

Abstract
The continued rise of antibiotic resistant bacterial infections has motivated alternative strategies for target discovery and treatment of infections. Antivirulence therapies function through inhibition of in vivo required virulence factors to disarm the pathogen rather than directly target viability or growth. This approach to treating bacterial-mediated diseases may have advantages over traditional antibiotics as it targets factors specific for pathogenesis, potentially reducing selection for resistance and limiting collateral damage to the resident microbiota. This review examines vulnerable molecular mechanisms used by bacteria to cause disease and the antivirulence compounds that sabotage these virulence pathways. By expanding the study of antimicrobial targets beyond those that are essential for growth, antivirulence strategies offer new, innovative opportunities to combat infectious diseases.
Keywords: Antivirulence therapies, Bacterial pathogenesis
Expanding the search for new antibiotic targets
The evolution and spread of drug resistant bacterial infections is an urgent threat to our ability to control infectious diseases [1, 2]. Antibiotic resistant infections are associated with increased mortality and are estimated to cause at least 2,000,000 illnesses annually in the United States and approximately 23,000 deaths [3]. Additionally, drug resistant illnesses are expensive to treat and often require extended time in the hospital and thus pose additional risks for acquiring secondary infections [4]. Confounding the issue further is the lack of new antibiotics entering the clinic that have novel targets and mechanisms of action. Drug discovery and development of novel antimicrobials often requires billions of dollars and longer than a decade before being launched as a commercial drug [5]. Together, the net result is a diminishing arsenal of antibiotics that can be used to treat microbial infections, prompting efforts to devise alternative strategies to treat bacterial diseases.
During the course of pathogenesis, invading bacteria need to overcome a variety of obstacles raised by the host immune system and the presence of the resident microbiota. Establishing a productive infection requires that the pathogen is able to sense and adapt to the changing environment within the host, including: pH changes, secretions from the host (e.g. mucus), physical barriers, reduced oxygen tension, as well as the active immune response functioning to prevent the pathogen from colonizing. Further, the ability of displacing or exploiting the resident microbiota is often required for establishing an infection. However, bacterial pathogens are equipped with a diverse array of strategies to subvert host defenses and cause disease [6]. Therefore, disrupting the pathogen’s mechanisms to thwart host defenses may serve as a therapeutic approach to combat bacterial infections.
Conventional drug discovery campaigns often seek to identify compounds with bactericidal or bacteriostatic activity by inhibiting targets essential for growth such as cell wall integrity, protein synthesis and DNA replication [7, 8]. Antivirulence compounds, sometimes referred to as anti-infective compounds, are an alternative approach to traditional therapeutic intervention of bacterial infections that have been actively investigated [9–12]. Antivirulence compounds disarm the pathogen by targeting in vivo required virulence factors as opposed to growth cycle or viability mechanisms. By sabotaging the function of virulence-associated factors during infection, the invading bacteria are potentially left in a susceptible state to immune clearance or enhanced susceptibility to antibiotic-mediated killing [13–15]. An additional advantage antivirulence compounds may possess is that by not directly targeting growth or viability, the inhibitor may not exert as much selective pressure as traditional antimicrobials, thus potentially slowing the evolution of resistance [14, 15]. During broad-spectrum antibiotic therapy, there is no discrimination between pathogen-associated targets and beneficial microbes, leading to a state of dysbiosis in the host microbiota. This can make the host susceptible to acute and chronic secondary infections [16, 17]. Anti-infective compounds can limit off-target effects against the resident microbial community by directly targeting a pathogen-specific virulence factor. Together, the increasing understanding of bacterial pathogenesis and sequencing-based approaches have yielded significant insights into the virulence requirements necessary during infection, revealing many potential targets to develop new treatments [9, 18–25].
This review provides a brief overview of selected mechanisms that bacteria use to cause disease and recently described antivirulence compounds that inhibit them. The discoveries reviewed here are of several newly identified antivirulence molecules and is not an exhaustive list; therefore we direct the reader to other reviews for additional examples [10, 12, 26–28]. Additional considerations are discussed regarding resistance mechanisms to anti-infective molecules and potential implications for future efforts to discover of virulence inhibitors.
Bacterial pathogenesis mechanisms targeted by antivirulence compounds
Two-component regulatory systems
Bacteria must sense environmental cues and co-ordinate adaptive responses to changes in the environment in order to survive in the host. A common sensing and response mechanism in bacteria is the two-component regulatory system (TCS) [29]. A prototypical TCS is composed of a sensor histidine kinase (HK) and a response regulator (RR). The HK is usually located within the bacterial membrane and is responsible for sensing the environmental signal. Once the signal has been sensed, the HK undergoes an activating conformation, leading to autophosphorylation activity through the ATPase domain. Phosphotransfer occurs through transfer of the phosphate from the HK at a conserved histidine residue to a conserved aspartic acid on the response regulator receiver domain. The response regulator will typically dimerize after phosphorylation and act as a transcription factor to modulate a regulatory cascade of genes involved in responding to the environmental cue (Figure 1) [29]. TCS represent a family of targets that are of particular interest to develop antivirulence therapies as they are not found in mammalian cells, limiting potential off target effects against host-associated factors [29]. Further, deletion of TCS have been shown to significantly attenuate pathogenesis, though many TCS are dispensable for in vitro growth, suggesting that screening for inhibitors of TCS in vitro requires a method alternative to growth inhibition, such as using a reporter system coupled to a gene regulated by the TCS [30, 31]. Inhibiting virulence-associated TCS “blinds” the pathogen from sensing and coordinating an adaptive response to host cues, potentially sensitizing it to antibiotic treatment and immune clearance.
Figure 1. Two-component regulatory sensor transduction systems.

A prototypical two-component sensor system (TCS) is composed of a histidine kinase (HK) and a response regulator (RR). Upon sensing the environmental signal, the HK undergoes autophosphorylation at a conserved histidine residue. The phosphate is transferred to the response regulator, which typically dimerizes and acts as a transcription factor to alter expression of virulence genes. All inhibitors are shown in red and associated steps at which they function to inhibit TCS signaling. Ethoxzolamide inhibits carbonic anhydrase activity in Mycobacterium tuberculosis, leading to downregulation of the virulence associated PhoPR regulon. LED209 directly binds to conserved lysine residues on the HK QseC and inhibits activation of virulence genes in multiple pathogens. Artemisinin targets heme carried by the Mtb DosS and DosT HK to inactivate the kinases. HC102A and HC103A inhibit DosS HK autophosphorylation and HC103A also inhibits DosT autophosphorylation. A collection of NSC inhibitors inhibit the formation of the Salmonella enterica PhoP-DNA complex in vitro. This model is derived and modified from Rasko et al. [39]
LED209
Many HKs are conserved throughout bacteria and respond to similar environmental cues, suggesting potential for broad-spectrum antivirulence inhibitors. For example, the HK QseC contributes to virulence in at least 25 animal and plant pathogens including: Salmonella enterica serovar Typhimurium, enterohemorrhagic E. coli (EHEC), uropathogenic E. coli (UPEC), Haemophilus influenzae, and Francisella tularensis [32–40]. As a bacterial receptor of epinephrine, norepinephrine, and the quorum sensing autoinducer-3 (AI-3), QseC contributes to transducing both host-derived stress signals and interkingdom signaling (Figure 1) [41]. In response to these cues, QseC controls the regulation of several virulence-associated genes by undergoing autophosphorylation and transfer of the phosphate to three RR: QseB, QseF, and KdpE. In EHEC, KdpE and QseF regulate induction of the locus of enterocyte effacement (LEE) and stxAB genes encoding Shiga toxin production, respectively. QseB regulates genes involved in flagella and motility [42]. Thus, QseC represents a conserved sensory transduction system that controls induction of virulence factors in many pathogens that could be targeted for development of broad-spectrum anti-infective therapeutics.
A high throughput screen (HTS) of approximately 150,000 small organic compounds using a LEE1::lacZ reporter in EHEC identified the lead compound LED209 as an inhibitor of QseC-mediated signaling in response to AI-3 (Figure 1) [39]. LED209 [N-phenyl-4-(3-phenylthioureido)benzenesulfonamide] (Figure 2) was discovered to be a pro-drug that modifies QseC lysine residues, impairing the function of the HK and inhibiting QseC-mediated induction of virulence factors in vitro and in vivo [39, 42]. Interaction of LED209 with QseC causes the release of isothiocyanate, in a reaction that was found to occur only inside the bacterium. Treatment of EHEC and Salmonella Typhimurium with isothiocyante in vitro and in ex vivo infected macrophage models was able to mimick treatment by LED209. Mass spectrometry studies of labeled isothiocyanate revealed direct binding to two conserved QseC lysine residues (K256 and K427). Site-directed mutagenesis of each of these residues to arginine lead to a loss of function of QseC, suggesting that isothiocyante binding these amino acids is the direct mechanism leading to inactivation of the HK. To assess in vivo potency of LED209 against Salmonella Typhimurium, mice were orally administered LED209 or a vehicle control 30 minutes prior to bacterial challenge. Pre-treatment with LED209 led to at least 50% survival against S. Typhimurium [42]. F. tularensis encodes a QseC homolog that is the sole HK in the genome. Previous work on LED209 demonstrated reduced F. tularensis survival in macrophages, suggesting that F. tularensis may also be attenuated in vivo by LED209 treatment [39]. Indeed, mice infected with a lethal dose of F. tularensis and treated either pre- or post-infection with LED209 had at least 50% survival 8 days post-infection.
Figure 2. Chemical structures of antivirulence compounds discussed in this review.

Small molecules that inhibit: A. Two-component regulatory systems; B. Bacterial adherence; C. Toxins and secretion systems; and D. Metabolic pathways required for virulence.
Ethoxzolamide
Mycobacterium tuberculosis (Mtb) remains a threat to global health. In 2015, approximately 10.4 million individuals developed new TB infections and 1.8 million individuals died, making TB the leading cause of death by an infectious disease [43]. The treatment course of antibiotic-susceptible Mtb consists of a multi-drug regimen that must be taken for at least six months. In areas where public health infrastructure is lacking, patients that do not complete the full treatment course may have a relapsing infection or develop a multi-drug resistant Mtb (MDR-TB) infection. However, patient non-compliance is not the only driver in the rise of MDR-TB. Mtb has a reasonably high mutation rate in vivo (~2–3x10−10 mutations/bp/generation) and naturally drug resistant strains can exist in susceptible populations [44]. MDR-TB infections were estimated to account for approximately 480,000 new TB infections in 2015 [43]. Thus, faster acting antibiotics and alternative treatment options are needed.
