

SUMMARY
Aging is the major risk factor for many human diseases. In vitro studies have demonstrated that cellular reprogramming to pluripotency reverses cellular age, but alteration of the aging process through reprogramming has not been directly demonstrated in vivo. Here, we report that partial reprogramming by short-term cyclic expression of Oct4, Sox2, Klf4, and c-Myc (OSKM) ameliorates cellular and physiological hallmarks of aging and prolongs lifespan in a mouse model of premature aging. Similarly, expression of OSKM in vivo improves recovery from metabolic disease and muscle injury in older wild-type mice. The amelioration of age-associated phenotypes by epigenetic remodeling during cellular reprogramming highlights the role of epigenetic dysregulation as a driver of mammalian aging. Establishing in vivo platforms to modulate age-associated epigenetic marks may provide further insights into the biology of aging.
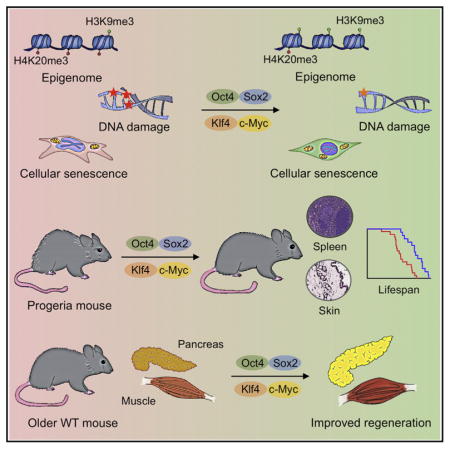
In Brief
Cellular reprogramming by transient expression of Yamanaka factors ameliorates age-associated symptoms, prolongs lifespan in progeroid mice, and improves tissue homeostasis in older mice.
INTRODUCTION
Embryonic development occurs as a unidirectional progression from a single-cell zygote to an adult organism. During embryogenesis and early stages of life, cells undergo a spatiotemporally orchestrated differentiation process, leading to the generation of all of the cell types that comprise an adult organism. These events take place within a stable environment that minimizes molecular and cellular damage. As an organism ages, however, there is a continuous and progressive decline in the mechanisms responsible for minimizing cellular damage. This eventually results in an organism’s inability to maintain homeostasis (López-Otín et al., 2013; 2016).
The last decade of scientific research has dramatically improved our understanding of the aging process (Johnson et al., 2013; Kenyon, 2010; Riera et al., 2016). The notion that cells undergo a unidirectional differentiation process during development was proved wrong by the experimental demonstration that a terminally differentiated cell can be reprogrammed into a pluripotent embryonic-like state (Gurdon, 1962; Takahashi and Yamanaka, 2006). Cellular reprogramming to pluripotency by forced expression of the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc [OSKM]) occurs through the global remodeling of epigenetic marks (Buganim et al., 2012, 2013; Hansson et al., 2012; Polo et al., 2012). Importantly, many of the epigenetic marks that are remodeled during reprogramming (e.g., DNA methylation, post-translational modification of histones, and chromatin remodeling) are dysregulated during aging (Benayoun et al., 2015; Liu et al., 2013b; Pollina and Brunet, 2011). In fact, epigenetic dysregulation has emerged as a key hallmark of the aging process (Sen et al., 2016). Several groups, including ours, have observed an amelioration of age-associated cellular phenotypes during in vitro cellular reprogramming (Lapasset et al., 2011; Liu et al., 2011; Mahmoudi and Brunet, 2012; Rando and Chang, 2012). Reprogramming of cells from centenarians or patients with Hutchinson-Gilford progeria syndrome, (HGPS) a disorder characterized by premature aging, resets telomere size, gene expression profiles, and levels of oxidative stress, resulting in the generation of rejuvenated cells (Lapasset et al., 2011; Liu et al., 2011; Zhang et al., 2011).
Although these in vitro studies have been informative, the physiological complexity of the aging process demands an in vivo approach to better understand how reprogramming may affect cellular and organismal aging. Breakthrough studies led by the Serrano and Yamada groups have shown that cellular reprogramming to pluripotency, although associated with tumor development (e.g., teratoma formation), can be achieved in vivo in mice by the forced expression of the Yamanaka factors (Abad et al., 2013; Ohnishi et al., 2014). In addition, we and other groups have demonstrated that partial reprogramming in vitro by transient expression of OSKM can induce a dedifferentiated progenitor-like state (Kurian et al., 2013; Thier et al., 2012). Together, these observations suggest that cellular reprogramming may be used to promote tissue regeneration and led us to hypothesize that in vivo partial reprogramming could slow or reverse the aging process and extend organismal lifespan. Here, we report that cyclic in vivo induction of OSKM in a mouse model of premature aging improves age-associated phenotypes and extends lifespan. In addition, we demonstrate the amelioration of cellular phenotypes associated with aging by short-term induction of the Yamanaka factors in mouse and human cells. Finally, we show that short-term expression of OSKM alleviates pancreatic and muscle injury in older wild-type (WT) mice. This in vivo platform for the reprogramming of epigenetic marks may be used to better understand physiological aging, as well as the role of epigenetics during mammalian aging.
RESULTS
Partial Reprogramming Ameliorates Cellular Phenotypes Associated with Aging
Based on our experience on the reprogramming of cells from patients with HGPS to study aging in vitro (Liu et al., 2011), we decided to use a premature aging mouse model to test the hypothesis that in vivo reprogramming can alter organismal aging. We chose the premature aging mouse model that carries a G609G mutation in the gene Lmna (LAKI). This mutation leads to the accumulation of a truncated form of lamin A (called progerin), which is responsible for human HGPS (Osorio et al., 2011; Scaffidi and Misteli, 2005). LAKI mice are short lived and exhibit accelerated onset of many age-associated physiological phenotypes, including weight loss and alterations associated with aging in multiple organs (e.g., skin, kidney, and spleen) (Osorio et al., 2011). To enable inducible expression of the Yamanaka factors upon doxycycline treatment, LAKI mice were crossed to mice carrying an OSKM polycystronic cassette (4F) and a rtTA trans-activator (Carey et al., 2010), thereby generating LAKI 4F mice. The fact that reprogramming proceeds in a stepwise manner allows for the induction of partial reprogramming without the complete loss of cellular identity by short exposure to the Yamanaka factors (Kurian et al., 2013). Partial reprogramming could potentially erase or delay the accumulation of aging phenotypes without leading to tumor formation. First, to determine whether partial reprogramming could ameliorate aging hallmarks in vitro, we isolated LAKI 4F tail tip fibroblasts (TTFs) and performed short-term induction of OSKM by doxycycline treatment for 2 or 4 days. Expression of OSKM was confirmed by qPCR and immunofluorescence (Figures 1A and S1A). Short-term induction of OSKM in TTFs did not lead to loss of cellular identity, as indicated by expression of the fibroblast-associated marker Thy1 and the absence of markers for intermediate cellular reprogramming, such as SSEA1 (Figure S1B). Moreover, induction of OSKM did not result in the expression of Nanog, a marker of pluripotency, even after 12 days of induction (Figure S1C), indicating that complete reprogramming to pluripotency was not attained. Interestingly, the number, intensity, and volume of foci for histone γ-H2AX, a marker of nuclear DNA double-strand breaks associated with aging (López-Otín et al., 2013), were significantly reduced by short-term induction of OSKM in LAKI 4F cells compared to untreated cells (Figures 1B and S1D). Similarly, levels of p53 binding protein 1 (53BP1), which participates in the DNA damage response, were reduced as a consequence of short-term induction of OSKM (Figure S1E). Furthermore, short-term induction of OSKM in LAKI 4F cells downregulated the expression of age-related stress response genes in the p53 tumor suppressor pathway, including p16INK4a, p21CIP1, Atf3, and Gadd45B, as well as the senescence-associated metalloprotease MMP13 and interleukin-6 (Figure 1C). Additionally, senescence-associated β-galactosidase activity was reduced in LAKI 4F cells by short-term induction of OSKM (Figure S1F). In agreement with the metabolic changes and improvements in mitochondrial function observed during cellular reprogramming, short-term induction of OSKM significantly reduced the production of mitochondrial reactive oxygen species (ROS), which are responsible for oxidative damage and considered as one of the drivers of physiological aging (Figure S1G). Finally, short-term induction of OSKM in LAKI 4F cells restored levels of H3K9me3 and H4K20me3, two epigenetic modifications involved in the maintenance of heterochromatin, which are downregulated and upregulated respectively during aging (Benayoun et al., 2015; Liu et al., 2013a) (Figures 1D and 1E).
Figure 1. Amelioration of Cellular Markers of Aging by Short-Term In Vitro Induction of Oct4, Sox2, Klf4, and c-Myc.

(A) Immunofluorescence of Oct4 and Sox2 in LAKI 4F TTFs. Scale bar, 10 μm.
(B) Immunofluorescence and quantification of γH2AX foci in LAKI 4F TTFs. Scale bar, 10 μm. **** p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(C) qPCR analysis of stress response genes in the p53 pathway, senescence-associated metalloprotease MMP13 and interleukin-6 in LAKI 4F TTFs. **p < 0.01, ***p < 0.001, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(D and E) Immunofluorescence of H3K9me3 and H4K20me3 in LAKI 4F TTFs. Scale bar, 10 μm. **p < 0.005 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(F) Immunofluorescence of Lamin A/C and quantification of nuclear abnormality in LAKI 4F TTFs. Arrows indicate blebbing in the nuclear envelope in Oct4 negative cells. Scale bar, 10 μm. **p < 0.005 according to one-way ANOVA with Bonferroni correction.
Data are presented as mean ± SEM. See also Figures S1 and S2.
Nuclear lamins maintain proper architecture of the nuclear envelope (Liu et al., 2005). Defects in the nuclear envelope resulting from the accumulation of progerin are the main drivers of premature aging in HGPS. Similar nuclear envelope abnormalities have been reported during physiological aging (Liu et al., 2005; Loópez-Otín et al., 2013). Importantly, short-term induction of OSKM in LAKI 4F cells significantly improved nuclear envelope architecture compared to untreated cells (Figure 1F). Lastly, in order to investigate the maintenance of the ameliorated aging-associated phenotypes after OSKM induction, we analyzed the level of DNA damage, H3K9me3, and nuclear envelope abnormalities 4 and 8 days after termination of doxycycline treatment. These analyses revealed a slow re-acquisition of age-associated phenotypes in LAKI 4F TTFs compared to untreated control cells (Figures 2A and 2B). Importantly, short-term re-induction of OSKM for an additional 4 days was able to revert the re-accumulation of aging-associated phenotypes (Figures 2A and 2B). These results suggest that cyclic induction of OSKM following an “on and off” scheme may have the capacity to prevent or reset the accumulation of age-associated phenotypes.
