially when the distance of inferred strains to their nearest reference was low (Figure S3).
To experimentally confirm our computational findings, we additionally selected 20 B. fragilis colonies isolated from two healthy adults and subjected these to whole genome sequencing. Consistent with our findings using metagenomic data, our sequencing showed that a single clonal strain of B. fragilis dominates the microbiome of these individuals (Figure S2D).
B. fragilis GA3 is important in the developing microbiome
The finding that the presence of singular GA3 genotypes within individuals is due to the dominance of one B. fragilis strain motivated us to investigate the role of this system in the microbiome. We reasoned that in this dense and competitive microbiome ecosystem an antagonistic pathway such as the T6SS might provide a fitness advantage (Ley et al., 2006),. The system could mediate antagonism against other B. fragilis strains, Bacteroides spp., Gram-negative inhabitants of the microbiome, or a combination of these. Assuming such a role for T6SS, we further reasoned that in the microbiome of infants, which is less stable than that of adults, the function of an antagonistic pathway like the T6SS might be more pronounced. To test this hypothesis, we obtained publically available metagenomic datasets derived from infant gut microbiomes (Backhed et al., 2015; Kostic et al., 2015; Vatanen et al., 2016; Yassour et al., 2016). We then identified samples that contain B. fragilis but lack GA3-associated structural genes (see Experimental Procedures) in both adult and infant datasets. Such samples indicate the presence of B. fragilis strains unable to intoxicate competitor bacteria using this pathway. We found that infant microbiomes containing B. fragilis are significantly less likely to lack GA3-associated structural genes relative to those of adults (Fisher’s exact test, P < 0.01, 8% infants, 23% adults; n = 276; Figures 3A and S4A–B).
Figure 3. E–I turnover and strain replacement in infant microbiomes.
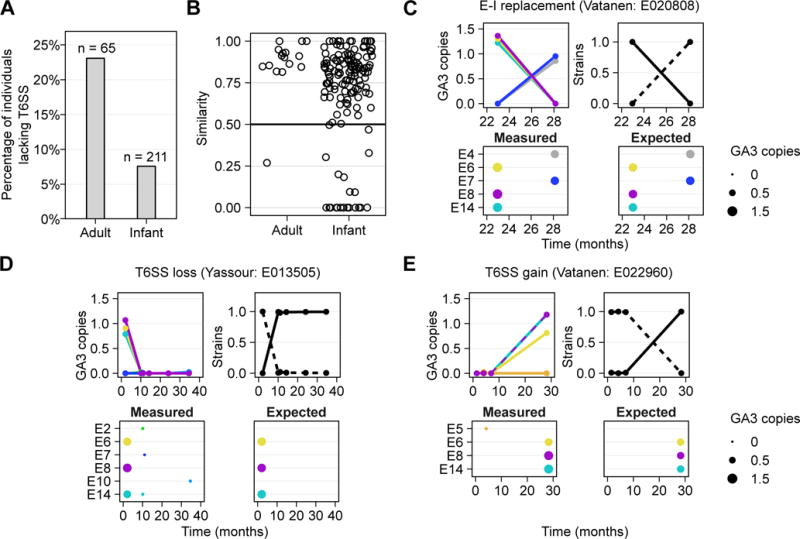
(A) The percentage of individuals of those harboring B. fragilis, that lack the GA3 T6SS across adult and infant datasets. (B) The minimal similarity (measured by the Jaccard similarity coefficient) in GA3 E–I gene content between the first time point and every subsequent time point in adults and infants. (C) Examples of E–I turnover events and corresponding strain replacement events are shown. The plots on the upper and bottom left in each panel illustrate the estimated abundance of GA3 effector genes (measured as copies per B. fragilis genome) over time, with the plot on the upper right illustrating the estimated frequency of inferred strains in these samples. Only samples in which B. fragilis is present are shown. The bottom right plot illustrates the expected abundance of the various effector genes based on the effector genes encoded by reference strains that are phylogenetically close to the inferred strains. See also Figure S4.