Mtb contains at least 11 complete TCS and several orphaned HK and RR [45]. pH is used as a cue for Mtb to modulate its physiology, and these adaptations play a significant role in pathogenesis [46]. The PhoPR TCS and its associated regulon have been studied extensively in Mtb. It has been implicated in sensing and responding to variety of signals including acidic pH and chloride, though the precise signal(s) remain unknown [47–50]. Further, a phoPR mutant is highly attenuated in animal models of infection [51–53]. Therefore, it follows that inhibitors of PhoPR-dependent pH-driven adaptation may function to attenuate Mtb pathogenesis.
Ethoxzolamide (ETZ) was identified as an inhibitor of PhoPR signaling in Mtb, presumably through inhibition of Mtb carbonic anhydrase activity [54]. ETZ (6-ethoxy-1,3-benzathiazole-2-sulfonamide) (Figure 2) was discovered through a HTS of approximately 220,000 compounds using an in vitro whole cell phenotypic screen, employing a fluorescent reporter that is inducible at acidic pH in a PhoPR-dependent manner [49]. Although PhoPR is required for replication and growth in vivo, it is dispensable for in vitro growth in rich medium. In this screen, compounds that inhibited the production of GFP fluorescence while leaving growth of Mtb relatively unaffected were hypothesized to be putative inhibitors of PhoPR-signaling. By taking an unbiased approach to identify Mtb PhoPR inhibitors, previously unrecognized regulators of the TCS at acidic pH could also be identified.
ETZ was identified as a candidate inhibitor of the PhoPR regulon through its ability to inhibit reporter fluorescence in a dose-dependent manner while not impacting growth. RNA-seq transcriptional profiling of Mtb treated with ETZ confirmed that ETZ inhibited the PhoPR regulon similar to a genetic mutant of PhoP. ETZ is an FDA-approved drug that was originally used to treat glaucoma, reduce the seizure threshold, and as a general dieuretic through its carbonic anhydrase (CA) inhibitory activity. ETZ was shown to be able to inhibit Mtb carbonic anhydrase activity using purified recombinant enzymes [55–65]. CA activity was assessed initially to define whether ETZ could act to inhibit Mtb carbonic anhydrases in whole cells and not just under cell free conditions. Indeed, a complete inhibition of CA activity was observed by ETZ treatment relative to mock treated (DMSO) cultures following 6 days incubation [54]. Together, these data established a previously unrecognized link between CA and PhoPR signaling in Mtb (Figure 1). However, it is possible that additional unknown targets of ETZ exist in Mtb that may lead to inhibition of PhoPR-dependent reporter fluorescence.
Phenotypes associated with genetic disruption of phoPR include altered virulence lipid production and the inability to secrete Esx-1 associated antigens ESAT-6/EsxA and CFP-10/EsxB [48, 66, 67]. Based on the transcriptional profiling data, it was hypothesized that ETZ could recapitulate these PhoPR-dependent defects in vitro. Indeed, treatment with ETZ leads to a reduction in the virulence-associated sulfolipid and a concomitant increase in long-chain fatty acids like triacylglycerol and phthiocerol dimycocerosate similar to a knockout of phoPR [49, 68, 69]. Additionally, under Esx-1 export inducing conditions, ETZ inhibited secretion of the ESAT-6 and CFP-10 proteins by Mtb, similar to phoPR or Esx-1 export system deficient mutant strains [66, 70]. Taken together, Mtb treated with ETZ exhibits defects similar to a phoPR mutant, suggesting that during infection, the compound will lead to an attenuated phenotype. To assess in vivo efficacy, mice were infected with a fluorescent reporter, similar to the strain used during the original screen. Mice were treated orally with either ETZ or vehicle 5 days a week for 4 weeks. ETZ treatment resulted in a 0.75 log reduction in bacterial burden within the lungs and significantly reduced GFP fluorescence relative to the mock treated control, demonstrating ETZ can effectively inhibit PhoPR in the infected mouse lung. Thus, this study represents the first proof-of-concept that this virulence pathway in Mtb could be targeted and manipulated using small molecule inhibitors. Additionally, the use of fluorescent reporters can be leveraged for both antivirulence compound discovery and also as biomarkers for in vivo, on-target drug exposure.
Artemisinin, HC102A, and HC103A
Mtb is a remarkably successful pathogen, in part, due to its ability to become dormant in response to host immune pressures, such as hypoxia and nitric oxide (NO)[71]. The two-component regulatory system DosRST is induced by hypoxia, NO and carbon monoxide and remodels Mtb physiology to promote non-replicating persistence (NRP)[72]. The presence of persisters is thought to drive the long course of treatment for TB[73], therefore, isolation of compounds that inhibit DosRST-dependent adaptation may identify new antivirulence therapies targeting the drug-tolerant persister population.
Similar to the approach described above to discover inhibitors of the PhoPR regulon, a reporter-based HTS was conducted to find inhibitors of the DosRST regulon. A DosRST-dependent, hypoxia-inducible fluorescent Mtb reporter strain was generated by fusing the DosR-regulated hspX promoter to GFP [50]. This reporter strain was used in to screen a >540,000 compound library for chemicals that inhibit hypoxia-dependent induction of reporter strain fluorescence and identified six new inhibitors of the DosRST regulon [74]. Mechanism of action studies were carried out on three prioritized inhibitors: artemisinin, HC102A and HC103A (Figure 2). RNAseq transcriptional profiling experiments show that the three compounds inhibit the DosR regulon when compared to a dosR mutant control strain. The transcriptional profiles indicated that Mtb treated with the inhibitors would be defective in persistence-associated triacylglycerol (TAG) metabolism as the TAG synthase gene tgs1 was significantly repressed. Indeed TAG accumulation was dramatically downregulated following treatment with each of the DosR regulon inhibitors. Using the hypoxic shiftdown model of Mtb persistence [75], the newly discovered inhibitors were shown to reduce Mtb survival by >50%, following 10 days of incubation with the inhibitors under NRP inducing conditions. Previously, disruption of tgs1 has been shown to reduce Mtb tolerance to the first-line Mtb drug isoniazid (INH) during NRP [76, 77]. In the hypoxic shiftdown model Mtb exhibits 100% survival following 10 days of treatment with 5 μM INH[74]. However, treatment of Mtb with 5 μM INH in combination with 40 μM of artemisinin, HC102A or HC103A, broke Mtb tolerance to INH and caused a ~25% enhancement of Mtb killing by INH. Together, these findings show that antivirulence compounds targeting DosRST can inhibit persistence and drug tolerance.
Mechanism of action studies revealed novel aspects of the DosS and DosT sensor kinases that are susceptible to chemical modulation. Using UV-visible spectroscopy, artemisinin was shown to modulate DosS and DosT sensor kinases by oxidizing and alkylating the heme group carried by DosS/T[74]. Using structural modeling DosS and DosT amino acid substitutions were identified that would interfere with artemisinin docking with heme and it was observed that these substitutions promoted resistance to artemisinin-mediated inhibition of DosS and DosT in biochemical assays. Additionally, the resistant alleles were transformed into Mtb and it was observed that one of the alleles provided artemisinin-resistance to Mtb, thus strongly supporting the sensor kinases are directly modulated by artemisinin. HC103A was shown to directly inhibit the autophosphorylation activity of the DosS and DosT sensor kinases, while HC102A was observed to inhibit DosS autophosphorylation, but not DosT autophosphorylation. Together, the mechanism of action studies support that the inhibitors are directly targeting DosS and DosT sensor kinases to inhibit the DosR regulon and Mtb persistence and antibiotic tolerance (Figure 1).
NSC inhibitors
The PhoPQ TCS is the major virulence regulator in Salmonella Typhimurium and related Gram-negative pathogens (e.g. Shigella flexneri and Neisseria meningitidis). This TCS belongs to the OmpR/PhoB family, which represents approximately a third of the known TCS. Further, PhoPQ has been shown to sense and respond to a variety of signals in Salmonella Typhimurium including acidic pH, magnesium stress, and antimicrobial peptides. Upon transducing an activating signal PhoQ undergoes autophosphorylation and transfers a phosphate to the response regulator, PhoP. PhoP then homodimerizes and binds to specific promoter regions, inducing the expression of virulence-associated genes. Due to the broad conservation of structural homology of the RR dimerization interface, which consists of an α4-β4-α5 motif, it was hypothesized that disrupting the formation of the PhoP homodimer would prevent the PhoP-DNA interaction and may be applied as a broad-spectrum antivirulence therapy. Thus, a hybrid screening approach, combining structure-guided in silico docking studies and in vitro experimental verification was applied to a subset of potential inhibitors from the NCI database [78]. Of the 1420 compounds virtually screened, 40 were examined experimentally. Using electrophoretic mobility shift assays, a series of 8 compounds were found to inhibit the Salmonella Typhimurium PhoP-DNA complex from forming in vitro (Figure 1). Unfortunately, none of the compounds tested showed activity to disrupt the formation of PhoP homodimers though several avenues of experimental approaches were examined. Though the direct mechanism remains to be determined, these compounds represent a proof-of-concept study that the broadly conserved RR PhoP can be inhibited through small molecules.
Bacterial adherence mechanisms
Attachment of bacteria to a host surface is often the first step in initiating and establishing an infection. However, the host possesses several mechanisms to prevent and remove bacteria that do not have specialized apparatuses to facilitate attachment, including the coordinated beating of cilia in the nasopharynx; peristaltic motion in the gastrointestinal tract; and, the presence of the resident microbiota blocking access to invading bacteria [79]. Further, pathogenic organisms will only attach and interact with a specific subset of cells that express the appropriate receptor for attachment [80–82]. Additional steps beyond attachment by bacterial adhesins are often required for infection to proceed including internalization (e.g. phagocytosis), deeper tissue penetration, biofilm formation, and for some pathogens, systemic distribution [80–82]. Pili are adherence structures that play a role in attachment and virulence of several pathogens including uropathogenic E. coli (UPEC) – the leading causative agent of urinary tract infections [82–86]. A pilus is a long multi-subunit appendage with binding specificity at the terminal subunits, typically through FimH or PapG, for a specific receptor on the host cell surface [81, 82, 85, 86]. Many pili and pili-like structures have been identified for several bacterial pathogens [80]. However, for the purposes of this review, we will focus on two common and well-studied types associated with UPEC infections: 1) type I and P pilus and 2) curli. Assembly of the type I and P pilus is accomplished through a chaperone in the periplasmic domain and an outer membrane usher protein [85, 86]. The chaperone facilitates the folding and transport of the pilin subunits and the usher aids in incorporation of each subunit into the growing appendage (Figure 3A) [85, 86]. Curli are another appendage involved in attachment and virulence. They are synthesized and secreted by an alternative mechanism relative to pili and appear as hair-like structures coating the cell surface [87, 88]. UPEC strains can use curli to form biofilms in the urinary tract to prevent washing out by the normal flow of urine [89, 90]. Curli are composed of the major and minor subunits CsgA and CsgB. Interestingly, CsgB acts as an extracellular nucleation factor for the amyloid-like deposit of CsgA leading to the growth of the curli structure. Two soluble accessory chaperone-like proteins called CsgE and CsgF, are implicated in the transport of the curli subunits (CsgA and CsgB) from the Sec system to the outer membrane assembly protein CsgG (Figure 3B) [87, 88].
