

Abstract
Background
Familial aggregation has been described for primary mitral regurgitation (MR) caused by mitral valve prolapse (MVP). We hypothesized that heritability of MR exists across different MR subtypes including non-primary MR.
Methods and Results
Study participants were Framingham Heart Study (FHS) Generation 3 (Gen 3) and Generation 2 (Gen 2) cohort participants and all adult Swedish siblings born after 1932 identified in 1997 and followed through 2010. MR was defined as ≥ mild regurgitation on color Doppler in FHS and from ICD codes in Sweden. We estimated the association of sibling MR with MR in Gen 2/Gen 3/Swedish siblings. We also estimated heritability of MR in 539 FHS pedigrees (7580 individuals). Among 5132 FHS Gen 2/Gen 3 participants with sibling information, 1062 had MR. Of siblings with sibling MR, 28% (500/1,797) had MR compared with 17% (562/3,335) without sibling MR (multivariable-adjusted odds ratio, 1.20; 95% CI,1.01–1.43; p = 0.04). When we combined parental and sibling data in FHS pedigrees, heritability of MR was estimated at 0.15 (95% CI, 0.07–0.23), 0.12 (95% CI, 0.04–0.20) excluding MVP, and 0.44 (95% CI, 0.15–0.73) for ≥ moderate MR only (all p < 0.05). In Sweden, sibling MR was associated with a hazard ratio of 3.57 (95% CI, 2.21–5.76; p < 0.001) for development of MR.
Conclusions
Familial clustering of MR exists in the community, supporting a genetic susceptibility common to primary and non-primary MR. Further studies are needed to elucidate the common regulatory pathways that may lead to MR irrespective of its etiology.
Journal Subject Terms: Valvular Heart Disease, Echocardiography, Epidemiology, Genetics
Keywords: mitral regurgitation, echocardiography, epidemiology, genetic epidemiology, genetics, mitral valve prolapse
Mitral regurgitation (MR) is the most common form of valve disease, affecting more than 2 million people in the US.1 MR is characterized by incomplete coaptation of valve leaflets, resulting in regurgitant flow across the valve and reduced effective cardiac output.1 When severe, MR is associated with onset and worsening heart failure and decreased survival.1
MR constitutes an etiologically heterogeneous set of conditions, similar to aortic stenosis, atrial fibrillation and heart failure. Whereas primary MR is most commonly caused by mitral valve prolapse (MVP), secondary or functional MR results from left ventricular (LV) dilation such as seen in dilated cardiomyopathies or isolated myocardial infarction causing papillary muscle displacement and leaflet tethering.1 Recent literature demonstrates that mitral valve leaflets are not innocent bystanders in functional MR, but are able to grow in response to tethering in both humans and in animal models.2–4 Interestingly, not all patients with coronary heart disease or other causes of LV dilatation develop significant MR, suggesting genetic variability in the ability to compensate for any potential leaflet tethering. Also, similar reactivation of embryonic development pathways has been demonstrated in primary MR due to MVP.5, 6 Moreover, mild- moderate degrees of MR are often observed in clinical practice without a clear etiology (absence of calcification, congenital conditions, or rheumatic involvement), and may be the result of leaflet response to subtle mechanical stress in the setting of hypertension or increased afterload.7
A familial component has been described for specific etiologies of MR such as MVP8, 9 and reported in a recent twin study,10 but has not been systematically studied across both primary and non-primary subtypes of MR in whole pedigrees. We postulate that genetic susceptibility and familial clustering of MR can be identified irrespective of etiology of MR in both the Framingham Heart Study (FHS) cohort and the entire Swedish population, two different, but complementary, datasets.
METHODS
Framingham Heart Study
Participants
The FHS is a multigenerational community-based cohort study including residents of the town of Framingham, Massachusetts. Beginning in 1948, 5209 men and women were enrolled into the Original cohort.11 Their offspring, and the offsprings’ spouses, were enrolled into the Offspring cohort (n=5124) starting in 1971. Examination cycles were performed at approximately 4 to 8 year intervals, with comprehensive echocardiograms and Doppler color flow imaging obtained at examination cycles 4,5,6, and 8. In our investigation, study participants included Generation 3 individuals (Gen 3; Examination 1, 2002–2005) with at least one parent identified in the Offspring cohort (Gen 2; Examinations 6 or 8, 1996–1998 and 2005–2008, respectively) or in the New Offspring Spouse cohort (Examination 1) (Figure 1). For a separate sibling analysis, study participants included Gen 2 and Gen 3 participants with at least one sibling at Gen 2 Examinations 6/8, and Gen 3 Examination 1, respectively (Figure 1). Participants were excluded if concomitant diagnoses of mitral or aortic stenosis (rheumatic or calcific, with or without history of surgery) were present (Gen 2/New Offspring Spouse cohort N = 32; Gen 3 N = 5). The study protocol was approved by the Institutional Review Board of Boston University Medical Center, and all participants provided written informed consent.
Figure 1.