This finding suggests that GA3 provides an advantage for B. fragilis in early life; however, the selective pressure underlying this advantage remained unclear. Several independent studies using gnotobiotic mice have shown that the GA3 T6SS can play a major role in the competition between B. fragilis strains in the gut (Chatzidaki-Livanis et al., 2016; Hecht et al., 2016; Wexler et al., 2016). However, B. fragilis is thought to be stable after acquisition from the mother, and inter-strain competition within the human gut microbiome has not been documented for this organism (Faith et al., 2013; Nayfach et al., 2016). Aiming to capture such processes in the developing microbiome, we estimated the abundance of GA3 E–I genes for individual infant samples as we did for adults. In general, the E–I landscape of infants mirrors that of adults, with generally a single genotype present in each sample. Moreover, many of the most prevalent E–I genotypes we observed in adults are also frequent in infants.
Notably, the infant microbiome datasets we analyzed include multiple samples per individual, thereby allowing us to examine the temporal dynamics of B. fragilis and of T6SS genes. Surprisingly, this analysis revealed many instances in which the E–I genotype of an individual changed between samples (Figure 3B). In total, we observed E–I turnover in 22 of the 117 infants for which longitudinal data was available. Such E–I turnover events include instances where one GA3 genotype is replaced by another (Figure 3C), but also gains and losses of the T6SS (Figure 3D–E). To further confirm these E–I dynamics, we used the strain inference method described above. We detected a corresponding strain replacement in 17 of the 22 individuals in which an E–I turnover event was observed (Figures 3C–E and S4C). Moreover, comparing the set of E–I genes detected in each sample to those encoded by the reference strains phylogenetically closest to the inferred strain, we further find overall agreement between observed and expected E–I turnover events. Notably, instances of one strain replaced by another with a similar E–I genotype (Figure S4C; Vatanen:T014827) or of transient co-existence of E–I genotypes (Figure S4C; Backhed:587) were also observed. Examination of the few HMP adult individuals for which data was available from multiple visits revealed one adult in which the E–I genotype similarly changed over time (Figure 3B). Similar analysis of GA1 and GA2 again revealed several individuals with non conserved E–I profiles over time, suggesting a general instability of these subtypes in the developing gut microbiome (Figure S4D).
B. fragilis GA3 T6SS is associated with shifts in community composition
Due to its lower frequency in adult microbiomes compared to those of infants, the GA3 T6SS is absent in many adult samples in which B. fragilis can be detected (23%), offering a unique opportunity to compare the community composition in samples with or without GA3. We hypothesized that such an analysis could identify potential competitors of B. fragilis targeted by the GA3 subtype. To this end, we obtained the taxonomic profile of all HMP samples (see Experimental Procedures) and identified associations between these profiles and the presence of GA3 T6SS structural genes. We first compared overall community composition between samples as measured by the Bray-Curtis distance. We found that samples harboring B. fragilis and GA3 genes (T6SS+) significantly differ in community composition from samples harboring B. fragilis but lacking these genes (T6SS−; P < 0.01 PERMANOVA; n = 51). Examining the abundance of each genera across samples, we further identified four genera whose abundance in T6SS+ versus T6SS− samples significantly differs (Wilcoxon rank sum test; FDR<0.05; Figure 4 and Table S1). Specifically, we found that the abundance of Bacteroides is positively correlated with the presence of GA3, which is consistent with experimental and theoretical work indicating that members of this genus are most likely to compete with B. fragilis for its niche (Trosvik and de Muinck, 2015). Furthermore, the genera Faecalibacterium, Oscillospira and Ruminococcus from the phylum Firmicutes were negatively correlated with GA3. Gram-positive organisms are not targets of the T6SS; therefore, the observed decreases in abundance of these genera in T6SS+ microbiomes are likely to be the indirect result of selection for GA3 occurring in communities with an increased ratio of Bacteroidetes to Firmicutes.
Figure 4. Differentially abundant genera between T6SS+ and T6SS− HMP samples.