Figure 2. Epigenetic Remodeling during Short-Term In Vitro Induction of Oct4, Sox2, Klf4, and c-Myc Reverses Cellular Markers of Aging.

(A) Immunofluorescence of γH2AX and Lamin A/C and quantification of γH2AX and nuclear abnormalities in LAKI 4F TTFs subjected to cyclic expression of OSKM for the indicated days. Scale bar, 10 μm. *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction compared to control.
(B) Immunofluorescence and quantification of H3K9me3 in LAKI 4F TTFs subjected to cyclic expression of OSKM for the indicated days. Scale bar, 10 μm. ***p < 0.0005 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(C) Immunofluorescence and quantification of H3K9me3 in LAKI 4F TTFs subjected to short-term expression of OSKM for 12 hr and 24 hr. Scale bar, 10 μm. **p < 0.005 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(D) Immunofluorescence of γH2AX and Lamin A/C, and quantification of γH2AX and nuclear abnormalities in LAKI 4F TTFs subjected to short-term expression of OSKM for 12 hr and 24 hr. Scale bar, 10 μm.
(E) Immunofluorescence and quantification of H3K9me3 in LAKI 4F TTFs subjected to short-term expression of OSKM in the presence of the H3K9 methyl-transferase inhibitor chaetocin. Scale bar, 10 μm. **p < 0.01 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(F) Immunofluorescence and quantification of γH2AX foci in LAKI 4F TTFs subjected to short-term expression of OSKM in the presence of the H3K9 methyl-transferase inhibitor chaetocin. Scale bar, 10 μm. ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
Data are presented as mean ± SEM.
To gain further insight into the mechanism behind our observations, we first decided to evaluate the effect of short-term OSKM induction in the expression of lamin A/C and the accumulation of progerin. Although we have previously shown that reprogramming HGPS cells to pluripotency causes a switch in nuclear lamin composition (Abad et al., 2013; Liu et al., 2011; Ohnishi et al., 2014), the improvement of aging phenotypes in LAKI 4F cells after short-term induction of OSKM did not depend on changes in the expression of nuclear lamin A/C or B1 or on reduced accumulation of progerin (Figures S2A and S2B). Moreover, selective induction of programmed cell death upon short-term OSKM induction in cells with high levels of age-associated phenotypes was not responsible for the amelioration of aging hallmarks in LAKI 4F cells (Figure S2C). Furthermore, none of the cellular aging hallmarks analyzed were ameliorated in LAKI cells not carrying an OSKM polycystronic cassette, indicating that OSKM expression was responsible for the rescue of age-associated phenotypes (Figures S2D–S2H). In addition, infection of LAKI cells with single reprogramming factors (i.e., Oct4, Sox2, or c-Myc alone) was insufficient to improve nuclear envelope architecture (Figure S2I), suggesting that cellular reprogramming, but not the induction of proliferation alone, restored the aberrant nuclear envelope in these cells. Finally, based on the epigenetic nature of the reprogramming process, we decided to confirm the role of epigenetic remodeling as a driver of the amelioration of age-associated phenotypes by short-term induction of OSKM. For this purpose, we carried out a detailed time-course analysis of the amelioration of DNA damage and nuclear envelope abnormalities in LAKI 4F TTFs, as well as the restoration of H3K9me3. Analysis of LAKI 4F TTFs at 12 and 24 hr after induction of OSKM revealed that restoration of H3K9me3 precedes the amelioration of DNA damage and nuclear envelope defects, suggesting that epigenetic remodeling during cellular reprogramming acts as a driver of the improvement of age-associated phenotypes (Figures 2C and 2D). In addition, in order to further confirm this hypothesis, we analyzed the amelioration of aging phenotypes in LAKI 4F TTFs upon short-term induction of OSKM in the presence of 10nM chaetocin, an H3K9 methyltransferase inhibitor. Importantly, induction of OSKM, in the presence of 10 nM chaetocin, prevented the restoration of H3K9me3 and abolished the amelioration of DNA damage and defects in nuclear envelope induced by cellular reprogramming (Figures 2E and 2F). These observations reinforce the role of epigenetic remodeling during cellular reprogramming as a driver of the improvement of age-associated phenotypes by short-term expression of OSKM.
Collectively, these results demonstrate that short-term in vitro induction of OSKM in cells derived from a premature aging mouse model ameliorates multiple age-associated hallmarks observed during physiological aging, including the accumulation of DNA damage, cellular senescence, epigenetic dysregulation, and nuclear envelope defects. Thus, epigenetic remodeling induced by partial reprogramming may potentially modify the aging process.
Cyclic In Vivo Induction of Reprogramming Factors Does Not Lead to Tumor Formation
Previous studies have shown that the in vivo induction of OSKM leads to teratoma formation and the development of cancer (Abad et al., 2013; Ohnishi et al., 2014). To establish a safe protocol for the in vivo induction of OSKM that does not result in teratomas or cancer, we tested multiple induction protocols, varying the dose of OSKM expression by modifying transgene copy number or the length of induction. Our initial experiments revealed that in vitro induction of OSKM in cells from multiple tissues of mice carrying a single copy of the OSKM polycystronic cassette and the rtTA trans-activator (4F) failed to generate induced pluripotent stem cells (iPSCs) compared to cells carrying two copies (data not shown). For this reason, and to prevent the development of teratomas in vivo, we focused on using 4F mice carrying a single copy of the OSKM cassette.
Continuous induction of OSKM in 4F mice by administration of doxycycline in drinking water resulted in significant weight loss and high mortality after 4 days (Figures 3A and 3B). This likely resulted from the dedifferentiation of cells in vital organs and subsequent loss of organ function. High levels of mortality were avoided by switching to a cyclic OSKM induction protocol, consisting of 2 days of doxycycline administration followed by 5 days of doxycycline withdrawal (Figure 3C). Analysis of blood samples from 4F mice following 2 days of doxycycline administration confirmed the induction of OSKM (Figure 3D). Cyclic induction of OSKM using this protocol did not result in weight loss or mortality of 4F mice compared to WT controls, even after 35 cycles of doxycycline administration (Figures S3A and S3B). We detected no signs of dysplasia, cancer development, or induction of pluripotency in any organs analyzed from these 4F mice (Figures S3C and S3D). In contrast, cyclic induction of OSKM for 8 weeks in mice carrying two copies of the OSKM and rtTA cassettes led to increased cell proliferation (Figure S3E) and teratoma formation in the liver, kidney, and pancreas (Figures 3E and 3F), thereby confirming the capacity of this protocol to induce in vivo cellular reprogramming. These experiments demonstrate that the negative consequences of in vivo reprogramming can be avoided by reducing levels of OSKM induction via the cyclic administration of doxycycline. In addition, based on our previous in vitro observations regarding the amelioration of aging phenotypes by cyclic induction of OSKM, we hypothesized that cyclic in vivo reprogramming may confer equivalent benefits at the organismal level.
Figure 3. Establishment of In Vivo Induction Protocol in 4F Mice.

(A) Body weight of 4F mice upon continuous administration of doxycycline (−Dox n = 11; +Dox n = 26).
(B) Survival of 4F mice upon continuous administration of doxycycline (−Dox n = 11; +Dox n = 26). ***p < 0.0005 according to log-rank (Mantel-Cox) test.
(C) Schematic representation of cyclic doxycy-cline administration protocol.
(D) qPCR analysis of Oct4, Sox2, Klf4, and c-Myc in blood samples of 4F mice after 2 days of doxycycline administration. **p < 0.005, ***p < 0.0005, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(E) Teratomas (arrows) in 4F mice carrying two copies of OSKM and rtTA cassette after 8 weeks of cyclic doxycycline administration.
(F) Histological analysis of teratoma with ectoderm, mesoderm, and endoderm. Scale bar, 50 μm.
Data are presented as mean ± SEM. See also Figure S3.
In Vivo Reprogramming Ameliorates Organismal Phenotypes Associated with Premature Aging
To test whether partial in vivo reprogramming could improve age-related phenotypes and extend organismal lifespan, we tested the effect of cyclic OSKM induction in LAKI 4F mice beginning at 8 weeks of age. Although in vivo OSKM induction did not significantly prevent the progressive loss of body weight characteristic of LAKI 4F mice (Figure 4A), we observed a dramatic increase in median and maximal lifespan in animals subjected to partial in vivo reprogramming (Figure 4B). On the other hand, doxycycline treatment in LAKI mice not carrying the 4F cassette did not lead to any improvement in the lifespan compared to untreated controls (Figure 4B). Significant improvements in external appearance, including a reduction in spine curvature, were observed in LAKI 4F mice subjected to cyclic OSKM induction (Figures 4C and S4A). Detailed necropsy analyses performed at 14 weeks of age after 6 weeks of cyclic induction of OSKM revealed gross improvement in the appearance of the gastrointestinal tract in doxycycline-treated LAKI 4F mice compared to untreated mice (Figure 4D). Age-associated histological changes are normally observed during physiological aging in multiple organs, including the skin, spleen, kidneys, and stomach (Cesta, 2006; Khanna et al., 1988; Kurban and Bhawan, 1990; Zhou et al., 2008). Importantly, many of these histological changes were improved in 14-week-old LAKI 4F mice subjected to cyclic induction of OSKM during 6 weeks compared to untreated mice (Figure 4E). Doxycycline-treated LAKI 4F mice showed an increased epidermal and dermal thickness and decreased keratinization of the skin compared to untreated controls (Figure 4E). Macroscopic involution of the spleen and lymphoid depletion of the white pulp, visualized by the reduced size of germinal centers, was significantly rescued in LAKI 4F mice after cyclic induction of OSKM (Figures 4E and S4B). Moreover, we observed a decrease in tubular atrophy and interstitial volume in kidneys of doxycycline-treated LAKI 4F mice compared to untreated mice (Figure 4E). Lastly, age-associated loss of parietal cells and thinning of gastric epithelium in the stomach were also rescued by cyclic induction of OSKM (Figure 4E).