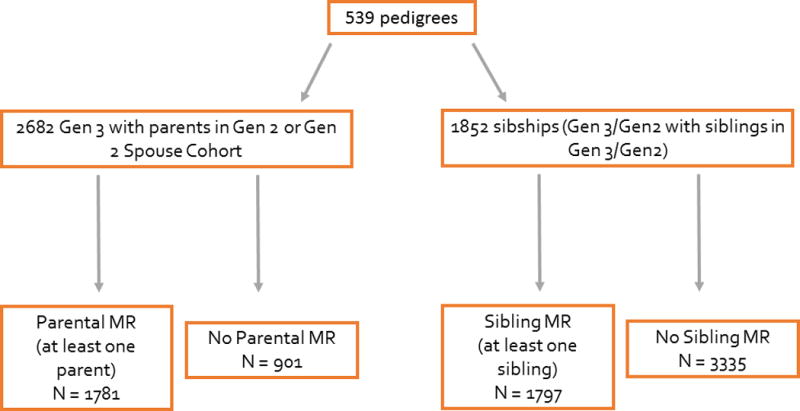
Schematic overview of the Framingham Heart Study cohorts according to parental or sibling mitral regurgitation (MR). Gen 2/Gen 3 = Generation 2/Generation 3.
Clinical Characteristics
Clinical variables used in the present investigation included: age, sex, and body mass index (BMI). History of smoking (using chronic obstructive pulmonary disease as a surrogate of significant tobacco use), diabetes, hypertension treatment, and systolic/diastolic blood pressure were included in the analysis as potential valve “stressors” that may influence the progression of MR. Risk factors for cardiovascular disease such as diet (total fat, protein, calories), physical activity index (calculated as described in a previous FHS investigation),12 and lipid levels (total cholesterol/high density lipoprotein) were also included among the clinical variables. Moreover, we determined if any of the study participants had a history of heart failure or myocardial infarction to account for any potential common genetic substrate for myopathy (ischemic or non-ischemic) and the development of secondary MR.
Echocardiographic characteristics
All study participants in the Gen 3, Gen 2, and New Offspring Spouse cohorts underwent standard two-dimensional echocardiography with a commercially available system (Sonos 1000, Hewlett–Packard Medical Products, Andover, MA) that used a 2.5-MHz transducer. Images included complete parasternal, apical, and subcostal views and color Doppler assessment of MR; they were stored on VHS and digitized for subsequent review. All measurements were performed with an off-line cardiac analysis system (Digiview, Houston, TX).
MR was assessed qualitatively by 2D color Doppler in a long-axis view and graded as trace, mild, moderate, moderate-severe or severe. Gen 3 MVP was diagnosed as leaflet displacement >2 mm beyond the mitral annulus in a parasternal or apical 3-chamber long-axis view.13 Gen 2 MVP was diagnosed using similar criteria at Examinations 6 or 8 (if 6 not available), and in the Offspring Spouse cohort at Examination 1.
Left atrial dimension was calculated by M-mode as the antero-posterior maximal left atrium diameter in systole. Left ventricular internal diameters were obtained in diastole and systole (LVIDd and LVIDs) by use of a leading-edge technique and averaging of M-mode measurements from at least three cardiac cycles. The fractional shortening percentage was calculated as (LVIDd − LVIDs)/ LVIDd × 100.
Case ascertainment
MR was defined regardless of etiology as ≥ mild regurgitation on Doppler color flow imaging. Etiology of MR was adjudicated as follows. If MVP was present, MR was considered primary. If no MVP was present and there was a clinical history of myocardial infarction or echocardiographic evidence of a regional wall motion abnormality or LV dilatation, MR was considered secondary and related to coronary heart disease or other etiology of LV dilatation. If none of the above conditions was present, MR was classified as idiopathic.
Finally, we determined if any of the participants with MR had asymmetric hypertrophy (as a surrogate for hypertrophic cardiomyopathy) or dilated cardiomyopathy (individuals with left ventricular cavity dilatation but no history of myocardial infarction), as both these conditions can be inherited and co-segregate with MR.
Nationwide Swedish Hospital Registers
Participants
All subjects born after 1932 and living in Sweden in 1997 with at least one sibling alive in 1997 were identified from nation-wide registers and included in this study (Figure 2). Siblings and spouses were identified from the Swedish Multigeneration Register.14 We protected anonymity by replacing the personal identification number with a serial number when linking data to hospital registers. The ethics committee at Lund University approved the study.
Figure 2.
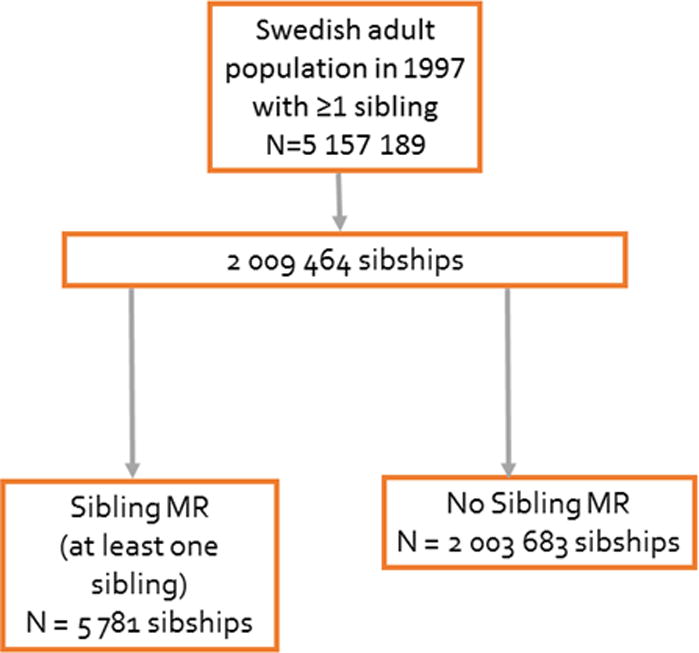
Schematic overview of the Swedish sibling cohort according to sibling mitral regurgitation (MR).