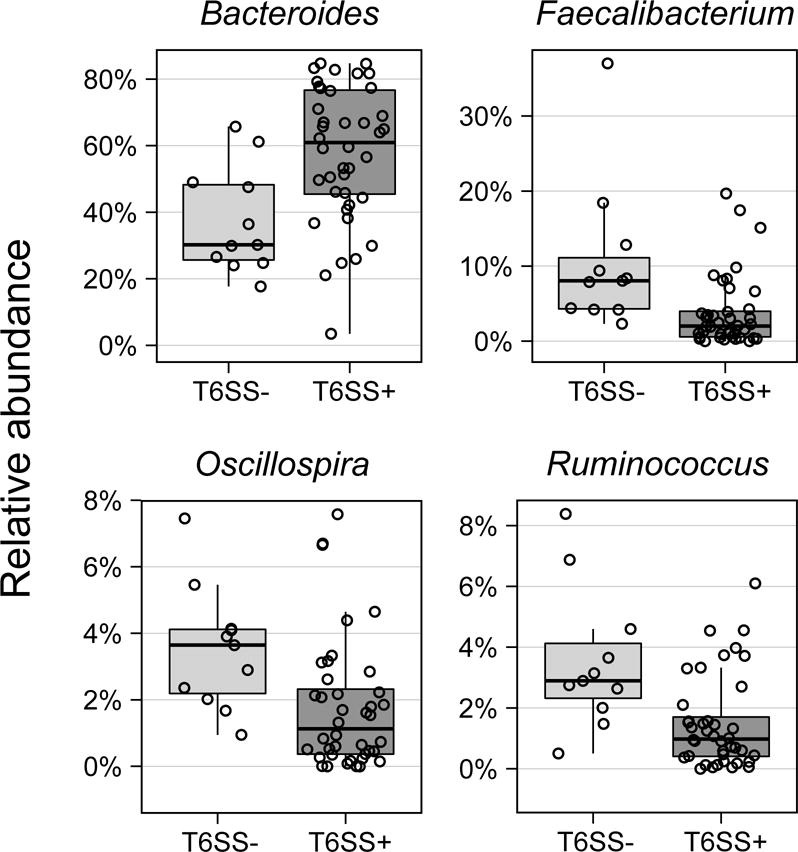
Abundances are based on a 16S rRNA survey and only genera whose abundances are significantly different in T6SS+ vs. T6SS− samples (at FDR < 0.05) are plotted. See also Table S1.
Discussion
Despite the wide distribution of T6S in Gram-negative bacteria, little is known about its role in natural communities. Here, we surveyed human microbiome samples and discovered that these communities are replete with bacteria containing the T6SS. We focused our analyses on genes specific to the order Bacteroidales; thus, our findings are an underestimate of the prevalence and impact of this system in gut communities. Nonetheless, our characterization of T6SS E–I gene distribution in human gut microbiomes suggests that this contact-dependent pathway plays a role in competition and selection at multiple levels.
We observed markedly low diversity of T6SS E–I genes in human microbiome samples. Specifically, a single genotype of GA1 and GA2 E–I genes is found in each microbiome, yet the abundance of these genes does not correlate with that of any one species in the Bacteroides genus. This supports a model in which antagonism via GA1 and GA2 exerts selective pressure for compatibility between Bacteroides spp. in the gut. We postulate that in the case of GA1 and GA2, horizontal transfer facilitates E–I compatibility. Indeed, theory predicts that strongly selected traits are most likely to be horizontally transferred in an environment in which ecological competition is strong, like the gut (Coyte et al., 2015; Niehus et al., 2015). Moreover, Comstock and colleagues found that transfer of GA1 and GA2 can occur between Bacteroidales species within the microbiome of an individual (Coyne et al., 2014).
Similar to GA1 and GA2, we found a single genotype of GA3 in each microbiome. However, GA3 is restricted to B. fragilis and our data are explained by the presence of a single B. fragilis strain in each individual. This is consistent with experimental studies demonstrating a role for GA3-dependent competition between B. fragilis strains (Chatzidaki-Livanis et al., 2016; Hecht et al., 2016; Wexler et al., 2016). Whether the infant microbiome strain replacements and accompanying E–I turnover events we observe are a consequence of GA3 activity cannot be determined from our current data. Nevertheless, these findings in conjunction with our observation that B. fragilis strains lacking the GA3 T6SS are more common in adults suggest that, early in life, B. fragilis strains compete for dominance. The infant microbiome may accordingly represent a particularly dynamic ecosystem in which the GA3 T6SS facilitates B. fragilis strain competition.
We find that strains of B. fragilis lacking GA3 are more commonly found in adults than infants. This could arise either by the replacement of T6SS+ with T6SS− strains, or by the loss of the T6SS system from a previously T6SS+ strain of B. fragilis. This decline in B. fragilis GA3 prevalence in adulthood may reflect a change in its selective advantage. Indeed, there is precedent for the lability of T6S in bacteria undergoing strong shifts in environmental context, such as Burkholderia mallei and Bordetella spp. (Schwarz et al., 2010). It is likely that community effects buffering the B. fragilis niche develop with the maturation of the more stable adult gut community. Stabilization over development appears to render GA3 dispensable in certain contexts, for instance within those microbiomes that contain lower populations of potential B. fragilis competitors and known targets of the GA3 pathway, other Bacteroides spp.