Figure 4. Extension of Lifespan and Prevention of Age-Associated Phenotypes by In Vivo Induction of Oct4, Sox2, Klf4, and c-Myc.

(A) Body weight of LAKI and LAKI 4F mice upon cyclic doxycycline administration. LAKI (−Dox n = 20; +Dox n = 13) and LAKI 4F (−Dox n = 18; +Dox n = 15).
(B) Survival of LAKI and LAKI 4F mice upon cyclic doxycycline administration. LAKI (−Dox n = 20; +Dox n = 13) and LAKI 4F (−Dox n = 18; +Dox n = 15). ****p < 0.0001 according to log-rank (Mantel-Cox) test.
(C) Representative photograph of 16-week-old LAKI 4F mice upon cyclic doxycycline administration.
(D) Necropsy analysis of 14-week-old LAKI 4F mice upon cyclic doxycycline administration.
(E) Histological analysis of indicated organs of 14-week-old LAKI 4F mice upon cyclic doxycycline administration. Arrows depict decreased epidermal thickness and increased keratinization, small lymphoid nodules in the splenic white pulp, tubular atrophy in the kidney, and loss of parietal cells in the stomach of untreated LAKI 4F mice. Scale bar, 50 μm (skin and kidney) and 100 μm (spleen and stomach). *p < 0.05, ***p < 0.001, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(F) Histology of aortic arch of LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 50 μm. *p < 0.05 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(G) ECG analysis in LAKI 4F mice upon cyclic doxycycline administration (−Dox n = 4; +Dox n = 4). Heart rate represented as beats per minute (bpm). *p < 0.05, **p < 0.001, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
Data are presented as mean ± SEM. See also Figure S4.
Cardiovascular alterations observed in patients with HGPS and during normal aging are also manifested in LAKI mice and are thought to be a major contributor to their premature death (Osorio et al., 2011; Stehbens et al., 1999). Importantly, cyclic induction of OSKM in LAKI 4F mice partially rescued the degeneration of vascular smooth muscle cells compared to untreated mice, as indicated by an increase in the number of nuclei in the medial layer of the aortic arch (Figure 4F). In addition, electrocardiographic (ECG) analysis of LAKI 4F mice revealed that the progressive development of bradycardia normally seen in these mice was partially rescued by cyclic induction of OSKM (Figure 4G). Analysis of additional organs, including the liver, heart, and skeletal muscle, did not reveal gross histological differences between untreated LAKI 4F and WT mice (Figure S4C). These results indicate that cyclic induction of OSKM in vivo significantly improved age-associated histological changes in multiple organs of LAKI 4F mice, which may ultimately extend their lifespan.
Analysis of skin, liver, stomach, and kidney from LAKI 4F mice after doxycycline treatment confirmed expression of Oct4 and Sox2 in these tissues (Figure 5A). Interestingly, cyclic OSKM induction during 6 weeks in LAKI 4F mice restored proliferation rates to WT levels in the stomach, kidney, and skin (as indicated by Ki67 staining) compared to untreated mice (Figure 5B). Importantly, the restoration of cell proliferation in these organs was not accompanied by the presence of dysplastic lesions or Nanog-positive cells (Figure S5A). Downregulation of stress response genes in the p53 pathway, as well as age-associated interleukin-1α, was observed in the liver, stomach, and kidneys of doxycycline-treated LAKI 4F mice compared to untreated controls (Figure S5B). Cyclic induction of OSKM also restored normal levels of the histone marks H3K9me3 and H4K20me3 involved in the maintenance of heterochromatin (Benayoun et al., 2015; Zhang et al., 2015) in the kidney and spleen of LAKI 4F mice (Figures 5C and 5D). We observed significant reduction in senescence-associated β-galactosidase expression in the liver of doxycycline-treated LAKI 4F mice compared to untreated animals (Figure 5E). Moreover, analysis of adult stem cells populations revealed a partial restoration of the number of satellite cells (i.e., Pax7-positive cells) in skeletal muscle and hair follicle stem cells (Cytokeratin-15-positive cells) in LAKI 4F mice subjected to cyclic induction of OSKM compared to untreated animals (Figures 5F and 5G). Lastly, rescue of these age-associated phenotypes in LAKI 4F mice was independent of changes in lamin A/C or progerin, as cyclic OSKM induction did not alter the levels of lamin A/C or progerin in any of the analyzed tissues (Figure S5C).
Figure 5. Amelioration of Cellular Markers of Aging by In Vivo Induction of Oct4, Sox2, Klf4, and c-Myc.

(A) Expression of Oct4 and Sox2 in the indicated organs of LAKI 4F upon cyclic doxycycline administration. *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(B) Immunostaining and quantification of Ki67 positive cells in stomach, kidney, and skin of LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 50 μm.
(C and D) Immunofluorescence of H3K9me3 and H4K20me3 in kidney and spleen of LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 10 μm.
(E) Immunostaining of β-galactosidase in liver of LAKI 4F mice upon cyclic doxycycline administration.
(F) Immunostaining and quantification of Pax7-positive cells in muscle of LAKI 4F mice upon cyclic administration of doxycycline. *p < 0.05, ***p < 0.001, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(G) Immunostaining and quantification of Cytokeratin-15 in skin of LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 20 μm. **p < 0.005, ***p = 0.0001.
Data are presented as mean ± SEM. See also Figure S5.
Decline in regenerative capacity and subsequent loss of tissue homeostasis is one of the most striking hallmarks of aging. Taken together, the results described above suggest that in vivo OSKM induction may slow the aging process by preventing molecular changes associated with aging, including epigenetic alterations, activation of cellular senescence pathways, and the exhaustion of adult stem cell populations. Molecular alterations induced by in vivo reprogramming may potentially lead to a better maintenance of tissue homeostasis and lifespan extension.
Improvement of Aging Hallmarks in Mouse and Human Cells by Partial Reprogramming
Although the age-associated accumulation of progerin is well documented, the true role of this protein variant during physiological aging has not been ultimately demonstrated (Burtner and Kennedy, 2010; Scaffidi and Misteli, 2006). Therefore, to evaluate the effects of the Yamanaka factors on cellular phenotypes observed during physiological aging, we performed short-term induction of OSKM in late-passage WT 4F TTFs. OSKM induction in WT 4F cells was confirmed by qPCR and immunofluorescence after doxycycline treatment (Figures 6A and S6A). Similar to LAKI 4F cells, analysis of DNA damage in late-passage WT 4F cells upon short-term induction of OSKM revealed a reduced number of foci of histone γ-H2AX per cell compared to untreated cells (Figures 6B and S6B). Moreover, short-term induction of OSKM in WT 4F cells downregulated the expression of age-related stress response genes in the p53 tumor suppressor pathway, as well as the senescence-associated metalloprotease MMP13 and interleukin-6 (Figure 6C). These results are in agreement with previous studies on global analysis of gene expression at early stages of cellular reprogramming (Hansson et al., 2012; Polo et al., 2012). Likewise, short-term induction of OSKM significantly reduced the production of mitochondrial ROS in WT 4F cells (Figure S6C) and restored levels of H3K9me3 compared to untreated cells (Figure 6D).
Figure 6. Amelioration of Aging Hallmarks in Wild-Type Mice and Human Cells by Short-Term In Vitro Induction of Oct4, Sox2, Klf4, and c-Myc.

(A) Immunofluorescence of Oct4 and Sox2 in WT 4F TTFs. Scale bar, 5 μm.
(B) Immunofluorescence and quantification of γH2AX foci in late-passage cells from WT 4F mice. Scale bar, 10 μm. ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(C) qPCR analysis of stress response genes in the p53 pathway, senescence-associated metalloprotease MMP13, and interleukin-6 in late-passage cells from WT 4F mice. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction.
(D) Immunofluorescence and quantification of H3K9me3 in late-passage WT 4F cells. Scale bar, 10 μm. *p < 0.05, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction compared to control.
(E) Immunofluorescence and quantification of γH2AX foci in late-passage human 4F cells. Scale bar, 10 μm. ***p < 0.0005 and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction compared to control.
(F) Immunofluorescence and quantification of H3K9me3 in late-passage human 4F cells. Scale bar, 10 μm. *p < 0.05, **p < 0.005, and ****p < 0.0001 according to one-way ANOVA with Bonferroni correction compared to control.
Data are presented as mean ± SEM. See also Figure S6 and Table S1.
To further explore whether the effect of short exposure to the Yamanaka factors on aging hallmarks is conserved among species, we next used human cells to test this hypothesis. For this purpose, and to avoid the use of viruses for the introduction of the Yamanaka factors, we chose a human secondary system for OSKM expression. We and others have previously generated human iPSCs carrying doxycycline-inducible Yamanaka factors to better study the process of cellular reprogramming in homogeneous populations of human cells (Hockemeyer et al., 2008; Ruiz et al., 2012). Using this system, we differentiated human 4F iPSCs into fibroblasts and cultured these cells for several weeks to allow the accumulation of age-associated phenotypes. Subsequently, we performed short-term induction of OSKM in late-passage human 4F cells and analyzed the effect on age-associated phenotypes. OSKM induction was confirmed by qPCR after doxycycline treatment (Figure S6D). As was seen with mouse WT 4F TTFs, short-term OSKM induction in human 4F cells significantly reduced the number of foci of histone γ-H2AX per cell compared to untreated cells (Figure 6E). In addition, short-term induction of OSKM in human 4F cells significantly restored the levels of H3K9me3 (Figure 6F).
Collectively, these results suggest that short-term induction of OSKM can ameliorate multiple hallmarks of aging in late-passage WT mouse and human cells, including the accumulation of DNA damage, cellular senescence, and epigenetic dysregulation, thus demonstrating the potential of partial reprogramming for the rejuvenation of phenotypes observed during physiological aging.
Increased Resistance to Metabolic Disease and Muscle Injury in Physiologically Aged Animals by Partial In Vivo Reprogramming
Lastly, to investigate whether in vivo partial reprogramming could have a beneficial effect on aging in WT organisms, we tested the effect of cyclic OSKM induction on the regenerative capacity of aged WT 4F mice. Aging is characterized by the decreased ability of an organism to resist stress, damage, and disease. In addition, aging represents the major risk factor for many chronic diseases, including neurodegeneration, cancer, and diabetes. Alteration in glucose homeostasis represents one of the leading causes of morbidity and mortality. While insulin resistance and decreased secretory capacity of pancreatic beta cells play crucial roles in the development of diabetes, the loss of the proliferative capacity of beta cells in response to pancreatic injury has emerged as a fundamental mechanism of pancreatic dysfunction during aging (Rankin and Kushner, 2009; Teta et al., 2005; Tschen et al., 2009).