Figure 3. Bacterial adherence mechanisms through Type I, P pili, and curli biogenesis.
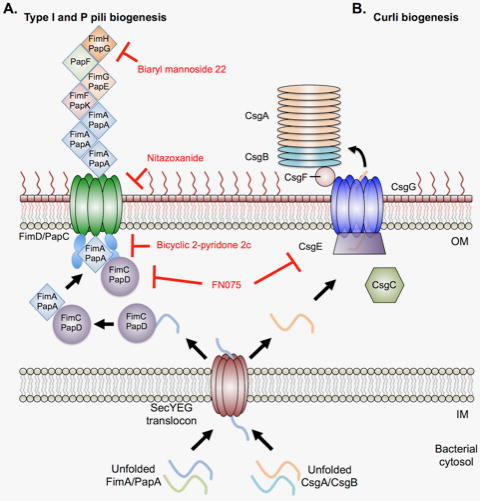
A. Graphical representation of type I and P pili biogenesis in Gram-negative pathogens, such as uropathogenic E. coli (UPEC). Unfolded pilin subunits are translocated through the SecYEG translocon into the periplasm. A chaperone (FimC/PapD) folds and stabilizes the subunit, passing it to the secretion machinery for incorporation into the growing appendage. FimD/PapC receives the folded subunit from the chaperone and adds the given subunit to the actively polymerizing pilus. Biaryl mannoside 22 inhibits the mannose binding capacity of the terminal subunit (FimH/PapG) in type I and P pili. Nitazoxanide inhibits pore formation of the secretion/polymerization complex (FimD/PapC). Biaryl 2-pyridone 2c binds to the surface of the chaperone (FimC/PapD) and prevents the subunit transfer to the secretion/polymerization complex (FimD/PapC). B. Graphical representation of curli biogenesis in UPEC. Unfolded curli subunits (CsgA/CsgB) are translocated into the periplasm by the SecYEG secretion machinery. CsgC is proposed to act as a chaperone, preventing premature amyloid formation by the unfolded curli subunits [186]. CsgE and CsgF act as soluble chaperones to stabilize CsgA/CsgB and transfer the unfolded subunits to the outer membrane assembly protein CsgG. CsgB acts as a nucleation factor for CsgA amyloid formation and deposition onto the growing appendage. FN075 inhibits CsgA polymerization and due to its parent structure, also acts as an inhibitor similar to the biaryl 2-pyridone 2c. All inhibitors are denoted in red. OM – bacterial outer membrane; IM – bacterial inner membrane. This model is derived and modified from Costa et al. [85]
Pilicides
The type I and P pili are assembled through what is known as the chaperone-usher pathway (CUP) and are encoded on the fim and pap operons, respectively [85]. The pilus appendage itself is composed of two parts: 1) the structural major subunit and 2) the flexible N-terminal tip required for receptor specificity and binding. For type I pili the major subunits are composed of FimA, while the minor subunits are made up of FimF, FimG, and the lectin containing terminal subunit FimH. The structural components of P pili are comprised of PapA, while the flexible tip is composed of PapE, PapF, PapK, and the lectin containing terminal subunit PapG [85]. All subunits are first secreted in an unfolded state past the inner membrane into the periplasmic space via the SecYEG machinery. The chaperones FimC and PapD proceed to fold and stabilize each subunit for type I and P pili, respectively. Chaperone-subunit complexes are then transferred to the usher (FimD or PapC) that facilitates subunit assembly and elongation of the pili (Figure 3A) [85].
These systems have been extensively studied and crystal structures exist for each component of the type I and P pili machinery [85]. Thus, leveraging the available structural data, a series of compounds known as pilicides were rationally designed to directly inhibit type I and P pili biogenesis and belong to the bicyclic 2-pyridones class of molecules (Figure 2) [91–93]. Pilicides do not impact bacterial growth but instead interfere with UPEC attachment [92]. Co-crystallization of pilicide 2c with PapD revealed binding on the surface of the chaperone known to interact with the usher PapC. Additional surface-plasmon resonance (SPR) studies showed that the pilicide prevents FimC-FimH from interacting with the usher (FimD). Together, the proposed mechanism for pilicide inhibition of type I and P pili biogenesis is by preventing the chaperone from transferring the pili subunit to the usher (Figure 3B) [92].
To develop novel pillicides, structure-guided rational design was used to synthesize FimH antagonists, resulting in the promising group of inhibitors known as biphenyl mannosides [94, 95]. These initial compounds were efficacious in treating acute, chronic, and antibiotic-resistant UPEC infections in vivo [96–98]. However, this first round of FimH antagonists suffered from low bioavailability. Thus, additional co-crystal structures of lead mannosides bound to the FimH lectin domain were obtained. Using a rational design strategy, new FimH antagonists were developed with improved drug-like properties (Figure 3A). This new group of inhibitors are known as biaryl mannosides [99]. In particular, isoquinolone 22 (2-methyl-4-(1-oxo-1,2-dihydroisoquinolin-7-yl)phenyl α-D-mannopyranoside) (Figure 2) was shown to exhibit low nanomolar potency against FimH, improved oral bioavailability and a 10-fold enhanced reduction of bacterial load in a chronic UTI mouse model of infection. The biaryl mannoside 22 analog is a promising lead molecule for further development as an antivirulence therapeutic to treat UTIs [99].
Another compound that inhibits bacterial adherence is the antiparasitic drug nitazoxanide (NTZ) (Figure 2), which is broadly active against the CUP in both enteroaggregative E. coli (EAEC) and UPEC strains [100, 101]. NTZ does not directly target growth in EAEC or UPEC but rather inhibits assembly and biogenesis of aggregative adherence fimbriae, type I and P pili [100]. Additionally, NTZ inhibited type I pilus-mediated hemagglutination activity in both EAEC and UPEC strains, leading to a shortened duration of infection in EAEC infected animals due to bacterial wash out [100–102]. It was noted that NTZ treatment of UPEC reduced the total amount of both the PapC and FimD usher proteins in the outer membrane fraction in a dose dependent manner. To investigate the mechanism for reducing usher protein localization to the outer membrane by NTZ, a series of PapC truncation mutants were examined to identify the precise component of the usher that was disrupted. It was found that NTZ prevents proper folding of the transmembrane β-barrel pore (Figure 3A) [101]. Interestingly, NTZ inihibits growth of several pathogens including Clostridium difficile, Helicobacter pylori, and Campylobacter jejuni through inhibition of pyruvate ferreredoxin oxidoreductase [103, 104]. However, the Enterobacteriaceae lack this target and NTZ is effective as a pilicide below the growth inhibitory concentrations observed for growth inhibition in NTZ-susceptible pathogens [100, 101, 105, 106]. Repurposing NTZ as a putative antivirulence therapy may be possible as it is an approved treatment for giardiasis and cryptosporidiosis, potentially expediting the process for entering clinical use.
Pilicide discovery and characterization has provided additional insight into the molecular mechanisms governing pili biogenesis. Further, this group of compounds provides an attractive approach to treating pathogenic E. coli by inhibiting the ability to adhere to host surfaces and preventing the establishment of infection.
Curlicides
Curli are small, hair-like structures that are produced by UPEC strains and are implicated in biofilm formation [87, 88]. These amyloid-aggregative features have also been shown to act synergistically with cellulose to promote biofilm formation, host colonization, and survival in a variety of environments [107]. Taken together, identification of inhibitors that target both type I pili formation and curli production may provide a therapeutic approach to effectively disrupt colonization, invasion, and biofilm formation of UPEC.
The thiazolo ring-fused 2-pyridones have been shown to act as type I pilicides by preventing the chaperone-pili subunit complex from transferring the subunit to the usher for polymerization and secretion of the pilus (Figure 2) [92]. Through exchange of a cyclopropyl group for a CF3-phenyl moiety on a lead thiazolo ring-fused 2-pyridone pilicide, the analog gained the ability to inhibit Alzheimer’s-associated β-amyloid polymerization in vitro [108]. Encouraged by the ability of the pilicide analog (FN075) (Figure 2) to inhibit amyloid polymerization, it was hypothesized that this compound may also disrupt curli formation. Indeed, FN075 was able to inhibit formation of curli in the UPEC strain UTI89 and disrupt CsgA polymerization in vitro by directly binding to the curli major subunit (Figure 3B) [89]. To examine whether FN075 could inhibit biofilm formation, curli-dependent biofilm models were identified in UTI89. Pellicle formation was identified as a method to screen for curli-dependent biofilm inhibitors as a curli deficient mutant (UTI89ΔcsgA) could not form a pellicle and expressing csgA in trans could restore the wild-type biofilm. FN075 treated cultures resulted in loss of pellicle formation, similar to the csgA mutant, suggesting that the compound can inhibit curli-dependent biofilms. Further, FN075 was able to completely inhibit type I pili biofilm formation. To assess in vivo potency of the curlicide, mice were challenged with either UTI89 cultures pretreated with FN075 or an equivalent volume of DMSO (mock treated control) under type I pili inducing conditions. FN075 pretreated cultures resulted in a 10-fold reduction in bladder bacterial load six hours post infection relative to the DMSO control [89].
FN075 thus represents a unique anti-virulence therapy that also exhibits anti-biofilm activity against two adherence mechanisms of UPEC (Figure 3B). The power of structure-based rational inhibitor design is demonstrated in several examples of pilicide and curlicide discovery and subsequent characterization. This modified targeted screening approach can provide researchers molecular probes to dissect previously unknown molecular dynamics of a pathogenic processes like pili or curli biogenesis, and improve inhibitors to possess enhanced drug-like properties.