T6SS activity typically results in growth arrest or the lysis of competitor cells (Russell et al., 2014a). Thus, we anticipated that GA3-containing microbiomes would contain lower relative abundance of taxa antagonized by B. fragilis than those lacking GA3. Our counterintuitive finding that GA3 is enriched in microbiomes containing a large Bacteroides population suggests instead that B. fragilis primarily faces selective pressure from closely related species. Importantly, B. fragilis abundance is low compared to the Bacteroides consortium as a whole. It is therefore likely that the observed association between GA3 and community assembly reflects selection on GA3 mediated by community composition, rather than GA3-mediated impact on overall community assembly. While we do not detect other Gram-negative genera whose abundance are specifically lowered in GA3+ microbiomes, we cannot rule out that competition from less common or low abundance genera that fall below our detection limit might also select for the retention of GA3 in adults.
We showed here that a systematic characterization and large-scale computational analysis of metagenomic data can provide a means of linking the presence and abundance of T6 genes to microbial community composition. A caveat of our approach is that it does not account for differences in gene expression that could alter the phenotype elicited by the T6SS. Future work that integrates meta-transcriptomic data could provide a more sensitive measure of T6SS activity. For contact-dependent pathways like the T6SS, metagenomic analyses can provide a unique window into community biogeography. Indeed, Bacteroides spp. are thought to occupy a crowded niche proximal to the gut mucosa and our findings herein provide evidence of extensive cell–cell contacts between species of the genus (Whitaker et al., 2017). The T6SS is one of many antagonistic pathways whose operation is determined by the presence or absence of polymorphic toxins and corresponding antitoxins (Aoki et al., 2010; Whitney et al., 2017; Zhang et al., 2012). Thus, our study offers an analytical framework for more globally deciphering the forces that dictate the establishment and maintenance of bacterial communities.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elhanan Borenstein (elbo@uw.edu).
Experimental Model and Subject Details
All human studies were conducted with the permission of the Yale Human Investigation Committee and informed consent was collected from all volunteers prior to participation. Recruitment of healthy human volunteers, sample collection, anaerobic processing, and −80°C storage of fecal samples in cryoprotectant under anaerobic conditions was previously described (Cullen et al., 2015). Briefly, the sample size of 30 donors (20 male and 10 female) was selected to represent common microbiome variation among humans. The donors were all greater than 18 years of age and were recruited without regard to sex. Samples were not assigned into groups. 16S rRNA gene sequencing was performed and used to identify samples which were most likely to harbor Bacteroides fragilis. The two samples chosen for strain isolation were a 24 year old male and a 54 year old female.
Methods Details
Identifying T6SS effector and immunity genes
We sought to comprehensively catalog Bacteroidales T6SS E–I genes from reference genomes. In Bacteroidales, as in other bacteria, E–I genes are encoded adjacent to the genes for secreted structural proteins Hcp and VgrG. Accordingly, we manually curated genes adjacent to these structural genes across all publically available Bacteroidales genomes. Identified genes exhibited reduced GC content relative to the rest of the T6SS locus or the genome as a whole, and were encoded in bi-cistrons. Putative effectors always lacked characteristic signal peptides, consistent with transport via the T6SS apparatus, while putative immunity genes often encoded proteins with signal peptides. We used structural homology prediction (Phyre) (Kelley et al., 2015) and remote sequence homology search algorithms (Hmmer) (Finn et al., 2015) to predict functions for these genes, identifying many genes with functions associated with known T6SS toxin effectors. As in Coyne et al (Coyne et al., 2016), our list included predicted cell-wall degrading enzymes, lipases, and nucleases as well as putative effector domains fused to either PAAR domains (DUF4280) or Hcp (Table S2). We additionally searched for contigs assembled from HMP metagenomes that contained subtype-specific T6SS structural gene sequences but lacked any E–I gene curated from reference genomes. Gene prediction was performed on these contigs using Glimmer within Geneious R10.1.3 and candidate genes were defined using the criteria as for reference genomes (Delcher et al., 2007; Kearse et al., 2012).