To confirm the decrease in regenerative capacity of beta cells during aging, we ablated beta cells from the pancreas of 2-month-old and 12-month-old mice by low-dose administration of the beta cell toxin streptozocin (STZ). Three weeks after injury, glucose tolerance test (GTT) demonstrated that 2-month-old mice restored glucose levels more effectively than 12-month-old mice, suggesting that younger mice have a higher beta cell regenerative capacity (Figure S7A). Next, in order to investigate whether in vivo induction of the Yamanaka factors in aged mice could enhance their recovery from pancreatic injury, 12-month-old WT 4F mice were subjected to cyclic induction of OSKM in vivo for 3 weeks (2 days of doxycycline administration followed by 5 days of doxycycline withdrawal) followed by low-dose STZ administration (Figure 7A). Expression of Oct4 and Sox2 in the pancreas of old WT 4F mice was confirmed by qPCR analysis (Figure S7B). Importantly, GTT demonstrated higher glucose tolerance in doxycycline-treated old 4F mice compared to untreated controls 2 weeks after injury, indicating better pancreatic function (Figure 7B). STZ is a glucose analog that, after being transported into beta cells by the glucose transporter 2 (Glut2), induces DNA damage and apoptosis. Analysis of Glut2 by qPCR in old WT 4F mice following in vivo induction of OSKM did not reveal significant changes in Glut2 expression, indicating that a decrease in STZ uptake was not responsible for the improved pancreatic function of doxycycline-treated animals (Figure S7C). Moreover, doxycycline treatment in 12-month-old WT mice not carrying 4F did not lead to any improvement in glucose tolerance following pancreatic injury (Figure S7D). Lastly, histological analysis of the pancreas 2 weeks after STZ administration revealed an increase in the size of pancreatic islets in doxycycline-treated 12-month-old WT 4F mice compared to untreated controls (Figure 7C). These results indicate that cyclic induction of OSKM in vivo can promote the expansion of the beta cell population in older animals and increase their glucose tolerance upon pancreatic injury.
Figure 7. Improved Resistance to Metabolic Disease and Skeletal Muscle Injury in Aged WT Animals by In Vivo Reprogramming.

(A) Schematic representation of induction of pancreatic injury by low-dose (30 mg/kg) STZ following in vivo reprogramming in 12-month-old WT 4F mice.
(B) Glucose tolerance test (GTT) in 12-month-old WT 4F mice following beta cell ablation by low dose STZ (−Dox n = 6; +Dox n = 6). **p < 0.01 and ***p = 0.0005 according to two-tailed Student’s t test.
(C) Immunostaining of Insulin and quantification of pancreatic islet size in pancreas of 12-month-old WT 4F mice 2 weeks following STZ. **p < 0.005 according to two-tailed Student’s t test.
(D) Schematic representation of induction of muscle injury by CTX following in vivo reprogramming in 12-month-old WT 4F mice.
(E) Representative image of H&E staining of tibialis anterior (TA) muscle of 12-month-old WT 4F mice following muscle injury by CTX injection. Scale bar, 50 μm.
(F) Immunostaining of Laminin in muscle sections of 12-month-old WT 4F mice.
(G) Quantification of fiber cross-sectional area frequency distribution and percentage of central nucleated fibers in muscle sections of 12-month-old WT 4F mice following muscle injury by CTX injection (−Dox n = 4; +Dox n = 4). **p < 0.001 according to two-tailed Student’s t test.
(H) Immunostaining and quantification Pax7-positive cells in muscle sections of 12-month-old WT 4F mice following muscle injury by CTX injection (−Dox n = 4; +Dox n = 4). *p < 0.05 according to two-tailed Student’s t test.
Data are presented as mean ± SEM. See also Figure S7.
Sarcopenia is defined as the loss of skeletal muscle mass during aging and represents one of the major causes of physical incapacitation in individuals of advanced age (Marcell, 2003). The age-associated loss of muscle mass during aging results from a decline in the regenerative capacity of muscle stem cells, also known as satellite cells (Chakkalakal et al., 2012; Cosgrove et al., 2014). Satellite cells exist in a quiescence state during life and are activated upon muscle injury to generate new muscle fibers (Bjornson et al., 2012; Cheung et al., 2012). During aging and as a consequence of environmental and cell-intrinsic factors, there is a decrease in the number and functionality of satellite cells that switch from a quiescent to a pre-senescence state that hampers their regenerative capacity (García-Prat et al., 2016; Sousa-Victor et al., 2014). First, in order to evaluate the decline in the regenerative capacity of skeletal muscle during aging, we performed cardiotoxin (CTX)-induced muscular injury in the tibialis anterior (TA) muscle of 2-month-old and 12-month-old mice. Histological analysis of the TA muscle 10 days after injury revealed a decrease in the regenerative capacity of the muscle in 12-month-old mice compared to 2-month-old mice (Figures S7E and S7F). Based on our observations that short-term expression of OSKM preserves muscle satellite cells in a mouse model of premature aging (Figure 5F), we tested whether partial reprogramming in vivo could have a beneficial effect on the regenerative capacity of skeletal muscle in 12-month-old WT 4F mice. To maximize the in vivo expression of OSKM, we tested the induction of reprogramming factors upon intramuscular injection of doxycycline into the TA muscle of WT 4F mice. Analysis of expression by qPCR revealed strong induction of OSKM 3 days following doxycycline injection (Figure S7G). Next, 12-month-old WT 4F mice were subjected to cyclic induction of OSKM in vivo by intramuscular injection of doxycycline weekly for 3 weeks followed by CTX-induced muscle injury of the TA muscle (Figure 7D). Remarkably, histological analysis and laminin immunostaining of the TA muscle 10 days after injury demonstrated a significant improvement in the regenerative capacity of the muscle by cyclic OSKM induction (Figures 7E and 7F), as indicated by an increase in the cross-sectional area of fibers and a reduction in the number of central nucleated fibers in the injured area (Figure 7G). Intramuscular injection of doxycycline in 12-month-old WT mice not carrying 4F did not lead to any improvement in the regenerative capacity of the muscle following injury (Figures S7H and S7I). Lastly, analysis of satellite cells revealed an increase in the number of Pax7-positive satellite cells in the muscle of 12-month-old 4F mice treated with doxycycline compared to untreated controls (Figure 7H). These results suggest that cyclic intramuscular induction of OSKM is capable of inducing an expansion of muscle stem cells in older mice and improving muscle regeneration following injury.
Together, these data highlight the potential of partial in vivo reprogramming for the amelioration of age-associated phenotypes, such as the decreased ability to resist damage and the decline in the regenerative capacity of tissues and organs during life.
DISCUSSION
For humans living in modern societies, aging is the largest risk factor for most diseases (Partridge, 2014). However, despite decades of effort, the complexity of cellular and organismal aging has limited our understanding of this critical biological process. Epigenetic alterations (e.g., DNA methylation, post-translational modifications of histones, and chromatin remodeling) have recently emerged as one of the most conserved hallmarks of aging (Benayoun et al., 2015; Sen et al., 2016). Interestingly, cellular reprogramming to pluripotency occurs through a stepwise global epigenetic remodeling (Benayoun et al., 2015; Liu et al., 2013b; Polo et al., 2012). The rejuvenation of aging hallmarks has been extensively described during cellular reprogramming to pluripotency in vitro (Lapasset et al., 2011; Liu et al., 2011; Mahmoudi and Brunet, 2012; Rando and Chang, 2012), but the dynamics of this process remain poorly understood. In addition, until now, the amelioration of age-associated phenotypes by cellular reprogramming has not yet been demonstrated at the organismal level.
Our results demonstrate that partial reprogramming by short-term expression of the Yamanaka factors has the capacity to rejuvenate cellular phenotypes of aging in mouse and human cells. Although previous studies have indicated that expression of the Yamanaka factors in vivo can lead to cancer development or teratoma formation (Abad et al., 2013; Ohnishi et al., 2014), here, we demonstrate that tumor formation can be avoided by short-term induction of OSKM. Cyclic induction of OSKM in vivo ameliorated hallmarks of aging and extended the lifespan of a mouse model of premature aging. Additionally, short-term induction of OSKM improved the regenerative capacity of pancreas and muscle following injury in physiologically aged mice. Together, these results show that partial in vivo reprogramming might be used to modulate aging hallmarks and significantly benefit organismal health.
Our observations may reinforce the potential role of epigenetic changes as drivers of aging and highlight the plasticity of the aging process, which might be altered by cellular reprogramming in vivo. In addition, our results suggest that aged cells undergo a process of molecular rejuvenation during the initial stages of cellular reprogramming to pluripotency. Failure to erase critical hallmarks of aging may lead to refractory populations of cells and cellular senescence. Due to the complexity of the reprogramming and aging processes, future studies will be necessary to investigate whether partial reprogramming can ameliorate aging hallmarks during physiological aging and to better understand the molecular mechanisms behind this phenomenon. This information will be necessary if we are to develop accurate and efficient epigenetic remodeling strategies toward maximizing the beneficial effects of in vivo reprogramming while avoiding potential risks associated with the in vivo expression of the Yamanaka factors.