Clinical characteristics
Clinical variables included: age, sex, history of chronic obstructive pulmonary disease, hypertension, diabetes, obesity, and coronary heart disease. Such variables were identified from the National Patient Register (NPR) and International Classification of Diseases (ICD) diagnosis codes as described in the following section.
Case ascertainment
Ascertainment and validity of clinical diagnoses from nation-wide Swedish registers has been described previously.15, 16 All patients with a first diagnosis of MR were identified from the NPR. The NPR includes diagnosis codes from all hospital inpatient and outpatient visits in Sweden. Reporting to the NPR is mandatory and departmental reimbursements from the Swedish tax-financed healthcare system are based on ICD. The ninth version was used from 1987–1996 and the tenth version of ICD was used from 1997 and onwards. ICD definitions are shown in the Supplemental Material. Individuals with a diagnosis of MR and hypertrophic or dilated cardiomyopathy were also identified using ICD codes.
Statistical analysis
Framingham Heart Study
Clinical and echocardiographic characteristics were compared between Gen 3 participants with and without parental MR and, in separate analyses, Gen 2/Gen 3 participants with and without a sibling with MR. We performed t-tests to compare continuous variables and Chi-squared tests to compare binary variables (Fisher’s exact test for binary variables with low frequencies). We used logistic regression via generalized estimating equations (GEE) to estimate the associations of parental MR with the prevalence of MR in their Gen 3 offspring (pooling all etiologies). A similar regression model was fitted to estimate the association of MR in an individual with the prevalence of MR in a sibling in Gen 2/3. For each sibling pair, sibling 1 and sibling 2 were included twice in the analysis: once for sibling 1 as the outcome and sibling 2 as the risk factor, and once for sibling 2 as the outcome and sibling 1 as the risk factor. We used the GEE procedure to accommodate correlated responses. Sensitivity analyses were performed 1) excluding MVP and 2) including ≥ moderate MR cases only. Multivariable models were estimated adjusting for age, sex, BMI, systolic/diastolic blood pressure, diabetes, chronic obstructive pulmonary disease and history of myocardial infarction. All analyses were conducted using R (R Core Team [2014]. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org). We also estimated additive heritability of MR in full pedigrees (n pedigrees = 539, n individuals = 7580) using SOLAR liability threshold model. Sensitivity analyses were conducted 1) excluding MVP, 2) including ≥ moderate MR cases only, and 3) excluding MR cases with hypertrophic or dilated cardiomyopathy. Finally, separate heritability analyses were conducted for primary, secondary, and idiopathic MR. A two-sided p value < 0.05 was the criterion for statistical significance.
Nationwide Swedish Hospital Registers
Risk of MR with an affected sibling was evaluated using Cox regression, censoring at death or emigration, and with follow-up until December 31st, 2010. Adjustments were performed for age, sex, family size and cardiovascular risk factors (hypertension, diabetes, history of coronary heart disease). Two sensitivity analyses were performed, restricting sibling history to 1) siblings diagnosed with MR between 1997–2010 (the 10th version of the ICD was used during this time), and 2) history of surgery for MR. Variance estimation accounted for sibships according to Hemminki.17 Statistical analyses were performed in SAS version 9.3 (SAS Institute Inc. Cary, NC, USA). As information on twins was not available in the Swedish sample, heritability of MR was estimated using tetrachoric correlations between full siblings (n = 5,157,189) and half siblings (n = 832,507). This method has been shown to generate comparable heritability estimates to twin designs in Swedish data for other diseases.18 Similar to the FHS, a sensitivity analysis was conducted excluding MR cases with a diagnosis of hypertrophic or dilated cardiomyopathy in the MR heritability estimate. Statistical significance was defined by a two-sided p value < 0.05.
RESULTS
Framingham Heart Study
There were a total of 1761 cases of MR (138 primary, 223 secondary, and 618 idiopathic) among Gen2/Gen3 participants regardless of availability of sibling/parental MR data. Clinical and echocardiographic characteristics of the 3679 Gen 3 participants (53.2% women, mean age 40 years) with available parental information on MR status are summarized in Table 1. Gen 3 participants with parental MR (n=1781) were slightly older, had higher diastolic blood pressure and a greater number of participants with a history of prior myocardial infarction compared to the group without parental MR (n=901). Otherwise, the two groups were fairly similar with regards to sex, BMI, diet, physical activity, lipid levels, diabetes, and prior heart failure. Gen 3 participants with parental MR had a higher proportion of MR cases (mostly mild) and slightly larger mean left atrial diameter. They had a higher number of primary MR cases. Otherwise, the two groups were similar with regards to other etiologies and other grading of MR. They also had similar LV dimensions.
Table 1.
Baseline characteristics of study subjects according to parental or sibling mitral regurgitation in the Framingham Heart Study.