Toxins and Specialized Secretion Mechanisms
Toxin production and delivery to host tissues is essential for several pathogens to promote infection and dissemination. A variety of mechanisms exist to transport toxins and effectors from within the bacterial cytoplasm to the extracellular environment, or in some cases, directly injected through host membranes. Secreted toxins can actively disrupt host intracellular signaling cascades, lead to rearrangements of cytoskeletal architecture, loosen tight junctions, and promote rapid efflux of electrolytes in addition to numerous other effects [109]. Ultimately, the toxins enhance pathogen survival, invasion, attachment or transmission by hijacking or disrupting host processes (Figures 4, Figure 5) [110, 111]. Inhibiting toxin production, function or secretion is a promising avenue for the development of antivirulence compounds.
Figure 4. Proposed mechanisms for toxtazins A and B inhibition of cholera toxin and toxin co-regulated pilus production.

ToxT regulates transcription of genes responsible for producing cholera toxin (CT) and the toxin co-regulated pilus (TCP), which are two virulence factors involved in cholera infections. Toxtazin A inhibits toxT transcription and is proposed to act by causing a general stress response, feeding back to shut down transcription of toxT. Toxtazin B inhibits transcription of tcpPH and is proposed to act by reducing intracellular levels of TcpP, leading to reduced transcription of toxT. Inhibitors are in red. OM – bacterial outermembrane; IM – bacterial inner membrane. This model is derived and modified from Anthouard et al. [26]
Figure 5. Targeting C. difficile oxin processing in host cells.

TcdA and TcdB are the two toxins produced by disease-associated C. difficile. Host processing of the toxins are mediated by endocytosis of the full-length toxin into an acidified compartment, leading to surface exposure of the cysteine protease domain (CPD) and glucosyltransferase domain (GTD). Interaction of the CPD with 1D-myo-inositol hexakisphosphate (IP6) leads to activation and autocatalysis of the linker region between the CPD and GTD. GTD is released into the host cytoplasm and alters intracellular signaling through glucosylation of Rho/Rac GTPase activity. Ebselen inhibits the protease activity of CPD, preventing the release of GTD into the host cytoplasm. The Ebselen inhibitor is in red. This model is derived and modified from Bender et al. [132]
Several microbial pathogens possess specialized secretion machinery to deliver toxins directly into the host cytoplasm. Particular attention has been given to the type III secretion system (T3SS) which is broadly conserved in Gram-negative pathogens including Yersinia sp., Salmonella sp., Shigella sp., EHEC, and Pseudomonas aeruginosa (Figure 6) [109]. The T3SS is often required for virulence and inhibitors of this toxin delivery system are predicted to attenuate pathogenesis [28, 110–112]. Mtb uses a type VII secretion system (T7SS) to export several effectors involved in pathogenesis (Figure 7). Mtb possesses five T7SS termed ESX-1 to ESX-5, though active secretion has not yet been demonstrated for ESX-2 and ESX-4 [114]. The ESX-1 locus is required for transport of two mycobacterial effectors known as EsxA and EsxB (ESAT-6 and CFP-10, respectively). Importantly, the inability to secrete these proteins is associated with the attenuation of the live-vaccine strain Mycobacterium bovis BCG [114]. Thus, small molecules that target ESX-1 through either inhibiting the secretion machinery directly or its regulation will likely lead to decreased survival of Mtb in vivo.
Figure 6. Type III secretion system for delivery of bacterial effectors directly into the host cytoplasm.

The type III secretion system (T3SS) resembles a syringe-like structure that bacterial pathogens use to inject effectors directly into the host cytoplasm. The terminal tip of the T3SS interacts with the host cell, creating a pore for active (ATP-dependent) delivery of substrates that disrupt host signaling, lead to cytoskeletal rearrangements, and inflammation as a few examples. Yersina pestis uses effector proteins known as Yersinia outer proteins (Yops) to cause disease. Within the bacterial cytoplasm the chaperone known as specific Yersinia chaperone (Syc) stabilizes and partially folds Yops for delivery to the T3SS. The ATPase YscN is responsible for dissociating the chaperone-effector complex and failure to do so results in bacterial attenuation. The compounds 7086, 7812, and 7832 inhibit YscN ATPase activity in vitro. The HK QseC is a global virulence regulator in several Gram-negative pathogens including S. Typhimurium. QseC has been shown to globally regulate the pathogenicity island associated with T3SS production and the effector sifA [34]. LED209 is a potent regulator of QseC and attenuates S. Typhimurium during infection through inhibiting virulence gene induction, including the T3SS. Additionally, the S. Typhimurium PhoPQ two-component regulatory system is implicated in regulating several virulence-associated genes including the T3SS secreted effector SrfJ [187]. The NSC inhibitors have been shown to inhibit the PhoP-DNA complex formation in vitro, potentially disrupting virulence gene expression, including srfJ, in S. Typhimurium. Inhibitors are shown in red. HM – host membrane; OM – bacterial outer membrane; IM – bacterial inner membrane; HK – histidine kinase; RR – response regulator. This model is derived and modified from Rasko et al., and Costa et al.[10, 85]
Figure 7. Mycobacterium tuberculosis type VII secretion of EsxA/EsxB.

Mycobacterium tuberculosis (Mtb) possesses a type VII secretion system (T7SS) (ESX-1) for export of host effector proteins EsxA and EsxB. The current working mechanism is that T7SS substrates are targeted for secretion by an unstructured C-terminal signal sequence (similar to type IV secretion systems). The substrates EsxA and EsxB form a dimer for secretion and stabilized by the soluble chaperone EspG and potentially exported in an ATP-dependent manner. Further, the protease MycP is required for secretion, though the exact role it plays in secretion is unclear. Regulation of ESX-1 secretion is accomplished by several cellular systems including the two-component regulator MprAB through the espACD operon. BPT15 inhibits the kinase activity of the sensor kinase MprB, leading to reduced secretion of EsxA, attenuating Mtb-mediated phagosome maturation arrest. BBH7 inhibits an unknown factor leading to reduced general secretion of Mtb and survival in fibroblasts. Inhibitors are in red. MM – mycobacterial membrane; PM – plasma membrane. This model is derived and modified from Rasko et al., and Houben et al. [10] [114]
Toxtazins
Once in the gut, the human pathogen Vibrio cholerae secretes cholera toxin (CT), which causes the characteristic disease symptoms of watery diarrhea and dehydration. The virulence regulator ToxT regulates expression of two of the major virulence factors in V. cholerae: CT and the toxin co-regulated pilus (TCP) (Figure 4) [117, 118]. ToxT-mediated expression of CT and the TCP has been shown to be inversely regulated by environmental signals relevant to the human gut including bicarbonate (inducing) and bile (repressing) [119]. Thus, it follows that identification of compounds that inhibit ToxT will attenuate V. cholerae. Indeed, as reviewed elsewhere [9, 10, 12], the antivirulence compound virstatin was discovered as an inhibitor of ToxT dimerization, leading to reduced colonization in an infant mouse model of cholera infection [120, 121]. However, resistance to virstatin has been demonstrated, suggesting that new inhibitors with alternative targets will be useful [121].
Toxtazins A, B and B’ were identified in a whole cell screen for inhibitors of ToxT expression (Figure 2) [26]. Using a ToxT-dependent fluorescent reporter strain, approximately 63,000 small molecules were screened for compounds capable of reducing GFP fluorescence but not significantly affecting growth. Toxtazins were able to reduce the production of both CT and the TCP under multiple growth conditions and across tested biotypes (classical and El Tor), suggesting that the compounds were affecting toxT expression and thus, validating the screen. Toxtazin B was shown to be effective in an infant mouse model of V. cholerae infection, where the compound was able to reduce the bacterial populations approximately 100-fold relative to the DMSO control. Interestingly, toxtazin A had no appreciable effect on bacterial burden in vivo and it was hypothesized that this apparent lack of activity may be due to poor bioavailability at the concentrations tested. To identify the targets of toxtazins A and B, a systematic examination of the toxT regulatory cascade was performed. toxT expression is regulated by ToxR and TcpP. Additionally, tcpP expression is regulated by AphA and AphB. Toxtazin A was shown to not alter expression of toxR or tcpP, while toxtazin B exhibited reduced TcpP protein and transcript levels (Figure 4.). Toxtazin B did not modulate expression of aphA or aphB and neither compound affected AphA or AphB protein levels. Restoration of ToxT in toxtazin treated cultures by ectopic expression of the regulator resulted in restoration of CT, suggesting that both compounds act upstream of ToxT.
Ebselen
Clostridium difficile infections (CDI) are the leading cause of hospital-acquired diarrhea and the etiological agent of pseudomembraneous colitis [122]. Treatments for CDI include antibiotic therapy and fecal bacteriotherapy. However, recurrent infections are as high as 25% and are often associated with antibiotic resistant strains [123, 124]. Only toxigenic strains of C. difficile are implicated in CDI [127, 128]. Disease causing C. difficile strains encode either one or both toxins: TcdA and TcdB [127–129]. The toxins are composed of a putative receptor-binding domain, a transmembrane domain, a cysteine protease domain (CPD), and a glucosyltransferase domain (GTD) [130]. Upon endocytosis and exposure to acidic pH, the toxin undergoes membrane translocation and exposure to the host sugar 1D-myo-inositol hexakisphosphate (IP6). IP6 allosterically activates the CPD allowing cleavage and release of the GTD. The GTD glucosylates the Rho/Rac GTPases leading to cytoskeletal rearrangement, inflammation, and fluid loss associated with CDI (Figure 5.) [131]. Thus, inhibitors of activating steps in the process of releasing GTD may lead to reduced toxicity and better patient outcomes from CDI.
A targeted HTS using a fluorescent reporter of CPD activity identified 44 potential inhibitors of the protease [132]. To refine this pool of hits, cell rounding assays were performed to identify the cytoprotective effects of each inhibitor against full-length toxin. Ebselen (2-phenyl-1,2-benzoselenazol-3-one) (Figure 2) was discovered as a potent inhibitor of toxin-mediated cell death and as an immediate candidate for further development as it is in clinical trials for unrelated diseases [133]. Notably, ebselen exhibits glutathione peroxidase-like activity and has been identified as an inhibitor of additional bacterial virulence factors including: thioredoxin reductase, antigen 85, and diguanylate-cyclase enzymes [134–137]. Together, this suggests that ebselen may be applied as a broad-spectrum antivirulence therapy.