Thus, the data presented here may guide the establishment of experimental in vivo platforms for investigating the mechanisms of physiological aging, as well as for revealing the specific role that epigenetic dysregulation may play in this important biological process. Reprogramming, currently an experimental tool to study development and cellular differentiation, may provide additional insights into the mechanisms of aging. Proposed drivers of physiological aging include the accumulation of DNA damage, increased ROS production, telomere shortening, cellular senescence, and defects in nuclear envelope architecture (Bernardes de Jesus and Blasco, 2013; Guarente, 2008; Haigis and Sinclair, 2009; Kennedy and Lamming, 2016; Soultoukis and Partridge, 2016; Steffen and Dillin, 2016). Multiple studies using animal models have demonstrated that the manipulation of these aging drivers leads to the manifestation of molecular hallmarks of aging that are shared between premature aging models and physiological aging (García-Prat et al., 2016; Mitchell et al., 2015). We hypothesize that the emergence of these molecular hallmarks during organismal aging results from the translation of aging signals by a unique and universal epigenetic program. Our results suggest that this epigenetic program, which is reset during embryogenesis, can also be experimentally altered by partial cellular reprogramming at later stages of life. Resetting of the aging clock by epigenetic reprogramming, which is also observed during somatic nuclear transfer, might allow for a deeper understanding of the molecular and cellular mechanisms underlying the aging process. Eventually, it may, as well, lead to the development of therapeutic strategies with the goal of ameliorating age-related diseases and thus improving health and longevity.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3K9me3 | Abcam | Cat# ab8898; RRID: AB_306848 |
| Anti-Histone H3K20me3 | Abcam | Cat# ab9052; RRID: AB_1951942 |
| Anti-Insulin | Abcam | Cat# ab7842; RRID: AB_306130 |
| Anti-Cytokeratin 15 [EPR1614Y] | Abcam | Cat# ab52816; RRID: AB_869863 |
| Anti-cMyc | Abcam | Cat# ab32072; RRID:AB_731658 |
| Anti-Integrin a 6 (ITGA6) [EPR5578] | Abcam | Cat# ab124924; RRID: AB_11127843 |
| Anti-Oct-3/4 (C-10) | Santa Cruz Biotechnology | Cat# sc-5279; RRID: AB_628051 |
| Anti-Lamin A/C (346) | Santa Cruz Biotechnology | Cat# sc-7293; RRID: AB_627874 |
| Anti-Sox2 | Cell Signaling Technology | Cat# 2748; RRID: AB_823640 |
| Anti-Sox2 (C70B1) | Cell Signaling Technology | Cat# 3728; RRID: AB_2194037 |
| Anti-Ki67 (D2H10) | Cell Signaling Technology | Cat# 9027 |
| Anti-phospho-Histone H2A.X (Ser139) (20E3) | Cell Signaling Technology | Cat# 9718; RRID: AB_2118009 |
| Anti-Laminin | Sigma | Cat# L9393; RRID: AB_477163 |
| Anti-53BP1 | Novus Biologicals | Cat# NB100-904; RRID: AB_10002714 |
| Anti-Nanog | Novus Biologicals | Cat# NB100-58842; RRID: AB_877697 |
| Anti-Pax7 | DSHB | Cat# Pax7; RRID: AB_528428 |
| Anti-b-galactosidase | EMD Millipore | Cat# AB986; RRID: AB_92401 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma | Cat# D9891 |
| Streptozocin (STZ) | Sigma | Cat# S0130 |
| Cardiotoxin (CTX) | Latoxan | Cat# L8102 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| HistoVT One (10x, pH 7.0) | Nacalai Tesque | Cat# 06380-05 |
| Polybrene | Millipore | Cat# TR-1003 |
| BLOXALL | Vector Laboratories | Cat# SP-6000 |
| ImmPRESS HRP anti-Mouse IgG (Peroxidase) detection kit | Vector Laboratories | Cat# MP-7402 |
| ImmPRESS HRP anti-Rabbit IgG (Peroxidase) detection kit | Vector Laboratories | Cat# MP-7401 |
| ImmPACT DAB Peroxidase (HRP) substrate | Vector Laboratories | Cat# SK-4105 |
| Quick Start Bradford protein assay | Bio-Rad | Cat# 5000201 |
| NuPAGE Novex 4%–12% Bis-Tris protein gels | ThermoFisher Scientific | Cat# NP0321 |
| Immobilon-P Membrane, PVDF | Millipore | Cat# IPVH00010 |
| ECL SuperSignal | GE Healthcare | Cat# RPN2232 |
| Chaetocin | Sigma | Cat# C9492 |
| OneTouch Ultra2 Meter | OneTouch | Cat# 021098 |
| OneTouch Ultra Blue Strips | OneTouch | Cat# 020994 |
| Formalin solution | Sigma | Cat# HT501128 |
| DMEM GlutaMAX | ThermoFisher Scientific | Cat# 10566024 |
| Non-essential amino acids (NEAA) | ThermoFisher Scientific | Cat# 11140050 |
| FBS | Gemini Bio-products | Cat# 100-106 |
| Lipofectamine 3000 | ThermoFisher Scientific | Cat# L3000-015 |
| MitoSox Red | Molecular Probes | Cat# M36008 |
| iScript Reverse Transcription Supermix | Bio-Rad | Cat# 1708840 |
| SsoAdvanced SYBR Green Supermix | Bio-Rad | Cat# 1725270 |
| Hoechst 33342 | ThermoFisher Scientific | Cat# 62249 |
| TRIzol | ThermoFisher Scientific | Cat# 15596026 |
| X-Gal | Apex | Cat# 20-108 |
| Critical Commercial Assays | ||
| Alexa Fluor 488 Annexin V/Dead Cell Apoptosis kit | ThermoFisher Scientific | Cat# V13241 |
| Experimental Models: Cell Lines | ||
| Primary Tail Tip Fibroblasts (TTFs) WT | This paper | N/A |
| Primary Tail Tip Fibroblasts (TTFs) WT 4F | This paper | N/A |
| Primary Tail Tip Fibroblasts (TTFs) LAKI 4F | This paper | N/A |
| Human 4F iPSCs | Jaenisch lab | N/A |
| Experimental Models: Organisms/Strains | ||
| LAKI mice (LMNA mutation G609G) | Brian Kennedy, Buck Institute | N/A |
| R26rtTA;Col1a12lox–4F2A | The Jackson Laboratory | JAX: 011011 |
| C57BL/6J mice | The Jackson Laboratory | JAX: 000664 |
| Recombinant DNA | ||
| pMXs-Oct3/4 | Addgene | Cat#13366 |
| pMXs-Sox2 | Addgene | Cat#13367 |
| pMXs-Klf4 | Addgene | Cat#13370 |
| pMXs-c-Myc | Addgene | Cat#13375 |
| Gag/pol | Addgene | Cat#14887 |
| pCMV-VSV-G | Addgene | Cat#8454 |
| pMXs-GFP | This paper | N/A |
| Sequence-Based Reagents | ||
| See Table S1 for qPCR primer list | N/A | N/A |
| Software and Algorithms | ||
| Prism 6 | GraphPad Software | http://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Imaris 8.0 | Bitplane | http://www.bitplane.com |
| LabChart | AD Instruments | https://www.adinstruments.com/products/labchart |
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact Juan Carlos Izpisua Belmonte (belmonte@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal procedures were performed according to NIH guidelines and approved by the Committee on Animal Care at the Salk Institute.
Mouse Strains
The mouse model of Hutchinson-Gilford progeria syndrome (HGPS) carrying the LMNA mutation G609G (LAKI) was generated by Carlos López-Otín at the University of Oviedo, Spain and kindly donated by Brian Kennedy at the Buck Institute. Mice carrying the OSKM polycystronic cassette and the rtTA trans-activator (WT 4F) were obtained from The Jackson Laboratory (Stock No: 011011). C57BL/6J (WT) mice were obtained from The Jackson Laboratory (Stock No: 000664). LAKI 4F carrying one (4F/+; rtTA/+) copy of the OSKM polycystronic cassette and the rtTA transactivator with homozygous LMNA mutation were generated by crossing LAKI and WT 4F mice. All the mice were in C57BL/6 background. In vivo experiments were performed in WT 4F mice carrying one (4F/+; rtTA/+) or two copies (4F/4F; rtTA/rtTA) of the OSKM polycystronic cassette and the rtTA transactivator. Experiments with WT and WT 4F mice were performed in males at 2 and 12 months of age. Experiments with LAKI and LAKI 4F mice were performed with mice of both genders at 7–8 weeks of age. For lifespan experiments, mice of both genders from a litter were randomly assigned to control and experimental groups. For beta cell ablation and muscle injury experiments, male mice were randomly assigned to control and experimental groups. Any animals that appeared unhealthy before the start of experiments were excluded. No inclusion criterion was used. The mice were housed with a 12 hr light/dark cycle between 06:00 and 18:00 in a temperature controlled room (22 ± 1°C) with free access to water and food. Blood samples from mice were collected by submandibular bleeding.
Cell Culture and Maintenance
Tail tip fibroblasts (TTFs) were isolated from WT 4F and LAKI 4F mice and cultured at 37 °C in DMEM (Invitrogen) containing Gluta-MAX, non-essential amino acids, and 10% fetal bovine serum (FBS). TTFs of WT 4F (passage 8) and LAKI 4F (passage 6) were treated with doxycycline (2 μg/ml) for the indicated time periods in the culture medium. For inhibition of H3K9 methyltransferases, cells were treated with chaetocin (10 nM) for the indicated time periods in the culture medium. Human induced pluripotent stem cells carrying doxycycline-inducible transgenes for the expression of OCT4, SOX2, KLF4 and c-MYC (4F iPSCs) were kindly donated by Rudolf Jaenisch. Human 4F iPSCs were differentiated into fibroblast-like cells by embryonic body formation in fibroblast medium in DMEM (Invitrogen) containing GlutaMAX, non-essential amino acids and 10% fetal bovine serum (FBS) for 5 days and subsequently plated into adherent tissue culture dishes. Subsequently, 4F fibroblasts were passaged and culture according to standard protocols. Experiments with human 4F fibroblasts were performed at passage 10. All experiments were performed a minimum of three times.
METHOD DETAILS
In Vivo Induction of OSKM
Induction of OSKM was performed by administration of doxycycline (1 mg/ml) (Sigma) in the drinking water. The in vivo cyclic induction protocol consisted of 2 days of doxycycline administration followed by 5 days of doxycycline withdrawal. For lifespan experiments, cyclic doxycycline administration started at 8 weeks of age and continued until death. For beta cell ablation experiments, 12-month old WT 4F mice were subjected to 3 cycles of 2 days of doxycycline administration in drinking water at a concentration of 0.5 mg/ml. For muscle injury experiments, cyclic expression of OSKM was induced in the TA muscle by a weekly intramuscular injection of 30 μL of doxycycline (1 mg/ml) prepared in PBS during the course of 3 weeks.