| Gen 3 participants with parental MR (n=1781) | Gen 3 participants with no parental MR (n=901) | P value | Gen 2/Gen3 participants with sibling MR (n=1797) | Gen 2/Gen 3 participants with no sibling MR (n=3335) | P value | |
|---|---|---|---|---|---|---|
| Clinical characteristics | ||||||
| Age, mean (SD), years | 41 (9) | 37 (8) | < 0.001 | 52 (13) | 45 (12) | < 0.001 |
| Sex (male), n (%) | 826 (46) | 428 (48) | 0.58 | 870 (48) | 1550 (46) | 0.19 |
| Diabetes, n (%) | 43 (2) | 21 (2) | 0.89 | 118 (7) | 151 (5) | 0.003 |
| SBP, mean (SD), mmHg | 116 (14) | 114 (13) | < 0.001 | 123 (17) | 119 (16) | < 0.001 |
| DBP, mean (SD), mmHg | 75 (10) | 74 (9) | 0.002 | 75 (10) | 75 (10) | 0.99 |
| Hypertension Treatment, n (%) | 152 (9) | 44 (5) | < 0.001 | 409 (23) | 440 (13) | < 0.001 |
| BMI, mean (SD), kg/m2 | 26.4 (5.0) | 26.3 (5.1) | 0.83 | 27.3 (5.1) | 27 (5.3) | 0.08 |
| Fat intake, mean (SD), gm | 76 (36) | 79 (38) | 0.14 | 78 (36) | 77 (37) | 0.51 |
| Protein intake, mean (SD), gm | 97 (47) | 100 (46) | 0.14 | 98 (43) | 98 (47) | 0.95 |
| Calories, mean (SD), kcal | 2134 (893) | 2190 (903) | 0.15 | 2142 (867) | 2131 (890) | 0.75 |
| Physical activity index, mean (SD) | 38 (8) | 38 (8) | 0.84 | 37 (8) | 38 (7.9) | 0.59 |
| Total cholesterol/HDL | 3.75 (1.32) | 3.75 (1.6) | 0.93 | 3.79 (1.35) | 3.76 (1.44) | 0.66 |
| Heart failure, n (%) | 1 (0.06) | 2 (0.2) | 0.26 | 13 (0.7) | 19 (0.6) | 0.52 |
| History of myocardial infarction, n (%) | 10 (0.6) | 0 (0) | 0.02 | 46 (2.6) | 51 (1.5) | 0.02 |
| Chronic obstructive pulmonary disease, n (%) | 66 (4) | 26 (4) | 0.36 | 91 (6) | 147 (5) | 0.37 |
| Echocardiographic characteristics | ||||||
| MR (total), n (%) | 262 (15) | 90 (10) | < 0.001 | 500 (28) | 562 (17) | < 0.001 |
| Primary versus secondary versus idiopathic MR, n (%) | 27/2/150 (2/0.1/8) | 5/2/59 (0.6/0.2/7) | 0.03/0.61/0.09 | 38/68/192 (2/4/11) | 47/61/230 (1/2/7) | 0.075/<0.001/<0.001 |
| LVIDd, mean (SD), mm | 4.9 (0.40) | 4.9 (0.4) | 0.53 | 4.9 (0.5) | 4.9 (0.4) | 0.49 |
| LVIDs, mean (SD), mm | 3.2 (0.3) | 3.2 (0.3) | 0.24 | 3.1 (0.4) | 3.1 (0.4) | 0.27 |
| LADs, mean (SD), mm | 3.7 (0.5) | 3.6 (0.5) | 0.002 | 3.9 (0.6) | 3.8 (0.5) | < 0.001 |
| Mild/moderate/mod-severe/severe MR, n (%) | 247/15/0/0 (14/0.8/0/0) | 84/5/1/0 (9/0.6/0.1/0) | <0.001/0.41/0.36 /NA | 461/37/2/0 (26/2/0.1/0) | 537/21/4/0 (16/0.6/0.1/0) | < 0.001/< 0.001/0.93/NA |
MR = mitral regurgitation; SD = standard deviation; SBP/DBP = systolic diastolic blood pressure; BMI = body mass index; HDL = high density lipoprotein; LVIDd/LVIDs: left ventricular internal diameters in diastole/systole; LADs = left atrial dimension in systole.
There were 1852 sibships (average sibship size 2.8, 792 sibships with at least one affected sibling). Similarly to participants with parental MR, Gen 2/Gen 3 participants with sibling MR (n =1797) (Table 1) were slightly older, had higher blood pressure and a higher proportion of individuals with prior myocardial infarction compared to the group without sibling MR (n=3335). Diet, physical activity, and lipid levels were not significantly different between the two groups. The sibling MR group had a higher proportion of MR cases (mostly mild and moderate) and a slightly larger left atrial size. There were more participants with secondary MR compared to Gen 2/Gen 3 participants without sibling MR. The two groups were similar with regards to LV dimensions.
Presence of both parental and sibling MR was associated with a greater odds of prevalent MR in offspring and sibs, respectively: 14% (n=262) offspring with parental MR had MR compared with 10% (n=90) without parental MR (multivariable-adjusted odds ratio [OR], 1.31; 95% confidence interval [CI], 0.98–1.74; p = 0.07); 28% (n=500/1,797) of siblings with sibling MR had MR compared with 17% (n=562/3,335) without sibling MR (multivariable-adjusted OR, 1.20; 95% CI,1.01–1.43; p = 0.04). These results did not change significantly when adjusting for age and sex alone (without the covariate history of myocardial infarction) (model 1, Table 2) and after excluding MVP (model 3). Results were reinforced after restricting to ≥ moderate MR (model 4), even in non-primary cases only (model 5).
Table 2.