Ebselen inhibited recombinant CPD and full-length toxin in a dose-dependent manner, exhibiting an IC50 of 17.2 nM against TcdB (Figure 5). Mass spectrometry analysis confirmed covalent modification of the active-site cysteine and an additional cysteine on the opposing CPD surface. To investigate whether ebselen inhibited release of the GTD, western blot analysis revealed a dose-dependent reduction in glucosylation of Rac1, a Rho/Rac GTPase, consistent with CPD-mediated cleavage being inhibited. A non-cleavable mutant of TcdB (TcdB L543A) exhibited similarly delayed kinetics of toxin-induced cell death as ebselen treated wild type toxin, underscoring the proposed mechanism of ebselen inhibition of TcdB. Notably, new findings also support additional targets for ebselen, including the GTD[138], suggesting the mechanism of action of ebselen is multifactorial. To assess the in vivo effects of ebselen treatment, mice pre-treated with a lethal dose of TcdB were injected with either ebselen or DMSO. Ebselen reduced CDI associated tissue pathology in a dose-dependent manner, however, bacterial burden between DMSO and ebselen treated animals were similar. If ebselen concludes clinical trials for its alternative uses, repurposing this compound as an alternative, non-antibiotic therapy to neutralize the toxicity of TcdA and TcdB in CDI may be expedited. Additionally, due to inhibitory effects against virulence factors in other pathogens, ebselen is another promising candidate for exploration as a broad-spectrum antivirulence therapy.
7086, 7812, 7832
Recent reviews cover the discovery of antivirulence compounds against T3SS [28, 112], therefore this review will focus on one study from Y. pestis as an example of T3SS inhibitor development [113]. Y. pestis is a Gram-negative zoonotic pathogen that is the causative agent of plague in humans [139]. Y. pestis possesses a T3SS that is essential for virulence in vivo (Figure 6) [140, 141] and a mutant strain lacking the T3SS ATPase YscN is completely attenuated in a mouse model of infection[142]. Delivery of the Yersinia outer protein (Yop) effectors YopH (phosphatase) and YopE (Rho GTPase activator) via the T3SS, enable intracellular survival through disruption of cellular signaling [144, 145]. Within the bacterial cytosol, Yops are maintained in a partially unfolded state by the specific Yersinia chaperone (Syc). The partial unfolding of the protein-chaperone complex is necessary for stability and translocation through the T3SS as the pore is not large enough for fully folded proteins (Figure 6) [146, 147]. The ATPase YscN is responsible for removal of the chaperone from the complex in preparation for the subsequent secretion of the Yop effectors (Figure 6) [148]. Taken together, the T3SS ATPase domain of YscN provides an attractive target for development of small molecule inhibitors that could broadly abrograte secretion of type III effector proteins.
The ZINC database of commercially available compounds was screened computationally using a structure-guided rational design scheme against the active site of YscN [113]. Initially, 36 compounds were examined for inhibition of catalytic activity in an expression optimized YscN and the related ATPase from Burkholderia mallei. The compounds that showed appreciable ATPase inhibitory activity were tested for their ability to inhibit YopE secretion in Y. pestis infected macrophages. Notably, compounds 7086, 7812, and 7832 (Figure 2) exhibited at least 50% inhibition of the YscN catalytic domain and >90% inhibition of YopE secretion in an ex vivo model of infection, but did not affect Y. pestis growth in vitro. All three compounds exhibited minor or no cytotoxic effects against eukaryotic cells. These novel scaffolds represent potential foundations to build on and undergo structure activity relationship studies to identify related compounds with enhanced drug-like properties against YscN.
BPT15 and BBH7
Mtb is an intracellular pathogen that inhabits macrophage phagosomes during the course of pathogenesis. ESX-1 and the associated substrate EsxA have been proposed to enable Mtb to perforate phagosomal membranes and provide access to the cytosol [150, 151]. Deletion of the ESX-1 locus leads to an attenuated phenotype during infection [152]. However, ESX-1 is not required for growth in vitro, and it has proven difficult to identify conditions amenable to HTS to discover inhibitors of ESX-1 secretion.
A recent study developed a novel approach to identify small molecules that disrupt Mtb’s ability to secrete EsxA [153]. Previous work has shown that the cytolytic activity of Mtb at high multiplicities of infection (MOIs) is the result of EsxA intoxication in eukaryotic cells [154, 155]. HTS of a ~11,000 compound library was carried out to identify EsxA secretion inhibitors by infecting MRC-5 lung fibroblasts at a high MOI with Mtb and measuring cell survival. Additionally, to distinguish potential ESX-1 inhibitors from compounds generally toxic to bacterial growth, Mtb cultures were counter-screened for inhibition of growth in vitro. Compounds that promoted fibroblast cell survival and did not affect bacterial growth were predicted to be small molecules that target ESX-1 dependent secretion of EsxA (Figure 7). A total of 55 hits were identified as potential antivirulence inhibitors of EsxA secretion. Following cheminformatic analysis, several analogs belonging to the benzyloxybenzylidene-hydrazine (BBH) and benzothiophene (BTP) classes of molecules were identified from the screen suggesting some specificity against mycobacterial secretion by these scaffolds. Two particularly potent inhibitors, BBH7 and BTP15, were chosen for additional characterization (Figure 2). Interestingly, BBH7 inhibited intracellular growth to a degree similar to the antibiotic rifampicin, while BTP15 had no effect on Mtb intracellular survival, suggesting divergent mechanisms of action for each compound. To assess the inhibitory effects of protein secretion by BBH7 and BTP15, treated culture filtrates were collected and EsxA secretion was analyzed by immunoblotting. BBH7 generally inhibited Mtb protein secretion in a dose-dependent manner, whereas increasing levels of BTP15 exhibited specificity for decreasing EsxA secretory activity.
To characterize the targets of BBH7 and BTP15, traditional routes of identifying resistant mutants was not possible since these compounds do not directly target growth in vitro. Global transcriptional profiling by RNA-seq revealed unique signatures for each inhibitor. BTP15 treatment led to differential expression of genes almost exclusively associated with the DosR regulon – a group of genes shown to promote a state of NRP, enhancing long-term intracellular survival. The two-component systems PhoPR and MprAB have been implicated in linking the DosR regulon and regulation of ESX-1 secretion through the espACD locus [51, 156, 157]. Loss of mprAB leads to upregulation of espA and reduced secretion of EsxA [157]. Quantitative real-time PCR (qRT-PCR) revealed greater than two-fold upregulation of espA in BTP15 treated cultures, suggesting BTP15 may be inhibiting MprAB activity and in a biochemical assay BPT15 was determined to be a inhibitor of MprB autophosphorylation. RNA-seq analysis of BBH7 exhibited large changes in genes involved in cell wall processes and heavy metal detoxification and the compound was shown to modulate permeability and metal-ion homeostasis. Both BBH7 and BPT15 hamper Mtb’s ability to arrest phagosome maturation in infected THP-1 macrophages as shown by the enhanced colocalization of Mtb in acidified compartments relative to mock treated controls. Notably, as discussed earlier, ETZ also inhibits EsxA secretion and Mtb survival in macrophages [54], but by targeting a distinct carbonic anhydrase- and PhoPR-dependent signaling pathway. Thus, multiple environmental signaling pathways, including MprAB and PhoPR are converging to regulate Mtb intracellular survival and ESX secretion, supporting the value of targeting these pathways for novel antivirulence therapies.
Metabolic requirements during infection
The outcome of a bacterial infection is determined either by the host’s ability to control the infection through immune effectors meant to clear the pathogen or countermeasures produced by the invading microbe to subvert them [158]. However, the interplay between the host and bacterial pathogen’s metabolic requirements is often overlooked. Indeed, minor perturbations to metabolism of either system during infection can have a significant impact on the outcome in favor of either the host or pathogen. Intracellular pathogens have a unique challenge during the course of infection as they often occupy host membrane-encapsulated compartments, limiting access to amino acids and carbon sources for growth. Further, intracellular pathogens directly compete with the host cell for metabolic requirements with each attempting to tip the balance in favor of themselves. The topic of how pathogens adapt their metabolism to intracellular environments and specialized approaches to effectively acquire necessary metabolites to promote disease has been recently reviewed [159] and thus this review will focus specifically on a recent study of inhibitors of Mtb cholesterol metabolism.
V-13-009920, V-13-0110503 and V-13-012725
Whole cell phenotypic screens for inhibitors of metabolic requirements necessary in vivo often suffer from an inability to adequately recapitulate the environment that the pathogen encounters within the host. Thus, there is a significant attrition rate associated with in vitro identification of potential inhibitors and activity in subsequent assays to validate the compound during infection. Intracellular pathogens such as Mtb inhabit macrophages during pathogenesis in vivo, and infecting macrophage cell lines can be adapted to HTS. HTS of a proprietary library of ~340,000 compounds using J774 macrophages infected with an Mtb fluorescent reporter identified ~1,356 compounds that inhibit intracellular survival with an IC50 value <50 μM [179]. Identification of targets from this phenotypic screen is complicated, as the compounds could be targeting any aspect of physiology required for intracellular adaption. During growth in macrophages, Mtb is restricted in its access to available carbon sources, therefore, it was hypothesized that some compounds may be targeting Mtb macrophage-adapted metabolism. Mtb encodes an extensive suite of genes for import and degradation of sterols and requires cholesterol for intracellular growth (Figure 8) [165, 166]. Therefore, the hits were subjected to a counterscreen to test the hypothesis that the inhibitors may target cholesterol catabolism.
Figure 8. Inhibiting cholesterol metabolism in Mycobacterium tuberculosis.
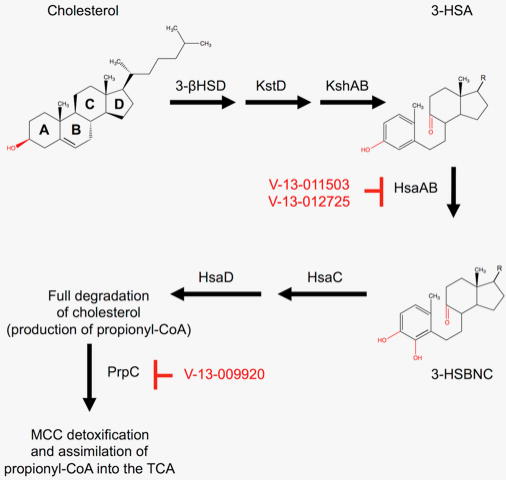
During the course of infection, Mycobacterium tuberculosis (Mtb) requires cholesterol as a carbon source for persistence and growth. Remarkably, Mtb is capable of fully catabolizing cholesterol through the proposed mechanism above. A/B ring catabolism is accomplished through a series of enzymatic and non-enzymatic steps. HsaAB likely catalyzes the hydroxylation of 3-HAS to 3-HSBNC and is inhibited by V-13-0110503 and V-13-012725. Propionyl-CoA is produced during cholesterol catabolism and is detoxified and assimilated into the TCA cycle via the methyl-citrate cycle (MCC). The small molecule V-13-009920 inhibits the first enzyme, PrpC, in the MCC. This inhibition results in attenuated survival in Mtb infected macrophages. Inhibitors are shown in red. This model is derived and modified from Capyk et al. [188].