Strain sequencing
Stool samples from four healthy individuals frozen in sterile glycerol (Cullen et al., 2015) were plated onto Bacteroides Bile Esculin or Brucella Blood Agar plates (BD Biosciences), to select for Bacteroidales colonies. Single colonies were picked into Mega Medium (Wu et al., 2015) and grown to stationary phase in anaerobic conditions at 37°C before freezing in 10% glycerol in 96-well plates. PCR was performed directly from the frozen glycerol stocks using primers to amplify the V1–V4 region of the 16S rRNA gene. PCR products were then Sanger sequenced. Sanger sequencing reads were converted to fastq format and NCBI Blast 2.2.31+ was used to align sequences to the SILVA 123 and GreenGenes 2011-1 16S rRNA gene databases in order to identify Bacteroides fragilis. To verify the B. fragilis-positive colonies, a second round of PCR was performed using primers to amplify and sequence the gyrB gene. Two of the four donors were confirmed to have B. fragilis. Twenty confirmed colonies from each B. fragilis-positive donor were then grown to stationary phase in TYG medium under anaerobic conditions at 37°C. Genomic DNA was isolated using the Qiagen DNeasy Blood and Tissue Kit and prepared for whole genome sequencing using the MiSeq V3 Reagent Kit. Sequencing was performed in the Nickerson lab core facility in the UW Department of Genome Sciences. Sequencing reads were mapped to the set of B. fragilis-specific marker genes to generate alignments. Samples under 10× mean alignment read coverage were then discarded. Consensus sequence for each remaining sample was generated using the GATK FastaAlternateReferenceMaker. Subsequently, we constructed multi-alignments for all the samples using MAFFT 7.237, concatenated them, and then inferred a phylogenetic tree using the GTRGAMMAI model from RAxML 8.2.8 (Stamatakis, 2014).
Quantification and Statistical Analysis
Metagenomic and genomic data
Our analysis utilizes short read metagenomic data from several large-scale microbiome datasets. For adult microbiomes we downloaded 147 shotgun samples from HMP (Human Microbiome Project, 2012), and 99 healthy human shotgun samples from MetaHIT (Qin et al., 2010). Since an excessive fraction of human DNA will likely not markedly impact our ability to quantify B. fragilis abundance, HMP samples which failed QC were nonetheless included in our analysis. For infant microbiomes, we downloaded 300 samples from a study of development of the microbiome in the first year of life (Backhed et al., 2015), 769 samples from a study of autoimmune diseases (Vatanen et al., 2016), 237 samples from a study of antibiotic usage (Yassour et al., 2016), and 126 samples from a study of the development of Type 1 Diabetes (Kostic et al., 2015). Several of these datasets include multiple longitudinal samples from the same individuals, which were used for temporal analysis.
We downloaded all available B. fragilis genomes from RefSeq. Sequences from 3 strains were found to be contaminated with contigs matching species other than B. fragilis, and were discarded. A group of 8 strains appeared to be very distant in sequence homology from the rest of the strains, and were also discarded. We additionally downloaded from RefSeq all genomes of other Bacteroides species for which at least 5 strain genomes were available.
Identifying species-specific marker genes
We compiled a list of marker genes that could be used for strain-level inference. Our marker gene approach is similar to that used by MetaPhlAn (Truong et al., 2015), but relies on a more stringent selection of marker genes, supporting a more robust comparison at the strain level. Specifically, for our analysis, we identified a subset of the MetaPhlAn marker genes that are found in the genome of every sequenced strain in a single copy. To this end, for each of the MetaPhlAn marker genes associated with a given species, we used BLASTn to find every homolog (>60% identity) in all the strains of that species. We used Usearch (Edgar, 2010) to cluster this larger set of genes into groups with >90% identity and if a cluster with exactly one gene in each strain could be found, the marker gene was included in our list.
Estimating gene abundance in microbiomes
We aligned shotgun reads single end using Bowtie2 (using parameters –a –N 1) to the set of genes of interest. Alignments with less than 97% identity, a quality score below 20, or multiple hits were discarded. To quantify the abundance of each gene, the number of reads aligned to this gene was normalized by the length of the gene and the total number of reads in the sample.