Risk of mitral regurgitation according to parental or sibling mitral regurgitation in the Framingham Heart Study.
| Mitral Regurgitation | ||||
|---|---|---|---|---|
| OR | 95% CI | P value | ||
| Parental mitral regurgitation | ||||
| Model 1: sex + age | 1.34 | 1.00 | 1.79 | 0.04 |
| Model 2: model 1 + risk factors* | 1.31 | 0.98 | 1.74 | 0.07 |
| Model 3: model 2 excluding MVP | 1.25 | 0.93 | 1.67 | 0.14 |
| Model 4: model 2 + ≥ moderate MR only | 1.42 | 0.97 | 2.06 | 0.07 |
| Model 5: model 3 + ≥ moderate MR only | 1.35 | 0.92 | 1.99 | 0.13 |
| Sibling mitral regurgitation | ||||
| Model 1: sex + age | 1.25 | 1.06 | 1.48 | 0.01 |
| Model 2: model 1 + risk factors* | 1.20 | 1.01 | 1.43 | 0.04 |
| Model 3: model 2 excluding MVP | 1.23 | 1.02 | 1.48 | 0.03 |
| Model 4: model 2 + ≥ moderate MR only | 1.78 | 1.25 | 2.53 | 0.002 |
| Model 5: model 3 + ≥ moderate MR only | 1.67 | 1.10 | 2.54 | 0.02 |
OR = odds ratio; CI = confidence interval; risk factors* = body mass index, systolic/diastolic blood pressure, diabetes, chronic obstructive pulmonary disease, history of myocardial infarction; MVP = mitral valve prolapse.
Multivariable-adjusted heritability of MR was estimated at 0.15 (95% CI, 0.07–0.23; p = < 0.001) in the FHS– based pedigree analysis. Heritability of MR was similar at 0.12 (95% CI, 0.04–0.20; p < 0.001) in the sensitivity analysis excluding individuals with MVP and more prominent at 0.44 (95% CI, 0.15–0.73; p = 0.001) when including ≥ moderate MR cases only. Multivariable-adjusted heritability of MR did not change (0.15; 95% CI, 0.07–0.23; p = < 0.001) when we excluded the 7 participants with available parental/sibling MR data and a diagnosis of hypertrophic cardiomyopathy (N = 3) or dilated cardiomyopathy (N = 4). Separate multivariable-adjusted heritability estimates for the 3 MR categories were as follows: 0.47 (95% CI, 0.20–0.74; p < 0.001) for primary MR, 0.18 (95% CI, −0.25–0.43; p = 0.21) for secondary MR, and 0.47 (95% CI, 0.20–0.74; p < 0.001) for idiopathic MR.
Nationwide Swedish Hospital Registers
Between 1997 and 2010 8,628 subjects were diagnosed with MR from a population of 5,157,189 subjects from Sweden (Table 3). The proportions of MR cases with hypertrophic and dilated cardiomyopathy were 1% and 5%, respectively. The group with sibling MR (N = 18,891) had larger family sizes and a nominally significant higher proportion of individuals diagnosed after 50 years of age (65%) compared to the group without sibling MR (n = 5,138,298). The two groups were similar with regards to sex distribution and cardiovascular risk factors (including history of myocardial infarction). A sibling history of MR was present in 0.4% (18,891/5,157,189) of all Swedish individuals, and 2.8% (239/8,628) of MR cases. One percent (n= 239/18891) of siblings with sibling MR had MR compared with 0.2% (n = 8,389/5,138,298) without sibling MR, corresponding to a hazard ratio of 4.00 (95% CI, 2.48–6.44; p<0.001) (Figure 3) adjusted for age and sex. Additional adjustment for family size and cardiovascular risk factors resulted in a slight risk attenuation (HR 3.57; 95% CI, 2.21–5.76; p<0.001) (Figure 3). The increased long-term MR risk in Swedish siblings in the presence of a sibling history of MR is shown in Figure 4. Results were similar in sensitivity analyses restricted to siblings diagnosed with MR between 1997–2010 (reflective of ICD10 coding of MR) and siblings undergoing valve surgery for MR, respectively (Figure 3). Of 2 million Swedish families, only 2 had 2 or more affected siblings. Among the 7 subjects originating from these 2 families, 6 were diagnosed with MR. Overall, the risk of MR was driven by the majority of families with one affected sibling (233 MR cases among 18,891 subjects with sibling MR; HR = 3.62, 95% CI, 3.18–4.13; p<0.001).
Table 3.
Baseline characteristics of study subjects according to sibling history of mitral regurgitation (MR) in the Swedish population.
| No sibling history of MR | Sibling history of MR | P value | |||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Population | 5,138,298 | 18,891 | |||
| Mitral regurgitation | 8,389 | 0.16 | 239 | 1.2 | <0.001 |
| Valvular surgery | 2,233 | 0.04 | 83 | 0.4 | <0.001 |
| Sex | 0.51 | ||||
| Men | 2,627,510 | 51.1 | 9,522 | 50.4 | |
| Women | 2,510,788 | 48.9 | 9,369 | 49.6 | |
| Age at MR diagnosis (years) | 0.09 | ||||
| <20 | 836 | 10.5 | 25 | 10.7 | |
| 20–29 | 351 | 4.4 | 5 | 2.1 | |
| 30–39 | 673 | 8.4 | 11 | 4.7 | |
| 40–49 | 1,509 | 1.9 | 41 | 17.5 | |
| 50–59 | 3,038 | 38.0 | 100 | 42.7 | |
| 60–69 | 1,579 | 19.8 | 52 | 22.2 | |
| Family size | <0.001 | ||||
| Two children | 2,433,566 | 47.4 | 3,823 | 20.2 | |
| Three children | 1,608,473 | 31.3 | 5,005 | 26.5 | |
| Four children | 629,813 | 12.2 | 3,764 | 20.0 | |
| Five or more children | 466,446 | 9.1 | 6,299 | 33.3 | |
| Chronic Obstructive Pulmonary Disease | 0.93 | ||||
| No | 4,968,928 | 96.7 | 18,007 | 95.3 | |
| Yes | 169,370 | 3.3 | 884 | 4.7 | |
| Diabetes | 0.71 | ||||
| No | 4,999,508 | 97.3 | 17,741 | 93.9 | |
| Yes | 138,790 | 2.7 | 1,150 | 6.1 | |
| Obesity | 0.15 | ||||
| No | 5,081,788 | 98.9 | 18,683 | 98.9 | |
| Yes | 56,510 | 1.1 | 208 | 1.1 | |
| Hypertension | 0.12 | ||||
| No | 4,873,515 | 94.8 | 16,274 | 86.1 | |
| Yes | 264,783 | 5.2 | 2,617 | 13.9 | |
| Coronary Heart Disease | 0.89 | ||||
| No | 4,998,073 | 97.3 | 17,135 | 90.7 | |
| Yes | 140,225 | 2.7 | 1,756 | 9.3 | |
Figure 3.