Mtb isocitrate lyase (Icl1) is a bifunctional enzyme acting in both the glyoxylate shunt of the TCA cycle and as a methyl-isocitrate lyase in the methylcitrate cycle (MCC) [180, 181]. The MCC is used by Mtb to integrate propionyl-CoA into central carbon metabolism by producing pyruvate and succinate [180, 182]. Notably, propionyl-CoA is generated during cholesterol catabolism and if steps in the MCC are disrupted, such as an Mtb Icl1 mutant (Δicl1), it is hypothesized that toxic intermediates accumulate leading to an growth inhibition. Thus, a novel chemical rescue assay was used to select for potential inhibitors of cholesterol catabolism by growing a Mtb Δicl1 mutant strain in rich medium supplemented with cholesterol. If enzymatic steps were inhibited during cholesterol degradation, toxic intermediates of the MCC would not accumulate and consequently allow for growth of Δicl1. Indeed, three candidate compounds were identified, V-13-009920, V-13-012725, and V-13-011503 (Figure 2). To distinguish between which compounds inhibit processes in the MCC versus cholesterol catabolism, each candidate inhibitor was subjected to the same chemical rescue assay described above but was supplemented with propionate as opposed to cholesterol. V-13-012725 and V-13-011503 were confirmed to rescue growth in the presence of cholesterol but not propionate, suggesting that these molecules were acting by disrupting cholesterol degradation. In contrast, V-13-00920 was capable of rescuing growth of Δicl1 in both cholesterol and propionate, suggesting that this compound may be targeting an enzyme in the MCC.
Detoxification of the propionyl-CoA produced during cholesterol degradation requires the MCC [180, 182]. The first step in the MCC is the condensation of oxaloacetate with propionyl-CoA by the enzyme PrpC [182]. Thus, to identify whether V-13-009920 inhibits PrpC, recombinant enzyme was incubated in the presence of the compound. Indeed, V-13-009920 inhibited purified PrpC with an IC50 of approximately 4.0 μM [179]. Notably, this compound exhibited an IC50 of 3.0 μM in Mtb infected macrophages and a 10-fold more potent IC50 in minimal medium containing cholesterol (0.3 μM). Additionally, growth in minimal medium containing cholesterol revealed that V-13-009920 is bacteriostatic against Mtb. Disruption of Mtb intracellular growth by inhibiting PrpC supports a previous report that has established a prpCD double mutant exhibits an attenuated phenotype in macrophages To determine if V-13-012725 and V-13-011503 inhibit cholesterol degradation, Mtb was grown in the presence of radiolabeled cholesterol and the evolution of 14CO2 was monitored via radiorespirometry. Both compounds reduced the total amount of 14CO2 produced, suggesting inhibition of catabolic steps in the degradation of cholesterol. Mass spectrometry analysis of treated culture lysates revealed a single peak not observed in mock treated controls that corresponded to 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione (3-HSA), which is a known metabolite of A/B ring degradation of cholesterol (Figure 8.) [185]. In vitro enzymatic assays of the enzymes HsaA-HsaD, which are required for degradation of the A/B rings of cholesterol, showed that V-13-012725 and V-13-011503 directly inhibit HsaAB (Figure 9). In vitro killing kinetics assays of Mtb grown in minimal medium containing cholesterol as the sole carbon source revealed that both compounds were bacteriostatic.
This study identified three compounds that inhibit the MCC and cholesterol catabolism during infection, respectively (Figure 9). Additional animal studies are needed to verify whether these compounds function during an in vivo model of infection. An additional advantage of inhibiting cholesterol degradation or the MCC is Mtb potentially relies on cholesterol as a carbon source for extracellular growth in the granuloma caseum. Taken together, V-13-009920, V-13-012725 and V-13-011503 represent the first known inhibitors of PrpC and cholesterol catabolism in Mtb and can be leveraged as chemical probes to further define the mechanisms underlying the requirement for cholesterol during Mtb pathogenesis.
Concluding Remarks
Antivirulence strategies aim to sabotage pathogen virulence, placing bacteria in a state of increased susceptibility to clearance at the site of infection. One of the theoretical advantages of antivirulence approaches is that by targeting virulence factors and not directly inhibiting growth, these compounds may exert reduced selective pressure for resistance [15] (Outstanding Questions box). However, bacteria have always evolved resistance to antibiotics and it reasons that resistance will also emerge to antivirulence therapies. Recent studies have shown potential mechanisms for resistance to antivirulence compounds in vitro [42, 120]. As with traditional antibiotics, combination therapies are likely the appropriate strategy to combat resistance to antivirulence therapies. Careful consideration should also be given to whether the pathogen inhabits additional environmental niches where the virulence factor is either not expressed or is required for non-pathogenic processes (e.g. UPEC attachment in the intestines) potentially leading to resistance during systemic treatment [15]. Therefore, to develop more evolutionarily robust therapeutics, it will be useful to consider pathogen fitness in multiple environments and the subsequent impact of inhibiting specific virulence factors in those niches.
Outstanding Questions.
Will the evolution of antimicrobial resistance happen more gradually with antivirulence therapies as compared to traditional antibiotics? Resistance has evolved to most antimicrobials and once an antivirulence compound is used clinically it will be interesting to see if, how and when resistance evolves.
Could antivirulence therapies be used as adjunct therapies to potentiate traditional antibiotics? Blocking virulence pathways may sensitize pathogens to clearance by the immune system. The multifactorial antimicrobial response raised by the immune system may weaken bacteria and enhance susceptibility to traditional antibiotics.
Will non-traditional approaches to antibiotic discovery and development be more widely adopted by industry? Traditional antibiotics are generally bacteriocidal or static in vitro and often have a relatively broad spectrum. Antivirulence therapies may only function in the host, which requires new ways of thinking about characterizing and validating the activity of antivirulence compounds. Additionally, virulence pathways are often highly adapted to specific pathogens and therefore antivirulence therapies may have a narrower spectrum of activity than traditional antibiotics. Given these perceived challenges, it will probably take several examples of successfully deployed antivirulence therapies before this strategy of drug development is more broadly adopted.
Once antivirulence therapies are deployed in the clinic, how will the evolution of resistance to these compounds be monitored, given that many of these compounds do not cause growth inhibition in culture? Many of the inhibitors block expression of virulence genes, therefore, it is possible that new transcriptional profiling-based diagnostics or toxin production assays will be required to assess antivirulence compound resistance.
Antivirulence therapies are not traditional antibiotics and therefore new issues arise when considering their clinical development and application. For example, in many cases antivirulence therapies may be used as adjunct therapies in combination with traditional antibiotics, therefore potential drug interactions or differences in pharmacokinetic profiles and the impact on dosing schedules must be carefully considered. Additionally, in many cases, antivirulence therapies on their own may not result in a traditional measure of efficacy, such as reduced bacterial numbers. Rather, by design, the compounds may function by reducing host tissue damage or improving host survival, without exhibiting strong bacteriocidal activity. Preclinical and clinical characterizations of antivirulence compounds will therefore require specific measures to evaluate their therapeutic value.
The world currently faces an antibiotic resistance crisis and a key strategy to combatting drug resistant strains is the development of novel antibiotics. By expanding the study of antimicrobial targets beyond those that are essential for growth antivirulence strategies offer new, innovative opportunities to combat infectious diseases. Considerable efforts have been made by scientists to understand the molecular mechanisms of pathogenesis and translation of these findings to develop virulence targeted therapeutics will fulfill the potential of these basic research studies and help combat antimicrobial resistant diseases.
Trends.
The evolution and spread of antibiotic resistant microbes represents a major threat to global health. Combatting antibiotic resistance will require new strategies for treating infectious diseases, including the development of new antibiotics and drug targets.
An antivirulence therapy is a compound that targets a virulence pathway required for microbial pathogenesis in the host, but that is not for essential growth in standard in vitro growth conditions.
Experimental antivirulence compounds have been developed that target distinct virulence mechanisms in bacteria including two component regulatory systems, adherence, toxins, specialized secretion systems, metabolism, and quorum sensing.
Antivirulence therapies may have advantages over traditional antibiotics as these compounds target factors specific for pathogenesis, potentially reducing selection for resistance and killing of the resident microbiota.
The development of antivirulence strategies enables the translation of basic microbial pathogenesis research into new treatments for infectious diseases.
Acknowledgments
Research in the Abramovitch lab is supported by start-up funding from Michigan State University and AgBioResearch and grants from the NIH-NIAID (R01AI116605, R21AI105867 and R21AI117018), and Grand Challenges Explorations awards (OPP1059227 and OPP1119065) from the Bill & Melinda Gates Foundation.