The average abundance of species-specific marker genes identified above was used as a proxy for the abundance of that species in the sample. We defined samples as having B. fragilis present if at least 100 reads could be aligned to B. fragilis-specific marker genes. When characterizing strain replacement, for which higher coverage of B. fragilis genes is required, we used instead a threshold of 500 reads. Because GA1 and GA2 are not restricted to a single species, and because Bacteroidales composition can vary dramatically, we considered samples with 100 reads mapping to the GA1 and GA2 E–I genes. GA1 and GA2 effector sequences contain repeats in the Rhs repeats which could interfere with read mapping, and therefore, we only used the toxin region (450bp from the C terminus) when quantifying GA1 or GA2 effector gene abundance. For GA3 we defined vgrG, TssP, TssN, TssK, TssB, TssO, TssG, TssF, and TssR as the Type VI structural genes, for GA1 we used TagB and for GA2 we used TagA. We selected these structural genes because based on reference genomes it was clear that a 97% identity similarity would unambiguously distinguish between subtypes.
Nucleotide diversity calculation
To estimate nucleotide diversity across species-specific marker genes, we again aligned all short reads in each sample to these genes. The obtained alignments were converted into a pileup using mpileup from samtools (parameters –excl-flags UNMAP,QCFAIL,DUP -A -q0 -C0 –B), and finally into an allele count matrix. The first and last 10 bases of each gene were discarded from the allele count matrix as we found they contained many poor quality alignments. We focused on high-coverage loci only, ignoring all loci where the coverage was less than 5×. If the number of high coverage sites was <10% of the total length of the sequence, the sample was excluded from further consideration. Variable sites were defined as those having at least 2 counts of the minor allele. Nucleotide diversity was then calculated at these variant sites according to:
Where n is the total length of the genes, i corresponds to the variable sites, and p and q correspond to the frequency of the major and minor allele at site i.
Inferring B. fragilis strains
To infer strain diversity in each sample, we used StrainFinder (https://github.com/cssmillie/fmt), a previously introduced method for inferring the most likely set of strains in metagenomic samples based on nucleotide variants. To this end, we again aligned the short read in each sample to the set of B. fragilis-specific marker genes identified above, and converted the alignment to a count matrix describing the number of counts of each nucleotide at every position along the genes. As when calculating nucleotide diversity, we discarded the first and last 10 bases and only considered sites with at least 5 counts. We also discarded samples where the high coverage sites were less than 10% of the total length as they resulted in poor quality trees. When running StrainFinder we reduced the data to only those sites with population variability, defined, as above, as sites with at least 2 counts of the minor allele. As noted above, we only considered samples with a sufficient coverage on the B. fragilis marker genes to enable robust strain inference.
StrainFinder determines the relative strain abundance and genotypes at variable sites by considering the likelihood of the observed allele counts and using an expectation maximization approach. An optimal number of strains between 1 and 10 was determined using AIC. For each run of StrainFinder we used 5 independent runs of 200 expectation maximization iterations and selected the best fit; these parameters yield reproducible inference of strains. When analyzing temporal data with StrainFinder, we combined allele counts from all samples of an individual into a single 3-dimensional matrix. Using the genotypes from StrainFinder we then reconstructed strain-specific versions of each marker gene and then created subsequences consisting of only the high coverage sites we used in the analysis. Inferred strains were then further examined using a phylogenetic analysis as described below.
Phylogenetic analysis
A phylogenetic tree of the reference B. fragilis strains was constructed based on their species-specific marker genes. Specifically, we aligned the strains’ versions of each marker gene using MAFFT, concatenated the alignments of all genes, and then constructed a tree using the GTRGAMMAI model from RAxML (Stamatakis, 2014), as has been done previously (Wexler et al., 2016).
To determine whether the strains inferred by StrainFinder are monophlyletic, we combined the sequences from the inferred strains (as determined by StrainFinder), with the sequences from the available reference B. fragilis genomes, and recreated the strain phylogeny using the same method as described above. We defined inferred strains as monophyletic if their common ancestor does not have any descendent outside the set of inferred strains or if the distance was less than 0.001 substitutions per site.