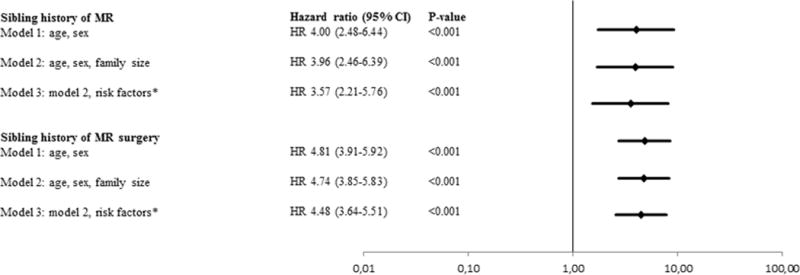
Sibling risk of mitral regurgitation (MR) in the Swedish population. *Risk factors = history of obesity, hypertension, diabetes, chronic obstructive pulmonary disease, coronary heart disease.
Figure 4.
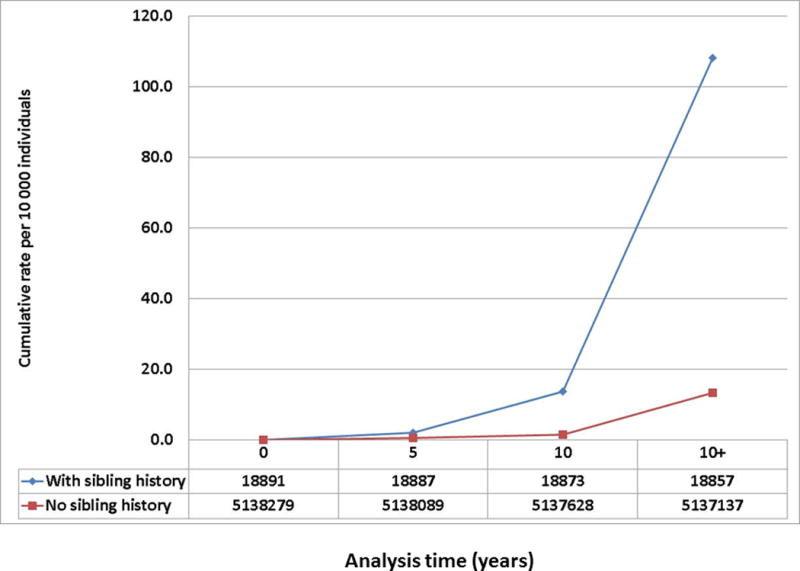
Risk of mitral regurgitation in Swedish siblings by sibling history.
Multivariable-adjusted heritability of MR was estimated at 0.52 (95% CI, 0.48–0.56) in the Swedish full sibling and half sibling analysis. The estimated heritability did not change after excluding individuals with hypertrophic or dilated cardiomyopathy (0.51; 95% CI, 0.48–0.56).
DISCUSSION
Our study demonstrates that familial clustering of MR exists in the community, supporting a genetic susceptibility common to primary and non-primary MR. Our findings are particularly relevant, as they originate from two different but complementary community-based studies: the FHS and the nationwide Swedish registries. In the former, diagnosis of MR was based on routine echocardiography and comprised mostly mild-moderate MR cases. In Swedish registries, MR diagnosis was based on nation-wide clinical/inpatient ICD coding and largely included moderate and severe cases. Accordingly, the risk ratios for familial clustering were lower in FHS, and considerably higher in Swedish registry data, but were comparably high in a FHS sensitivity analyses restricted to at least moderate cases.