Footnotes
Conflicts of Interest
The authors have no competing interests in the presented work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiology and molecular biology reviews: MMBR. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Costa VM, et al. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 3.CDC. Antibiotic Resistance Threats in the United States, 2013. Centers for Disease Control and Prevention; Atlanta, GA: 2013. [Google Scholar]
- 4.CDC. Antimicrobial Resistance Posing Growing Health Threat. Center for Disease Control and Prevention; Atlanta, GA: 2011. [Google Scholar]
- 5.Payne DJ, et al. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nature reviews Drug discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 6.Staskawicz BJ, et al. Common and contrasting themes of plant and animal diseases. Science. 2001;292:2285–2289. doi: 10.1126/science.1062013. [DOI] [PubMed] [Google Scholar]
- 7.Silver LL. Challenges of antibacterial discovery. Clinical microbiology reviews. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aminov RI. A brief history of the antibiotic era: lessons learned and challenges for the future. Frontiers in microbiology. 2010;1:134. doi: 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anthouard R, DiRita VJ. Chemical biology applied to the study of bacterial pathogens. Infect Immun. 2015;83:456–469. doi: 10.1128/IAI.02021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nature reviews Drug discovery. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 11.Lee YM, et al. Targeting virulence for antimicrobial chemotherapy. Curr Opin Pharmacol. 2003;3:513–519. doi: 10.1016/j.coph.2003.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Cegelski L, et al. The biology and future prospects of antivirulence therapies. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Then RL, Sahl HG. Anti-infective strategies of the future: is there room for species-specific antibacterial agents? Current pharmaceutical design. 2010;16:555–566. doi: 10.2174/138161210790361407. [DOI] [PubMed] [Google Scholar]
- 14.Ternent L, et al. Bacterial fitness shapes the population dynamics of antibiotic-resistant and -susceptible bacteria in a model of combined antibiotic and anti-virulence treatment. Journal of theoretical biology. 2015;372:1–11. doi: 10.1016/j.jtbi.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen RC, et al. Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol. 2014;12:300–308. doi: 10.1038/nrmicro3232. [DOI] [PubMed] [Google Scholar]
- 16.Sekirov I, et al. Gut microbiota in health and disease. Physiological reviews. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 17.McFarland LV. Antibiotic-associated diarrhea: epidemiology, trends and treatment. Future Microbiol. 2008;3:563–578. doi: 10.2217/17460913.3.5.563. [DOI] [PubMed] [Google Scholar]
- 18.Schnappinger D. Genomics of host-pathogen interactions. Progress in drug research. Fortschritte der Arzneimittelforschung. Progres des recherches pharmaceutiques. 2007;64:311, 313–343. doi: 10.1007/978-3-7643-7567-6_12. [DOI] [PubMed] [Google Scholar]
- 19.Rosamond J, Allsop A. Harnessing the power of the genome in the search for new antibiotics. Science. 2000;287:1973–1976. doi: 10.1126/science.287.5460.1973. [DOI] [PubMed] [Google Scholar]
- 20.Nagaraj NS, Singh OV. Using genomics to develop novel antibacterial therapeutics. Critical reviews in microbiology. 2010;36:340–348. doi: 10.3109/1040841X.2010.495941. [DOI] [PubMed] [Google Scholar]
- 21.Haney SA, et al. Genomics in anti-infective drug discovery--getting to endgame. Current pharmaceutical design. 2002;8:1099–1118. doi: 10.2174/1381612023394845. [DOI] [PubMed] [Google Scholar]
- 22.Wilson DJ. Insights from genomics into bacterial pathogen populations. PLoS Pathog. 2012;8:e1002874. doi: 10.1371/journal.ppat.1002874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roemer T, et al. Bugs, drugs and chemical genomics. Nature chemical biology. 2012;8:46–56. doi: 10.1038/nchembio.744. [DOI] [PubMed] [Google Scholar]
- 24.Pucci MJ. Novel genetic techniques and approaches in the microbial genomics era: identification and/or validation of targets for the discovery of new antibacterial agents. Drugs in R&D. 2007;8:201–212. doi: 10.2165/00126839-200708040-00001. [DOI] [PubMed] [Google Scholar]
- 25.Amini S, Tavazoie S. Antibiotics and the post-genome revolution. Curr Opin Microbiol. 2011;14:513–518. doi: 10.1016/j.mib.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 26.Anthouard R, DiRita VJ. Small-molecule inhibitors of toxT expression in Vibrio cholerae. mBio. 2013:4. doi: 10.1128/mBio.00403-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaSarre B, Federle MJ. Exploiting quorum sensing to confuse bacterial pathogens. Microbiology and molecular biology reviews: MMBR. 2013;77:73–111. doi: 10.1128/MMBR.00046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charro N, Mota LJ. Approaches targeting the type III secretion system to treat or prevent bacterial infections. Expert opinion on drug discovery. 2015;10:373–387. doi: 10.1517/17460441.2015.1019860. [DOI] [PubMed] [Google Scholar]
- 29.Gao R, Stock AM. Biological insights from structures of two-component proteins. Annu Rev Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shestov M, et al. Encyclopedia of bacterial gene circuits whose presence or absence correlate with pathogenicity--a large-scale system analysis of decoded bacterial genomes. BMC genomics. 2015;16:773. doi: 10.1186/s12864-015-1957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr Opin Microbiol. 2006;9:143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Weiss DS, et al. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A. 2007;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Unal CM, et al. QseC controls biofilm formation of non-typeable Haemophilus influenzae in addition to an AI-2-dependent mechanism. International journal of medical microbiology: IJMM. 2012;302:261–269. doi: 10.1016/j.ijmm.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 34.Moreira CG, et al. QseC mediates Salmonella enterica serovar typhimurium virulence in vitro and in vivo. Infect Immun. 2010;78:914–926. doi: 10.1128/IAI.01038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreira CG, Sperandio V. Interplay between the QseC and QseE bacterial adrenergic sensor kinases in Salmonella enterica serovar Typhimurium pathogenesis. Infect Immun. 2012;80:4344–4353. doi: 10.1128/IAI.00803-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mokrievich AN, et al. Biological properties and structure of the lipopolysaccharide of a vaccine strain of Francisella tularensis generated by inactivation of a quorum sensing system gene qseC. Biochemistry Biokhimiia. 2010;75:443–451. doi: 10.1134/s0006297910040073. [DOI] [PubMed] [Google Scholar]
- 37.Kostakioti M, et al. QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol Microbiol. 2009;73:1020–1031. doi: 10.1111/j.1365-2958.2009.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kostakioti M, et al. Distinguishing the contribution of type 1 pili from that of other QseB-misregulated factors when QseC is absent during urinary tract infection. Infect Immun. 2012;80:2826–2834. doi: 10.1128/IAI.00283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasko DA, et al. Targeting QseC signaling and virulence for antibiotic development. Science. 2008;321:1078–1080. doi: 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hadjifrangiskou M, et al. A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol Microbiol. 2011;80:1516–1529. doi: 10.1111/j.1365-2958.2011.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clarke MB, et al. The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci U S A. 2006;103:10420–10425. doi: 10.1073/pnas.0604343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curtis MM, et al. QseC inhibitors as an antivirulence approach for Gram-negative pathogens. mBio. 2014;5:e02165. doi: 10.1128/mBio.02165-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.WHO. Global Tuberculosis Report. 2016. [Google Scholar]
- 44.Ford CB, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bretl DJ, et al. Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiology and molecular biology reviews: MMBR. 2011;75:566–582. doi: 10.1128/MMBR.05004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandal OH, et al. Acid-Susceptible Mutants of Mycobacterium tuberculosis Share Hypersusceptibility to Cell Wall and Oxidative Stress and to the Host Environment. J Bacteriol. 2009;191:625–631. doi: 10.1128/JB.00932-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohde KH, et al. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe. 2007;2:352–364. doi: 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Walters SB, et al. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol. 2006;60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 49.Abramovitch RB, et al. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol. 2011;80:678–694. doi: 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan S, et al. Mycobacterium tuberculosis Responds to Chloride and pH as Synergistic Cues to the Immune Status of its Host Cell. PLoS Pathog. 2013;9:e1003282. doi: 10.1371/journal.ppat.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalo-Asensio J, et al. PhoP: A Missing Piece in the Intricate Puzzle of Mycobacterium tuberculosis Virulence. Plos One. 2008;3:e3496. doi: 10.1371/journal.pone.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin C, et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine. 2006;24:3408–3419. doi: 10.1016/j.vaccine.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 53.Perez E, et al. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. 2001;41:179–187. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 54.Johnson BK, et al. The Carbonic Anhydrase Inhibitor Ethoxzolamide Inhibits the Mycobacterium tuberculosis PhoPR Regulon and Esx-1 Secretion and Attenuates Virulence. Antimicrob Agents Chemother. 2015;59:4436–4445. doi: 10.1128/AAC.00719-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carta F, et al. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg Med Chem Lett. 19:6649–6654. doi: 10.1016/j.bmcl.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 56.Nishimori I, et al. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem. 2009;52:3116–3120. doi: 10.1021/jm9003126. [DOI] [PubMed] [Google Scholar]
- 57.Maresca A, et al. Carbonic anhydrase inhibitors. Inhibition of the Rv1284 and Rv3273 beta-carbonic anhydrases from Mycobacterium tuberculosis with diazenylbenzenesulfonamides. Bioorg Med Chem Lett. 2009;19:4929–4932. doi: 10.1016/j.bmcl.2009.07.088. [DOI] [PubMed] [Google Scholar]
- 58.Maresca A, et al. Dihalogenated sulfanilamides and benzolamides are effective inhibitors of the three beta-class carbonic anhydrases from Mycobacterium tuberculosis. Journal of enzyme inhibition and medicinal chemistry. 2013;28:384–387. doi: 10.3109/14756366.2011.645539. [DOI] [PubMed] [Google Scholar]
- 59.Guzel O, et al. Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial beta-carbonic anhydrases Rv1284 and Rv3273. J Med Chem. 2009;52:4063–4067. doi: 10.1021/jm9004016. [DOI] [PubMed] [Google Scholar]
- 60.Pacchiano F, et al. Inhibition of beta-carbonic anhydrases with ureido-substituted benzenesulfonamides. Bioorg Med Chem Lett. 2011;21:102–105. doi: 10.1016/j.bmcl.2010.11.064. [DOI] [PubMed] [Google Scholar]
- 61.Buchieri MV, et al. Inhibition of the beta-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: identification of the first inhibitor with anti-mycobacterial activity. Bioorg Med Chem Lett. 2013;23:740–743. doi: 10.1016/j.bmcl.2012.11.085. [DOI] [PubMed] [Google Scholar]
- 62.Maresca A, et al. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. Journal of enzyme inhibition and medicinal chemistry. 2013;28:392–396. doi: 10.3109/14756366.2011.650168. [DOI] [PubMed] [Google Scholar]
- 63.Minakuchi T, et al. Molecular cloning, characterization, and inhibition studies of the Rv1284 beta-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J Med Chem. 2009;52:2226–2232. doi: 10.1021/jm9000488. [DOI] [PubMed] [Google Scholar]
- 64.von Gnielinski N, et al. Non-classical β-carbonic anhydrase inhibitors-towards novel anti-mycobacterials. Med Chem Commun. 2014;5:1563–1566. [Google Scholar]
- 65.Ceruso M, et al. Sulfonamides incorporating fluorine and 1,3,5-triazine moieties are effective inhibitors of three beta-class carbonic anhydrases from Mycobacterium tuberculosis. Journal of enzyme inhibition and medicinal chemistry. 2014;29:686–689. doi: 10.3109/14756366.2013.842233. [DOI] [PubMed] [Google Scholar]