Predicting E–I genes of inferred B. fragilis
We predicted the E–I gene content of an inferred B. fragilis strain by examining the E–I content in the genome of its nearest neighbors on a phylogenetic tree that contains the inferred strains from a given sample and the reference strains (as described above). Specifically, for every strain identified from StrainFinder, we identified the most recent ancestor that have both the inferred strain and at least one reference strain as descendants. We then used the average E–I content of all reference strains descendant from this ancestor as the predicted E–I content of the inferred strain. To then estimate the predicted E–I content in the sample, we combined the predicted E–I content of each inferred strain weighted by their relative abundance. To determine the confidence of the predicted E–I content we determined the average phylogenetic distance of these reference strains to the ancestor identified above.
Identifying T6SS in microbiome samples
For every sample, we estimated the number of reads expected to map to the B. fragilis GA3 T6SS structural genes based on the number of reads mapped to B. fragilis-specific marker genes in that sample and the ratio between the total length of B. fragilis-specific marker genes and B. fragilis T6SS structural genes. We define samples to be T6SS+ if B. fragilis was present (as defined above) and the number of reads mapped to T6SS structural genes was more than 10% of the expected number (and see Figure S4A). We define samples to be T6SS− if B. fragilis was present and the number of reads mapped to T6SS structural genes was less than 10% of the expected number.
Community composition analysis
To obtained independent estimate community composition in each sample, we downloaded the v35 16S OTU abundance table for human gut microbiomes from HMP (ftp://public-ftp.hmpdacc.org/HMQCP/otu_table_psn_v35.txt.gz), summed the counts from all OTUs in the same genus, and calculated the relative abundance of each genus. Importantly, because 16S sequencing depth is independent from the depth of shotgun samples used to determine T6SS+ vs. T6SS− classification, using these 16S-based data allows us to compare T6SS presence with community taxonomic profiles without potential coverage-related biases. Samples were classified into T6SS+ and T6SS− as described above. GA1 and GA2 lack uniquely identifying structural genes so we defined T6SS+ vs. T6SS− as samples with vs. without 100 counts mapping to the GA1 or GA2 E–I genes respectively. The distance between samples was defined by the Bray-Curtis distance at the genus level, and significance of separation between T6SS+ and T6SS− samples was evaluated using PERMANOVA. For the subset of genera whose average abundance across samples was > 0.1%, we used a Wilcoxon rank sum test to compare their abundance in T6SS+ vs. T6SS− samples using a 5% FDR.
Data and Software Availability
All sequences were deposited into NCBI SRA under BioProject ID PRJNA375094.
Supplementary Material
Comprehensive metagenomic mapping of Bacteroidales T6SS genes in the gut microbiome
Evidence for selection for T6SS compatibility at the species and strain levels
Bacteroides fragilis strains compete for dominance in the infant microbiome
B. fragilis T6SS provides a selective advantage in Bacteroides-rich microbiomes
Acknowledgments
We thank Eric Alm and Chris Smillie for sharing StrainFinder code and for their support in running it, UW Genome Sciences ITS for high-performance computing resources, S. Brook Peterson for careful review of the manuscript, and Borenstein and Mougous lab members for helpful discussions. This work was supported by National Institutes of Health grants GM118159 (ALG), AI080609 (JDM), and New Innovator Award DP2AT00780201 (to EB), the Pew Scholars Program (ALG). AJV was supported by a postdoctoral fellowship from the Natural Sciences and Engineering Research Council of Canada. BDR was supported by a Simons Foundation-sponsored Life Sciences Research Foundation postdoctoral fellowship. JDM and ALG hold Investigator in the Pathogenesis of Infectious Disease Awards from the Burroughs Wellcome Fund (BWF 1010010 and BWF 1014860) and JDM is an HHMI investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
BDR and JDM conceived the study. AJV, BDR, JDM, and EB designed the study. AJV conducted the computational analysis. BDR, MCR, YB, and ALG conducted experimental work. AJV, BDR, JDM, and EB wrote the paper.