Currently, a distinct separation between primary and secondary MR etiologies exists with regards to pathophysiology and genetic substrate. MVP (primary MR) is typically considered a problem of “excessive leaflet growth” and has a strong heritable component.8 Recent genetic studies have identified mutations in genes involved in the organization, assembly and alignment of valvular interstitial cells and extra-cellular matrix into a trilaminar architecture (filamin A, DCHS1, TNS1, and LCMD1) with consequent dysregulation of extra-cellular matrix in functional models.19–21 Conversely, secondary MR is the result of “insufficient leaflet growth” with less MR seen in tethered valves with more prominent leaflet elongation, and a similar dysregulation of extra-cellular matrix observed in MVP.3 Despite the individual variability in developing MR in the setting of LV systolic dysfunction/dilatation, a genetic susceptibility to secondary MR has never been postulated. Moreover, mild-moderate MR in the absence of MVP or other primary etiologies, and without papillary muscle displacement or leaflet tethering has traditionally been considered “idiopathic” and without a clear pathophysiologic or genetic background. In our study, presence of sibling MR in FHS was associated with greater odds of prevalent MR in siblings. The association with parental MR was weaker, likely due to lower statistical power to detect a significant difference compared to the no parental MR group. Full pedigree heritability analysis demonstrated that 15% of the total variation of the MR trait in the FHS sample was due to genetic variation. This proportion did not change significantly after excluding participants with MVP in a sensitivity analysis, suggesting that familial clustering of MR is not exclusively explained by a traditionally inherited condition such as MVP. These findings were confirmed in the Swedish population (over 5 million individuals), in which the hazard of MR was almost four times higher in the presence of a sibling history of MR. Furthermore, primary results were reinforced after restricting to individuals with ≥ moderate MR (FHS) or treated surgically (Swedish sample), suggesting a spectrum of effect of sibling MR on development of MR based on severity of disease. We found substantially higher MR heritability estimates in Sweden (52%) than in the FHS (15%). This is consistent with the greater severity of MR observed in the Swedish study design (with at least moderate severity in >75% of cases as described in a recent validation study)16 as compared to FHS where cases were mostly mild (Table 1). Indeed, the estimate was comparable to the heritability estimate from the sensitivity analysis of FHS with ≥ moderate MR (44%). We also note that the magnitude of MR heritability in Sweden was comparable to that of other complex diseases including coronary artery disease (40–60%),22atrial fibrillation (62%),23 and venous thromboembolism (47%).24
Whereas multiple genetic variants have been associated with coronary heart disease,25 genetic predisposition to the development of secondary MR (independent of coronary heart disease inheritance) has not been established. In our investigation, risk estimates of sibling MR remained statistically significant after adjusting for history of myocardial infarction in the multivariable models in both FHS and Swedish samples. On the other hand, MR heritability was not statistically significant in a separate FHS pedigree analysis that included secondary MR only. The lack of statistical significance of secondary MR heritability was likely due to a small number of cases included in the pedigree analysis (only 4 of Gen 3 participants with available parental information on MR had secondary MR – 2 with and 2 without parental MR- see Table 1). As the diagnosis of secondary MR was not available in the Swedish registries, familial aggregation of secondary MR could not be explored in the Swedish sample. Larger studies in different populations are needed to better understand the heritability of secondary MR.
When we assessed the proportion of cases of MR based on etiology, the majority of Gen 3 subjects with either parental or sibling MR were classified as idiopathic. In a separate analysis assessing the heritability of idiopathic MR alone, heritability estimates were as high as those for primary (MVP-related) MR. In vitro studies have demonstrated histological changes in mitral valves in response to increased blood pressure or afterload.7 The ability of human mitral leaflets to grow in response to valve leaflet stressors (i.e. blood pressure, smoking or diabetes) and whether this ability is genetically determined remain to be determined.
Strengths and Limitations
The strengths of the FHS investigation include the unique availability of multi-generational clinical and echocardiographic data allowing detailed phenotyping and pedigree analysis. In addition, MR was diagnosed blinded to parental or sibling MR status, and risk factors potentially contributing to MR risk (blood pressure, age, sex, BMI, history of heart failure or myocardial infarction etc.) were routinely ascertained. The limitations of the FHS are as follows: first, our analysis was limited to a single sample of European ancestry and the results may not be generalizable to other races/ethnicities. Second, the parental MR sample size was small; hence, some of the statistically non-significant comparisons may have been underpowered. Third, not all siblings and parents were included in the FHS so there may be some misclassification of presence versus absence of MR in pedigrees. In the analysis of association between sibling MR and MR, a sibling may not have been recruited if he/she refused enrollment, moved out of town, or died. Among the mechanisms of missing data, death could be related to MR status. However, the relationship between MR and death may be substantially weakened after adjusting for age, sex, BMI, systolic/diastolic blood pressure, and history of myocardial infarction. A residual relationship between MR and death may be present despite adjusting for such risk factors. In this case, it is more likely that healthy siblings were ultimately included in the study, thus reducing the number of affected sibling pairs, and causing underestimation of the odds ratio. Fourth, as data on Generation 3 was available at only a single time point, we could not assess the association of parental or sibling MR with longitudinal development of MR in FHS offspring or siblings, respectively. Hence, survival analysis was not used in the FHS sample.
The major strengths of the Swedish population registries are the large sample size (> 5 million individuals) facilitating detection of small differences in effect size between comparison groups. Whereas the diagnosis of MR was based on routine echocardiography in the FHS, in the Swedish population ICD9 and ICD 10 coding included more severe, clinically apparent cases. Therefore, our study results were consistent across two different epidemiologic settings with different MR disease severities. Among the limitations of Swedish registries was the lack of detailed phenotyping as diagnoses were based on ICD 9 and 10 coding. The Swedish population is largely of European ancestry and is characterized by unique lifestyle and climate. Hence, findings may not be generalizable to individuals of other ancestries or with different environmental conditions. Finally, some diagnoses such as obesity or hypertension may not be routinely coded using ICD9 and ICD10 codes, hence their prevalence may be underestimated in Swedish registries.
Clinical and Research Implications
Our study demonstrates familial clustering of MR irrespective of etiology. “Upstream”, genetically determined regulatory mechanisms able to influence either excessive (primary MR) or insufficient (secondary MR) leaflet growth may be postulated and investigated in further studies. Similarly to other inherited cardiovascular conditions, we established that only a portion of variation in the MR phenotype is due to genetic variation. The role of environmental factors and the interaction between genes and environment in MR expression remains to be determined. Moreover, establishing heritability of MR in the community may clarify the utility and cost-effectiveness of screening family members, which to date has not been performed on a routine basis.
CONCLUSIONS
Familial clustering of MR exists in the community, supporting a genetic susceptibility common to primary and non-primary MR subtypes, which represents a novel finding. Further studies are needed to elucidate common regulatory pathways that may lead to MR irrespective of etiology.
Supplementary Material
Clinical Perspective.