Beta Cell Ablation
Induction of diabetes by low-dose streptozocin (STZ) treatment was performed in 2 and 12-month old WT mice, and 12-month old WT 4F mice. 12-month old WT 4F mice were subjected to 3 cycles of 2 days of doxycycline administration in drinking water at a concentration of 0.5 mg/ml. Following the last cycle of doxycycline treatment, mice were fasted for 5 hr before streptozocin administration. Subsequently, mice were treated with five daily injections of low-dose (30–50 mg/kg) streptozocin in 0.1 mol/l sodium citrate buffer (pH 4.5). Glucose tolerance test (GTT) was performed 2 weeks after STZ treatment. For GTT, animals were fasted for 16 hr and subsequently injected with 1.5 g of glucose per kg of body weight. Blood glucose levels were measured at 15, 30, 60 and 120 min with an OneTouch Ultra 2 glucometer (OneTouch) using blood from the tail vein. At the indicated times mice were sacrificed and the pancreas were dissected and processed for histological analysis.
Muscle Injury Model
Induction of muscle injury was performed in 2 and 12-month old WT mice, and 12-month old WT 4F mice by intramuscular injection of 50 μL of cobra venom (10 μM; ~97% carditoxin; CTX) from Naja Pallida (Latoxan) in the tibialis anterior (TA) muscle. Prior to CTX-induced muscle injury, cyclic expression of OSKM was induced in the TA muscle by a weekly intramuscular injection of 30 μL of doxycycline (1 mg/ml) prepared in PBS during the course of 3 weeks. Subsequently, 10 days after CTX-induced muscle injury, mice were sacrificed and the TA muscles were dissected and processed for histological analysis.
Heart Rate Analysis
For analysis of heart rate, mice were anesthetized with 2.5% isoflurane and heart rate was monitored using Power Lab data acquisition instrument (Ad Instruments). Data were processed and analyzed using LabChart (AD Instruments).
Histological Analysis
For histological analysis, tissue samples from a minimum of 5 mice were collected at 14 weeks of age after 6 weeks of cyclic induction of OSKM by doxycycline treatment. Mice were perfused with PBS and 10% buffered formalin solution. Subsequently, tissues were fixed overnight at 4°C in 10% buffered formalin solution and embedded in paraffin. Paraffin sections were used for hematoxylin and eosin staining (H&E) or for immunohistochemistry.
Immunohistochemistry
Paraffin tissues sections underwent antigen retrieval using HistoVT One (Nacalai Tesque). Endogenous peroxidase was blocked using Bloxall (Vector Laboratories). Subsequently, tissue sections were incubated overnight with primary antibody in donkey serum. Finally, tissue sections were incubated with ImmPress reagent for 30 min (Vector Laboratories), and positive cells were visualized with ImmPACT DAB substrate (Vector Laboratories).
Antibodies
Antibodies were obtained from the following sources. Abcam: anti-H3K9me3 (ab8898), anti-H4K20me3 (ab9052), anti-insulin (ab7842), anti-cytokeratin 15 (ab52816), anti-c-myc (ab32072), anti-integrin α 6 (ITGA6) (ab124924); Santa Cruz Biotechnology: anti-Oct-3/4 (sc-5279), anti-lamin A/C (sc-7293); Cell Signaling: anti-Sox2 (2748), anti-Sox2 (3728), anti-Ki67 (9027); anti-phospho histone H2AX (9718), Sigma: anti-laminin (L9393); Novus: anti-53BP1 (NB100-904) anti-Nanog (NB100-58842); DSHB: anti-Pax7 (pax7 s); Millipore: anti-β-galactosidasa (AB986).
Immunofluorescence Microscopy
Cells were fixed with 4% formaldehyde in PBS at room temperature (RT) for 10 min. After fixation, cells were treated with 0.5% Triton X-100 in PBS for 5 min at RT. After blocked with 1% BSA in PBS for 1 hr, cells were incubated at 4°C overnight with the primary antibody, followed by washing in PBS and incubation at RT for 1 hr with the corresponding secondary antibody. Nuclei were stained with Hoechst 33342 (Invitrogen). Cells were mounted using Fluoromount-G (SouthernBiotech). Confocal image acquisition was performed using a Zeiss LSM 780 laser-scanning microscope (Carl Zeiss Jena). Images were taken at z sections of 0.25 μm intervals using the adequate lasers (488-nm, 568-nm, 633-nm and 405-nm). The laser intensity was typically set to 3%–5% transmission of the maximum intensity, and the settings were established to avoid signal saturation for any of the lasers.
Western Blot Analysis
Protein samples from cells and tissues were isolated in RIPA buffer. Protein concentration was evaluated by Quick Start Bradford protein assay (Bio-Rad). Equal amounts of protein were loaded on 4%–12% SDS-PAGE polyacrylamide gels. After electrophoresis, gels were electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Millipore), and blocked with 5% nonfat milk. Subsequently, membranes were incubated overnight at 4°C with the indicated antibody. Finally, blots were incubated with horse-radish peroxidase secondary antibodies at room temperature for 1 hr. The ECL SuperSignal (GE Healthcare) was use for final detection.
RNA Analysis
Total RNA was extracted from cells, tissues, and blood samples using TRIzol (Invitrogen) followed by cDNA synthesis using iScript Reverse Transcription Supermix for RT-PCR (Bio-Rad). qPCR was performed using SsoAdvanced SYBR Green Supermix (Bio-Rad). Primer sequences are listed in Table S1.
Virus Preparation and Infection
For retrovirus preparation, plasmid encoding Oct4, Sox2, Klf4, and c–Myc were cotransfected into HEK293FT together with packing plasmids using Lipofectamine 3000 (Invitrogen). Retrovirus particles were collected 2 days after transfection and filtered. Retroviral transduction of tail tip fibroblasts (TTFs) was done overnight in 8 μg/ml polybrene (Millipore). Analysis of TTFs transduced with single factors was performed 4 days after transduction.
Analysis of Programmed Cell Death
Programmed cell death (apoptosis) was evaluated by flow cytometry using the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis kit (ThermoFisher Scientific). Briefly, cells were resuspended in 100 μL of 1x annexin-binding buffer. Subsequently, 5 μL of Alexa Fluor 488 annexin V and 1 μL 100 μg/ml PI working solution were added to 100 μL of cell suspension. The cells were incubated for 15 min at room temperature. Lastly, 400 μL of 1x annexin-binding buffer was added to cells and analyzed using flow cytometry.
Measurement of Reactive Oxygen Species
Mitochondrial reactive oxygen species (ROS) were measured using the mitochondrial superoxide indicator MitoSox Red (Molecular Probes). Briefly, cells were washed with PBS and incubated for 10 min at 37°C in HBSS buffer containing MitoSox at a concentration of 5 μM. Subsequently cells were washed and analyzed using flow cytometry.
Senescence-associated Beta-galactosidase Assay
Senescence-associated beta-galactosidase (SA-βgal) assay was performed as described (Debacq-Chainiaux et al., 2009). Briefly, first, the cells were fixed in 4% paraformaldehyde for 5 min at room temperature. Next, the cells were washed twice with PBS and incubated overnight 37°C in staining solution containing 40 mM citric acid/Na phosphate buffer, 5 mM K4[Fe(CN)6] 3H2O, 5 mM K3[Fe(CN)6], 150 mM sodium chloride, 2 mM magnesium chloride and 1 mg/ml X-gal. Finally, the cells were washed twice with PBS and once with methanol. The plate was dried and photos of cells were taken using bright field microscopy.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
For quantification of immunofluorescence microscopy images, a minimum of 50–100 cells were counted to determine the number, volume, and intensity of γH2AX, 53BP1, H3K9me3 and H4K20me3 foci per cell or cells with nuclear abnormalities. To ensure reliable quantification, image recording was performed under the same scanning conditions for all the samples in the same experiment in terms of: objective lens, illumination intensity, emission window, digital zoom, scanning speed, and offset and gain settings. The z sections were assembled using Imaris 8.0 software (Bitplane) and then used for further analysis using the Imariscell module. For quantification of histological and immunohistochemistry analysis, three sections per tissue from 3–5 animals were analyzed using ImageJ (NIH). All quantifications were performed blinded.
Statistical Analysis
All of the data are presented as the mean ± SEM and represent a minimum of three independent experiments. Statistical parameters including statistical analysis, statistical significance, and n value are reported in the Figure legends. For in vivo experiments n = number of animals. Statistical analyses were carried out using Prism 6 Software (GraphPad). For statistical comparison of multiple groups, we performed one-way ANOVA followed by Bonferroni correction. For statistical comparison of two groups, we performed two-tailed Student’s t test. A value of p < 0.05 was considered significant. For statistical analysis of survival curves, we performed log-rank (Mantel-Cox) test.
Supplementary Material
Figure S1. Characterization of LAKI 4F TTFs upon Short-Term Induction of OSKM, Related to Figure 1
(A) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in LAKI 4F tail tip fibroblasts (TTFs). **p < 0.005, ***p < 0.0005 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(B) Analysis of cellular identity of LAKI 4F TTFs following partial reprogramming analyzed by flow cytometry using Thy1 and SSEA1 staining.
(C) qPCR analysis of Nanog expression in LAKI 4F TTFs after short-term induction of OSKM by doxycycline treatment.
(D) Quantification of foci intensity and foci volume of γH2AX staining in LAKI 4F TTFs. ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(E) Immunostaining of 53BP1 in LAKI 4F TTFs following doxycycline treatment. The number, volume, and intensity of 53BP1 foci in LAKI 4F TTFs were quantified in Oct4 positive and negative cells. Scale bar, 10 μm. *p < 0.05, **p < 0.01, ***p < 0.0005 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(F) β-galactosidase activity in WT and LAKI 4F TTFs. Quantification of cells with β-galactosidase staining. Scale bar, 100 μm. ***p = 0.001 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(G) Levels of mitochondrial ROS in LAKI 4F TTFs after doxycycline treatment. **p < 0.01, according to one-way ANOVA with Bonferroni correction.
Data are presented as mean ± SEM.
Figure S2. Characterization of LAKI 4F and LAKI TTFs following Doxycycline Treatment, Related to Figure 1
(A) Detection of Lamin A/C and progerin levels in WT 4F and LAKI 4F TTFs after short-term doxycycline treatment by western blot. Arrowheads denote Lamin A and C. Asterisk denotes progerin.
(B) qPCR analysis of Lamin A and Lamin B1 levels in LAKI 4F TTFs.