References
- Anderson MC, Vonaesch P, Saffarian A, Marteyn BS, Sansonetti PJ. Shigella sonnei Encodes a Functional T6SS Used for Interbacterial Competition and Niche Occupancy. Cell Host Microbe. 2017;21:769–776 e763. doi: 10.1016/j.chom.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Benz J, Meinhart A. Antibacterial effector/immunity systems: it’s just the tip of the iceberg. Curr Opin Microbiol. 2014;17:1–10. doi: 10.1016/j.mib.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Bulgarelli D, Garrido-Oter R, Munch PC, Weiman A, Droge J, Pan Y, McHardy AC, Schulze-Lefert P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015;17:392–403. doi: 10.1016/j.chom.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki-Livanis M, Geva-Zatorsky N, Comstock LE. Bacteroides fragilis type VI secretion systems use novel effector and immunity proteins to antagonize human gut Bacteroidales species. Proc Natl Acad Sci U S A. 2016;113:3627–3632. doi: 10.1073/pnas.1522510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne MJ, Roelofs KG, Comstock LE. Type VI secretion systems of human gut Bacteroidales segregate into three genetic architectures, two of which are contained on mobile genetic elements. BMC Genomics. 2016;17:58. doi: 10.1186/s12864-016-2377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, Comstock LE. Evidence of extensive DNA transfer between bacteroidales species within the human gut. MBio. 2014;5:e01305–01314. doi: 10.1128/mBio.01305-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: Networks, competition, and stability. Science. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science. 2015;347:170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Egan F, Reen FJ, O’Gara F. Tle distribution and diversity in metagenomic datasets reveal niche specialization. Environ Microbiol Rep. 2015;7:194–203. doi: 10.1111/1758-2229.12222. [DOI] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–38. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR, Bubeck Wardenburg J. Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep. 2016;17:1281–1291. doi: 10.15252/embr.201642282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe. 2010;7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, Peet A, Tillmann V, Poho P, Mattila I, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17:260–273. doi: 10.1016/j.chom.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci U S A. 2013;110:12804–12809. doi: 10.1073/pnas.1300926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Research. 2016 doi: 10.1101/gr.201863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehus R, Mitri S, Fletcher AG, Foster KR. Migration and horizontal gene transfer divide microbial genomes into multiple niches. Nat Commun. 2015;6:8924. doi: 10.1038/ncomms9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser JI, Bohannan BJ, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP, Green JL, Green LE, Killham K, Lennon JJ, et al. The role of ecological theory in microbial ecology. Nat Rev Microbiol. 2007;5:384–392. doi: 10.1038/nrmicro1643. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533:255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley MA, Wertz JE. Bacteriocins: evolution, ecology, and application. Annu Rev Microbiol. 2002;56:117–137. doi: 10.1146/annurev.micro.56.012302.161024. [DOI] [PubMed] [Google Scholar]
- Russell AB, Peterson SB, Mougous JD. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol. 2014a;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, et al. A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe. 2014b;16:227–236. doi: 10.1016/j.chom.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sana TG, Flaugnatti N, Lugo KA, Lam LH, Jacobson A, Baylot V, Durand E, Journet L, Cascales E, Monack DM. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc Natl Acad Sci U S A. 2016;113:E5044–5051. doi: 10.1073/pnas.1608858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S, Hood RD, Mougous JD. What is type VI secretion doing in all those bugs? Trends Microbiol. 2010;18:531–537. doi: 10.1016/j.tim.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosvik P, de Muinck EJ. Ecology of bacteria in the human gastrointestinal tract–identification of keystone and foundation taxa. Microbiome. 2015;3:44. doi: 10.1186/s40168-015-0107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hamalainen AM, et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell. 2016;165:842–853. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- Wexler AG, Bao Y, Whitney JC, Bobay LM, Xavier JB, Schofield WB, Barry NA, Russell AB, Tran BQ, Goo YA, et al. Human symbionts inject and neutralize antibacterial toxins to persist in the gut. Proc Natl Acad Sci U S A. 2016;113:3639–3644. doi: 10.1073/pnas.1525637113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Shepherd ES, Sonnenburg JL. Tunable Expression Tools Enable Single-Cell Strain Distinction in the Gut Microbiome. Cell. 2017;169:538–546 e512. doi: 10.1016/j.cell.2017.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JC, Peterson SB, Kim J, Pazos M, Verster AJ, Radey MC, Kulasekara HD, Ching MQ, Bullen NP, Bryant D, et al. A broadly distributed toxin family mediates contact-dependent antagonism between gram-positive bacteria. Elife. 2017;6 doi: 10.7554/eLife.26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, et al. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science. 2015;350:aac5992. doi: 10.1126/science.aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Vatanen T, Siljander H, Hamalainen AM, Harkonen T, Ryhanen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8:343ra381. doi: 10.1126/scitranslmed.aad0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.