Mitral regurgitation (MR) is the most common form of valve disease, affecting more than 2 million people in the United States. A familial component has been described for specific etiologies of MR such as mitral valve prolapse leading to primary MR. However, it has not been systematically studied across both primary and non-primary subtypes of MR. We demonstrate that familial clustering of MR can be identified irrespective of etiology of MR in both the Framingham Heart Study cohort and the entire Swedish population, two different but complementary datasets. In the former, diagnosis of MR was based on routine echocardiography and comprised mostly mild-moderate MR cases. In Swedish registries, MR diagnosis was based on nationwide clinical/inpatient ICD coding and largely included more severe cases. Establishing heritability of MR in the community may clarify the utility and cost-effectiveness of screening family members, which to date has not been performed on a routine basis.
Acknowledgments
SOURCES OF FUNDING: Grant support from NIH K23 HL116652 (FND), contracts HHSN268201500001I, contract No1-HL25195, and R01HL126136-01A1 (VSR), the European Research Council, the Wallenberg Center for Molecular Medicine in Lund, the Swedish Heart-Lung Foundation, the Swedish Research Council, the Crafoord Foundation, governmental funding of clinical research within the Swedish National Health Service, and Skåne University Hospital in Lund (JGS).
Footnotes
DISCLOSURES: None
References
- 1.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–11. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.Chaput M, Handschumacher MD, Guerrero JL, Holmvang G, Dal-Bianco JP, Sullivan S, et al. Mitral leaflet adaptation to ventricular remodeling: prospective changes in a model of ischemic mitral regurgitation. Circulation. 2009;120:S99–103. doi: 10.1161/CIRCULATIONAHA.109.844019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–42. doi: 10.1161/CIRCULATIONAHA.108.846782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaput M, Handschumacher MD, Tournoux F, Hua L, Guerrero JL, Vlahakes GJ, et al. Mitral leaflet adaptation to ventricular remodeling: occurrence and adequacy in patients with functional mitral regurgitation. Circulation. 2008;118:845–52. doi: 10.1161/CIRCULATIONAHA.107.749440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geirsson A, Singh M, Ali R, Abbas H, Li W, Sanchez JA, et al. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation. 2012;126:S189–97. doi: 10.1161/CIRCULATIONAHA.111.082610. [DOI] [PubMed] [Google Scholar]
- 6.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–32. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 7.Butcher JT, McQuinn TC, Sedmera D, Turner D, Markwald RR. Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ Res. 2007;100:1503–11. doi: 10.1161/CIRCRESAHA.107.148684. [DOI] [PubMed] [Google Scholar]
- 8.Delling FN, Rong J, Larson MG, Lehman B, Osypiuk E, Stantchev P, et al. Familial clustering of mitral valve prolapse in the community. Circulation. 2015;131:263–8. doi: 10.1161/CIRCULATIONAHA.114.012594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delling FN, Vasan RS. Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation. 2014;129:2158–70. doi: 10.1161/CIRCULATIONAHA.113.006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakkestrom R, Larsen LA, Moller JE, Videbaek L, Hjelmborg JV, Christensen K. Mitral valve regurgitation in twins: Concordance and survival. Am Heart J. 2016;177:51–7. doi: 10.1016/j.ahj.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Dawber TR, Meadors GF, Moore FE., Jr Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–81. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shortreed SM, Peeters A, Forbes AB. Estimating the effect of long-term physical activity on cardiovascular disease and mortality: evidence from the Framingham Heart Study. Heart. 2013;99:649–54. doi: 10.1136/heartjnl-2012-303461. [DOI] [PubMed] [Google Scholar]
- 13.Levine RA, Stathogiannis E, Newell JB, Harrigan P, Weyman AE. Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol. 1988;11:1010–9. doi: 10.1016/s0735-1097(98)90059-6. [DOI] [PubMed] [Google Scholar]
- 14.Ekbom A. The Swedish Multi-generation Register. Methods Mol Biol. 2011;675:215–20. doi: 10.1007/978-1-59745-423-0_10. [DOI] [PubMed] [Google Scholar]
- 15.Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450. doi: 10.1186/1471-2458-11-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andell P, Li X, Martinsson A, Andersson C, Stagmo M, Zoller B, et al. Epidemiology of valvular heart disease in a Swedish nationwide hospital-based register study. Heart. 2017 doi: 10.1136/heartjnl-2016-310894. epub Apr 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 18.Frisell T, Holmqvist M, Kallberg H, Klareskog L, Alfredsson L, Askling J. Familial risks and heritability of rheumatoid arthritis: role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum. 2013;65:2773–82. doi: 10.1002/art.38097. [DOI] [PubMed] [Google Scholar]
- 19.Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–9. doi: 10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- 20.Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–13. doi: 10.1038/nature14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–11. doi: 10.1038/ng.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPherson R, Tybjaerg-Hansen A. Genetics of Coronary Artery Disease. Circ Res. 2016;118:564–78. doi: 10.1161/CIRCRESAHA.115.306566. [DOI] [PubMed] [Google Scholar]
- 23.Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–83. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoller B, Ohlsson H, Sundquist J, Sundquist K. A sibling based design to quantify genetic and shared environmental effects of venous thromboembolism in Sweden. Thromb Res. 2017;149:82–87. doi: 10.1016/j.thromres.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Pjanic M, Miller CL, Wirka R, Kim JB, DiRenzo DM, Quertermous T. Genetics and Genomics of Coronary Artery Disease. Curr Cardiol Rep. 2016;18:102. doi: 10.1007/s11886-016-0777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.