(C) Analysis by flow cytometry of programmed cell death in LAKI 4F TTFs following doxycycline treatment.
(D) Immunofluorescence of γH2AX in LAKI TTFs following doxycycline treatment.
(E) Immunofluorescence of H3K9me3 in LAKI TTFs following doxycycline treatment.
(F) qPCR analysis of stress response genes in the p53 pathway, senescence-associated metalloprotease MMP13 and interleukin-6 in LAKI TTFs following doxycycline treatment.
(G) Immunofluorescence of Lamin A/C in LAKI TTFs following doxycycline treatment.
(H) Levels of mitochondrial ROS in LAKI TTFs after doxycycline treatment.
(I) Immunofluorescence of Oct4, Sox2, c-Myc and Lamin A/C in LAKI TTFs infected with GFP, Oct4, Sox2 and c-Myc retroviruses. Scale bar, 10 μm.
Figure S3. Effect of Long-Term In Vivo Cyclic Induction of Oct4, Sox2, Klf4, and c-Myc in Mice Carrying One and Two Copies of OSKM and rtTA Cassette, Related to Figure 3
(A) Body weight of wild-type (WT) and 4F mice carrying single copy of OSKM and rtTA cassette upon cyclic administration of doxycycline. WT (n = 5) and 4F (n = 3).
(B) Survival of WT and 4F mice carrying single copy of OSKM and rtTA cassette upon cyclic administration of doxycycline. WT (n = 5) and 4F (n = 3).
(C and D) Immunostaining of Ki67 and Nanog in spleen, liver, and kidney of WT and 4F mice carrying single copy of OSKM and rtTA cassette. Scale bar, 100 μm (spleen) and 50 μm (liver and kidney).
(E) Immunostaining of Sox2 and Ki67 in spleen, liver, and kidney of 4F mice carrying two copies of OSKM and rtTA cassette. Scale bar, 50 μm.
Data are presented as mean ± SEM.
Figure S4. Characterization of LAKI 4F Mice Subjected to Cyclic Induction of OSKM, Related to Figure 4
(A) Representative picture of LAKI 4F mice at 16-weeks of age upon doxycycline administration. Curvature of spine can be observed in LAKI 4F not treated with doxycycline (−Dox).
(B) Partial rescue of spleen involution is observed in LAKI 4F mice upon doxycycline administration.
(C) Histological analysis of liver, heart, and muscle of WT and LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 50 μm.
Figure S5. Molecular Changes Induced by Cyclic In Vivo Induction of Oct4, Sox2, Klf4, and c-Myc, Related to Figure 5
(A) Immunohistochemistry of Nanog in skin and kidney of WT and LAKI 4F mice upon cyclic doxycycline administration. Teratoma was used as a positive control. Scale bar, 50 μm.
(B) qPCR analysis of stress response genes in the p53 pathway and interleukin-1α in liver, stomach, and kidney of LAKI 4F mice upon cyclic doxycycline administration. *p < 0.05, **p < 0.01, ***p < 0.0001 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(C) Detection of Lamin A/C and progerin levels in skin, spleen, and stomach of WT and LAKI 4F mice by western blot. Arrowheads denote Lamin A and C. Asterisk denotes progerin.
Data are presented as mean ± SEM.
Figure S6. Amelioration of Aging-Associated Phenotypes in Wild-Type Mice and Human Cells by Short-Term Expression of Reprogramming Factors, Related to Figure 6
(A) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in WT 4F TTFs. *p < 0.05, **p < 0.05, ***p < 0.005, ****p < 0.0001.
(B) Quantification of foci intensity and foci volume of γH2AX staining in WT 4F TTFs. ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(C) Analysis of ROS levels in LAKI 4F TTFs after doxycycline treatment. *p < 0.05, according to one-way ANOVA with Bonferroni correction.
(D) qPCR analysis of OCT4, SOX2, KFL4 and c-MYC in human 4F fibroblasts.
Data are presented as mean ± SEM.
Figure S7. Analysis of Pancreatic Function and Skeletal Muscle Regeneration in Wild-Type Mice, Related to Figure 7
(A) Glucose tolerance test (GTT) in 2-month and 12-month old WT mice following beta cell ablation by low dose (50 mg/kg) STZ (2-month WT n = 3; 12-month WT n = 3).
(B) qPCR analysis of Oct4, Sox2, in pancreas of 12-month old WT 4F mice upon administration of doxycycline. **p < 0.01, according to one-way ANOVA with Bonferroni correction.
(C) qPCR analysis of Glut2 in pancreas of 12-month old WT 4F mice upon administration of doxycycline.
(D) GTT in 12-month old WT mice following doxycycline and beta cell ablation by low dose (50 mg/kg) STZ (−Dox n = 5; +Dox n = 5).
(E) Representative image of H&E staining and immunostaining of Laminin of tibialis anterior (TA) muscle of 2-month and 12-month old WT mice following muscle injury by CTX injection. Scale bar, 50 μm.
(F) Quantification of fiber cross-sectional area frequency distribution and percentage of central nucleated fibers in muscle sections of 2-month and 12-month old WT mice following muscle injury by CTX injection. (2-month WT n = 3; 12-month WT n = 3). ****p < 0.0001, according to two-tailed Student’s t test.
(G) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in skeletal muscle of 12-month WT 4F mice upon administration of doxycycline. *p < 0.05, **p < 0.01, according to two-tailed Student’s t test.
(H) Representative low- and high-magnification images of H&E staining of TA muscle of 12-month-old WT mice following doxycycline treatment and muscle injury by CTX injection. Scale bar, 50 μm.
(I) Quantification of percentage of central nucleated fibers in muscle sections of 12-month old WT mice following doxycycline treatment and muscle injury by CTX injection (−Dox n = 3; +Dox n = 3).
Highlights.
Partial reprogramming erases cellular markers of aging in mouse and human cells
Induction of OSKM in progeria mice ameliorates signs of aging and extends lifespan
In vivo reprogramming improves regeneration in 12-monthold wild-type mice
Acknowledgments
We thank M. Schwarz for administrative support. We thank N. Varki from the Department of Pathology and K. McIntyre from the Moores Cancer Center at University of California San Diego. We thank R. Jaenisch for kindly donation of human induced pluripotent stem cells carrying transgenes for the inducible expression of the reprogramming factors. A.O. was partially supported by a US NIH Ruth L. Kirschstein National Research Service Award Individual Postdoctoral Fellowship. P.R. was partially supported by the Muscular Dystrophy Association. P.M.-R. was partially supported by Fundación Alfonso Martin Escudero. A.P.-L. was partially supported by the Hewitt Foundation. F.H. was partially supported by the Uehara Memorial Foundation. T.H. was partially supported by the Nomis Foundation. M.K. was partially supported by a JSPS Postdoctoral Fellowship for Research Abroad. D.L. was partially supported by the University of California, San Diego. This study was supported by the G. Harold and Leila Y. Mathers Charitable Foundation, The Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED002), The Moxie Foundation, The Glenn Foundation, Universidad Católica San Antonio de Murcia (UCAM), and Fundación Dr. Pedro Guillen.
Footnotes
Supplemental Information includes seven figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.11.052.
AUTHOR CONTRIBUTIONS
A.O., P.R., P.M.-R., P.G., and J.C.I.B. designed all experiments. A.O., P.R., P.M.-R., and J.C.I.B. prepared the figures and wrote the manuscript. A.O., P.R., P.M.-R., T.H., D.L., E.V.-F., C.R., and I.G. performed and analyzed in vitro experiments. A.O., P.R., P.M.-R., A.P.-L., F.M., M.L., D.L., M.K., E.B., T.A., D.D., J.L.R., J.X., G.N., E.N., and J.M.C. performed and analyzed in vivo experiments.
References
- Abad M, Mosteiro L, Pantoja C, Cañamero M, Rayon T, Ors I, Graña O, Megías D, Domínguez O, Martínez D, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340–345. doi: 10.1038/nature12586. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16:593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes de Jesus B, Blasco MA. Telomerase at the intersection of cancer and aging. Trends Genet. 2013;29:513–520. doi: 10.1016/j.tig.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232–242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat Rev Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection? Nat. Rev Mol Cell Biol. 2010;11:567–578. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Beard C, Hanna J, Jaenisch R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat Methods. 2010;7:56–59. doi: 10.1038/nmeth.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol. 2006;34:455–465. doi: 10.1080/01926230600867743. [DOI] [PubMed] [Google Scholar]
- Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482:524–528. doi: 10.1038/nature10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- Guarente L. Mitochondria–a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev Biol. 1962;4:256–273. doi: 10.1016/0012-1606(62)90043-x. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu Rev Pathol Mech Dis. 2009;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson J, Rafiee MR, Reiland S, Polo JM, Gehring J, Okawa S, Huber W, Hochedlinger K, Krijgsveld J. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep. 2012;2:1579–1592. doi: 10.1016/j.celrep.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, Jaenisch R. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell. 2008;3:346–353. doi: 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Lamming DW. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016;23:990–1003. doi: 10.1016/j.cmet.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Khanna PB, Davies I, Faragher EB. Age-related changes in the stomach of the laboratory mouse: a quantitative morphological study. Age Ageing. 1988;17:257–264. doi: 10.1093/ageing/17.4.257. [DOI] [PubMed] [Google Scholar]
- Kurban RS, Bhawan J. Histologic changes in skin associated with aging. J Dermatol Surg Oncol. 1990;16:908–914. doi: 10.1111/j.1524-4725.1990.tb01554.x. [DOI] [PubMed] [Google Scholar]
- Kurian L, Sancho-Martinez I, Nivet E, Aguirre A, Moon K, Pendaries C, Volle-Challier C, Bono F, Herbert JM, Pulecio J, et al. Conversion of human fibroblasts to angioblast-like progenitor cells. Nat Methods. 2013;10:77–83. doi: 10.1038/nmeth.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez J-M, De Vos J, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–2253. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang J-D, Li KM, Chau PY, Chen DJ, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang Z, Zhang L, Ghosh S, Zheng H, Zhou Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat Commun. 2013a;4:1868. doi: 10.1038/ncomms2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, Hurgo BMC, Leavitt T, Shih J, Brunet A, Rando TA. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013b;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Mahmoudi S, Brunet A. Aging and reprogramming: a two-way street. Curr Opin Cell Biol. 2012;24:744–756. doi: 10.1016/j.ceb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcell TJ. Sarcopenia: causes, consequences, and preventions. J Gerontol A Biol Sci Med Sci. 2003;58:M911–M916. doi: 10.1093/gerona/58.10.m911. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Scheibye-Knudsen M, Longo DL, de Cabo R. Animal models of aging research: implications for human aging and age-related diseases. Annu Rev Anim Biosci. 2015;3:283–303. doi: 10.1146/annurev-animal-022114-110829. [DOI] [PubMed] [Google Scholar]
- Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156:663–677. doi: 10.1016/j.cell.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Osorio FG, Navarro CL, Cadiñanos J, López-Mejía IC, Quirós PM, Bartoli C, Rivera J, Tazi J, Guzmán G, Varela I, et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra107. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- Partridge L. Intervening in ageing to prevent the diseases of ageing. Trends Endocrinol Metab. 2014;25:555–557. doi: 10.1016/j.tem.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Pollina EA, Brunet A. Epigenetic regulation of aging stem cells. Oncogene. 2011;30:3105–3126. doi: 10.1038/onc.2011.45. [DOI] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. doi: 10.1016/j.cell.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin MM, Kushner JA. Adaptive β-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–1372. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Merkwirth C, De Magalhaes Filho CD, Dillin A. Signaling Networks Determining Life Span. Annu Rev Biochem. 2016;85:35–64. doi: 10.1146/annurev-biochem-060815-014451. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Panopoulos AD, Montserrat N, Multon MC, Daury A, Rocher C, Spanakis E, Batchelder EM, Orsini C, Deleuze JF, Izpisua Belmonte JC. Generation of a drug-inducible reporter system to study cell reprogramming in human cells. J Biol Chem. 2012;287:40767–40778. doi: 10.1074/jbc.M112.384024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166:822–839. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultoukis GA, Partridge L. Dietary Protein, Metabolism, and Aging. Annu Rev Biochem. 2016;85:5–34. doi: 10.1146/annurev-biochem-060815-014422. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Steffen KK, Dillin A. A Ribosomal Perspective on Proteostasis and Aging. Cell Metab. 2016;23:1004–1012. doi: 10.1016/j.cmet.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol. 1999;8:29–39. doi: 10.1016/s1054-8807(98)00023-4. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of β-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Thier M, Wörsdörfer P, Lakes YB, Gorris R, Herms S, Opitz T, Seiferling D, Quandel T, Hoffmann P, Nöthen MM, et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell. 2012;10:473–479. doi: 10.1016/j.stem.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160–1163. doi: 10.1126/science.aaa1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Rakheja D, Yu X, Saxena R, Vaziri ND, Silva FG. The aging kidney. Kidney Int. 2008;74:710–720. doi: 10.1038/ki.2008.319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of LAKI 4F TTFs upon Short-Term Induction of OSKM, Related to Figure 1
(A) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in LAKI 4F tail tip fibroblasts (TTFs). **p < 0.005, ***p < 0.0005 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(B) Analysis of cellular identity of LAKI 4F TTFs following partial reprogramming analyzed by flow cytometry using Thy1 and SSEA1 staining.
(C) qPCR analysis of Nanog expression in LAKI 4F TTFs after short-term induction of OSKM by doxycycline treatment.
(D) Quantification of foci intensity and foci volume of γH2AX staining in LAKI 4F TTFs. ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(E) Immunostaining of 53BP1 in LAKI 4F TTFs following doxycycline treatment. The number, volume, and intensity of 53BP1 foci in LAKI 4F TTFs were quantified in Oct4 positive and negative cells. Scale bar, 10 μm. *p < 0.05, **p < 0.01, ***p < 0.0005 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(F) β-galactosidase activity in WT and LAKI 4F TTFs. Quantification of cells with β-galactosidase staining. Scale bar, 100 μm. ***p = 0.001 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(G) Levels of mitochondrial ROS in LAKI 4F TTFs after doxycycline treatment. **p < 0.01, according to one-way ANOVA with Bonferroni correction.
Data are presented as mean ± SEM.
Figure S2. Characterization of LAKI 4F and LAKI TTFs following Doxycycline Treatment, Related to Figure 1
(A) Detection of Lamin A/C and progerin levels in WT 4F and LAKI 4F TTFs after short-term doxycycline treatment by western blot. Arrowheads denote Lamin A and C. Asterisk denotes progerin.
(B) qPCR analysis of Lamin A and Lamin B1 levels in LAKI 4F TTFs.
(C) Analysis by flow cytometry of programmed cell death in LAKI 4F TTFs following doxycycline treatment.
(D) Immunofluorescence of γH2AX in LAKI TTFs following doxycycline treatment.
(E) Immunofluorescence of H3K9me3 in LAKI TTFs following doxycycline treatment.
(F) qPCR analysis of stress response genes in the p53 pathway, senescence-associated metalloprotease MMP13 and interleukin-6 in LAKI TTFs following doxycycline treatment.
(G) Immunofluorescence of Lamin A/C in LAKI TTFs following doxycycline treatment.
(H) Levels of mitochondrial ROS in LAKI TTFs after doxycycline treatment.
(I) Immunofluorescence of Oct4, Sox2, c-Myc and Lamin A/C in LAKI TTFs infected with GFP, Oct4, Sox2 and c-Myc retroviruses. Scale bar, 10 μm.
Figure S3. Effect of Long-Term In Vivo Cyclic Induction of Oct4, Sox2, Klf4, and c-Myc in Mice Carrying One and Two Copies of OSKM and rtTA Cassette, Related to Figure 3
(A) Body weight of wild-type (WT) and 4F mice carrying single copy of OSKM and rtTA cassette upon cyclic administration of doxycycline. WT (n = 5) and 4F (n = 3).
(B) Survival of WT and 4F mice carrying single copy of OSKM and rtTA cassette upon cyclic administration of doxycycline. WT (n = 5) and 4F (n = 3).
(C and D) Immunostaining of Ki67 and Nanog in spleen, liver, and kidney of WT and 4F mice carrying single copy of OSKM and rtTA cassette. Scale bar, 100 μm (spleen) and 50 μm (liver and kidney).
(E) Immunostaining of Sox2 and Ki67 in spleen, liver, and kidney of 4F mice carrying two copies of OSKM and rtTA cassette. Scale bar, 50 μm.
Data are presented as mean ± SEM.
Figure S4. Characterization of LAKI 4F Mice Subjected to Cyclic Induction of OSKM, Related to Figure 4
(A) Representative picture of LAKI 4F mice at 16-weeks of age upon doxycycline administration. Curvature of spine can be observed in LAKI 4F not treated with doxycycline (−Dox).
(B) Partial rescue of spleen involution is observed in LAKI 4F mice upon doxycycline administration.
(C) Histological analysis of liver, heart, and muscle of WT and LAKI 4F mice upon cyclic doxycycline administration. Scale bar, 50 μm.
Figure S5. Molecular Changes Induced by Cyclic In Vivo Induction of Oct4, Sox2, Klf4, and c-Myc, Related to Figure 5
(A) Immunohistochemistry of Nanog in skin and kidney of WT and LAKI 4F mice upon cyclic doxycycline administration. Teratoma was used as a positive control. Scale bar, 50 μm.
(B) qPCR analysis of stress response genes in the p53 pathway and interleukin-1α in liver, stomach, and kidney of LAKI 4F mice upon cyclic doxycycline administration. *p < 0.05, **p < 0.01, ***p < 0.0001 and ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(C) Detection of Lamin A/C and progerin levels in skin, spleen, and stomach of WT and LAKI 4F mice by western blot. Arrowheads denote Lamin A and C. Asterisk denotes progerin.
Data are presented as mean ± SEM.
Figure S6. Amelioration of Aging-Associated Phenotypes in Wild-Type Mice and Human Cells by Short-Term Expression of Reprogramming Factors, Related to Figure 6
(A) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in WT 4F TTFs. *p < 0.05, **p < 0.05, ***p < 0.005, ****p < 0.0001.
(B) Quantification of foci intensity and foci volume of γH2AX staining in WT 4F TTFs. ****p < 0.0001, according to one-way ANOVA with Bonferroni correction.
(C) Analysis of ROS levels in LAKI 4F TTFs after doxycycline treatment. *p < 0.05, according to one-way ANOVA with Bonferroni correction.
(D) qPCR analysis of OCT4, SOX2, KFL4 and c-MYC in human 4F fibroblasts.
Data are presented as mean ± SEM.
Figure S7. Analysis of Pancreatic Function and Skeletal Muscle Regeneration in Wild-Type Mice, Related to Figure 7
(A) Glucose tolerance test (GTT) in 2-month and 12-month old WT mice following beta cell ablation by low dose (50 mg/kg) STZ (2-month WT n = 3; 12-month WT n = 3).
(B) qPCR analysis of Oct4, Sox2, in pancreas of 12-month old WT 4F mice upon administration of doxycycline. **p < 0.01, according to one-way ANOVA with Bonferroni correction.
(C) qPCR analysis of Glut2 in pancreas of 12-month old WT 4F mice upon administration of doxycycline.
(D) GTT in 12-month old WT mice following doxycycline and beta cell ablation by low dose (50 mg/kg) STZ (−Dox n = 5; +Dox n = 5).
(E) Representative image of H&E staining and immunostaining of Laminin of tibialis anterior (TA) muscle of 2-month and 12-month old WT mice following muscle injury by CTX injection. Scale bar, 50 μm.
(F) Quantification of fiber cross-sectional area frequency distribution and percentage of central nucleated fibers in muscle sections of 2-month and 12-month old WT mice following muscle injury by CTX injection. (2-month WT n = 3; 12-month WT n = 3). ****p < 0.0001, according to two-tailed Student’s t test.
(G) qPCR analysis of Oct4, Sox2, Klf4 and c-Myc in skeletal muscle of 12-month WT 4F mice upon administration of doxycycline. *p < 0.05, **p < 0.01, according to two-tailed Student’s t test.
(H) Representative low- and high-magnification images of H&E staining of TA muscle of 12-month-old WT mice following doxycycline treatment and muscle injury by CTX injection. Scale bar, 50 μm.
(I) Quantification of percentage of central nucleated fibers in muscle sections of 12-month old WT mice following doxycycline treatment and muscle injury by CTX injection (−Dox n = 3; +Dox n = 3).