- 66.Frigui W, et al. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. 2008;4:e33. doi: 10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asensio JG, et al. The virulence-associated two component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem. 2006;281:1313–1316. doi: 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 68.Jain M, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007;104:5133–5138. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker JJ, et al. Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol Microbiol. 2014;94:56–69. doi: 10.1111/mmi.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stanley SA, et al. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gengenbacher M, Kaufmann SH. Mycobacterium tuberculosis: success through dormancy. FEMS microbiology reviews. 2012;36:514–532. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sivaramakrishnan S, de Montellano PR. The DosS-DosT/DosR Mycobacterial Sensor System. Biosensors. 2013;3:259–282. doi: 10.3390/bios3030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harms A, et al. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 2016:354. doi: 10.1126/science.aaf4268. [DOI] [PubMed] [Google Scholar]
- 74.Zheng H, et al. Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nature chemical biology. 2016 doi: 10.1038/nchembio.2259. [DOI] [PubMed] [Google Scholar]
- 75.Mak PA, et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS chemical biology. 2012;7:1190–1197. doi: 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deb C, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. Plos One. 2009;4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baek SH, et al. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011;9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang YT, et al. Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chemical biology & drug design. 2012;79:1007–1017. doi: 10.1111/j.1747-0285.2012.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ofek I, et al. Bacterial adhesion to animal cells and tissues. ASM Press; 2003. [Google Scholar]
- 80.Proft T, Baker EN. Pili in Gram-negative and Gram-positive bacteria - structure, assembly and their role in disease. Cellular and molecular life sciences: CMLS. 2009;66:613–635. doi: 10.1007/s00018-008-8477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pizarro-Cerda J, Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 82.Kline KA, et al. Bacterial adhesins in host-microbe interactions. Cell Host Microbe. 2009;5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 83.Thanassi DG, et al. Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS microbiology reviews. 2012;36:1046–1082. doi: 10.1111/j.1574-6976.2012.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sauer FG, et al. Bacterial pili: molecular mechanisms of pathogenesis. Curr Opin Microbiol. 2000;3:65–72. doi: 10.1016/s1369-5274(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 85.Costa TR, et al. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat Rev Microbiol. 2015;13:343–359. doi: 10.1038/nrmicro3456. [DOI] [PubMed] [Google Scholar]
- 86.Lillington J, et al. Reprint of “Biogenesis and adhesion of type 1 and P pili”. Biochim Biophys Acta. 2015;1850:554–564. doi: 10.1016/j.bbagen.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 87.Evans ML, Chapman MR. Curli biogenesis: order out of disorder. Biochim Biophys Acta. 2014;1843:1551–1558. doi: 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van Gerven N, et al. Bacterial amyloid formation: structural insights into curli biogensis. Trends Microbiol. 2015;23:693–706. doi: 10.1016/j.tim.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cegelski L, et al. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nature chemical biology. 2009;5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hung C, et al. Escherichia coli biofilms have an organized and complex extracellular matrix structure. mBio. 2013;4:e00645–00613. doi: 10.1128/mBio.00645-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Svensson A, et al. Design and evaluation of pilicides: potential novel antibacterial agents directed against uropathogenic Escherichia coli. Chembiochem: a European journal of chemical biology. 2001;2:915–918. doi: 10.1002/1439-7633(20011203)2:12<915::AID-CBIC915>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 92.Pinkner JS, et al. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci U S A. 2006;103:17897–17902. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berg V, et al. Design, synthesis and evaluation of peptidomimetics based on substituted bicyclic 2-pyridones-targeting virulence of uropathogenic E. coli. Bioorganic & medicinal chemistry. 2006;14:7563–7581. doi: 10.1016/j.bmc.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 94.Han Z, et al. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J Med Chem. 2010;53:4779–4792. doi: 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han Z, et al. Lead optimization studies on FimH antagonists: discovery of potent and orally bioavailable ortho-substituted biphenyl mannosides. J Med Chem. 2012;55:3945–3959. doi: 10.1021/jm300165m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Totsika M, et al. A FimH inhibitor prevents acute bladder infection and treats chronic cystitis caused by multidrug-resistant uropathogenic Escherichia coli ST131. J Infect Dis. 2013;208:921–928. doi: 10.1093/infdis/jit245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cusumano CK, et al. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Science translational medicine. 2011;3:109ra115. doi: 10.1126/scitranslmed.3003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guiton PS, et al. Combinatorial small-molecule therapy prevents uropathogenic Escherichia coli catheter-associated urinary tract infections in mice. Antimicrob Agents Chemother. 2012;56:4738–4745. doi: 10.1128/AAC.00447-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jarvis C, et al. Antivirulence Isoquinolone Mannosides: Optimization of the Biaryl Aglycone for FimH Lectin Binding Affinity and Efficacy in the Treatment of Chronic UTI. ChemMedChem. 2016;11:367–373. doi: 10.1002/cmdc.201600006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shamir ER, et al. Nitazoxanide inhibits biofilm production and hemagglutination by enteroaggregative Escherichia coli strains by blocking assembly of AafA fimbriae. Antimicrob Agents Chemother. 2010;54:1526–1533. doi: 10.1128/AAC.01279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chahales P, et al. Nitazoxanide Inhibits Pilus Biogenesis by Interfering with Folding of the Usher Protein in the Outer Membrane. Antimicrob Agents Chemother. 2016;60:2028–2038. doi: 10.1128/AAC.02221-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bolick DT, et al. Enteroaggregative Escherichia coli strain in a novel weaned mouse model: exacerbation by malnutrition, biofilm as a virulence factor and treatment by nitazoxanide. J Med Microbiol. 2013;62:896–905. doi: 10.1099/jmm.0.046300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Warren CA, et al. Amixicile, a novel inhibitor of pyruvate: ferredoxin oxidoreductase, shows efficacy against Clostridium difficile in a mouse infection model. Antimicrob Agents Chemother. 2012;56:4103–4111. doi: 10.1128/AAC.00360-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hoffman PS, et al. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrob Agents Chemother. 2007;51:868–876. doi: 10.1128/AAC.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sisson G, et al. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother. 2002;46:2116–2123. doi: 10.1128/AAC.46.7.2116-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dubreuil L, et al. In vitro evaluation of activities of nitazoxanide and tizoxanide against anaerobes and aerobic organisms. Antimicrob Agents Chemother. 1996;40:2266–2270. doi: 10.1128/aac.40.10.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saldana Z, et al. Synergistic role of curli and cellulose in cell adherence and biofilm formation of attaching and effacing Escherichia coli and identification of Fis as a negative regulator of curli. Environmental microbiology. 2009;11:992–1006. doi: 10.1111/j.1462-2920.2008.01824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aberg V, et al. Microwave-assisted decarboxylation of bicyclic 2-pyridone scaffolds and identification of Abeta-peptide aggregation inhibitors. Organic & biomolecular chemistry. 2005;3:2817–2823. doi: 10.1039/b503294f. [DOI] [PubMed] [Google Scholar]
- 109.Cornelis GR, Van Gijsegem F. Assembly and function of type III secretory systems. Annu Rev Microbiol. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- 110.Stavrinides J, et al. Host-pathogen interplay and the evolution of bacterial effectors. Cell Microbiol. 2008;10:285–292. doi: 10.1111/j.1462-5822.2007.01078.x. [DOI] [PubMed] [Google Scholar]
- 111.Keyser P, et al. Virulence blockers as alternatives to antibiotics: type III secretion inhibitors against Gram-negative bacteria. Journal of internal medicine. 2008;264:17–29. doi: 10.1111/j.1365-2796.2008.01941.x. [DOI] [PubMed] [Google Scholar]
- 112.Duncan MC, et al. Chemical inhibitors of the type three secretion system: disarming bacterial pathogens. Antimicrob Agents Chemother. 2012;56:5433–5441. doi: 10.1128/AAC.00975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Swietnicki W, et al. Identification of small-molecule inhibitors of Yersinia pestis Type III secretion system YscN ATPase. PLoS One. 2011;6:e19716. doi: 10.1371/journal.pone.0019716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Houben EN, et al. Take five - Type VII secretion systems of Mycobacteria. Biochim Biophys Acta. 2014;1843:1707–1716. doi: 10.1016/j.bbamcr.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 115.Angelichio MJ, et al. Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect Immun. 1999;67:3733–3739. doi: 10.1128/iai.67.8.3733-3739.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanchez J, Holmgren J. Cholera toxin - a foe & a friend. The Indian journal of medical research. 2011;133:153–163. [PMC free article] [PubMed] [Google Scholar]
- 117.Taylor RK, et al. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.DiRita VJ, et al. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci U S A. 1991;88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Almagro-Moreno S, et al. Intestinal Colonization Dynamics of Vibrio cholerae. PLoS Pathog. 2015;11:e1004787. doi: 10.1371/journal.ppat.1004787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shakhnovich EA, et al. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc Natl Acad Sci U S A. 2007;104:2372–2377. doi: 10.1073/pnas.0611643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hung DT, et al. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science. 2005;310:670–674. doi: 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- 122.Surawicz CM, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. The American journal of gastroenterology. 2013;108:478–498. doi: 10.1038/ajg.2013.4. quiz 499. [DOI] [PubMed] [Google Scholar]
- 123.Kelly CP. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2012;18(Suppl 6):21–27. doi: 10.1111/1469-0691.12046. [DOI] [PubMed] [Google Scholar]
- 124.Hu MY, et al. Prospective derivation and validation of a clinical prediction rule for recurrent Clostridium difficile infection. Gastroenterology. 2009;136:1206–1214. doi: 10.1053/j.gastro.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 125.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. The New England journal of medicine. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 126.Pamer EG. Fecal microbiota transplantation: effectiveness, complexities, and lingering concerns. Mucosal immunology. 2014;7:210–214. doi: 10.1038/mi.2013.117. [DOI] [PubMed] [Google Scholar]
- 127.Shim JK, et al. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet. 1998;351:633–636. doi: 10.1016/S0140-6736(97)08062-8. [DOI] [PubMed] [Google Scholar]
- 128.Kyne L, et al. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. The New England journal of medicine. 2000;342:390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 129.Drudy D, et al. Emergence and control of fluoroquinolone-resistant, toxin A-negative, toxin B-positive Clostridium difficile. Infection control and hospital epidemiology. 2007;28:932–940. doi: 10.1086/519181. [DOI] [PubMed] [Google Scholar]
- 130.Pruitt RN, Lacy DB. Toward a structural understanding of Clostridium difficile toxins A and B. Frontiers in cellular and infection microbiology. 2012;2:28. doi: 10.3389/fcimb.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shen A, et al. Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nature structural & molecular biology. 2011;18:364–371. doi: 10.1038/nsmb.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bender KO, et al. A small-molecule antivirulence agent for treating Clostridium difficile infection. Science translational medicine. 2015;7:306ra148. doi: 10.1126/scitranslmed.aac9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.trials, N.c. https://clinicaltrials.gov/ct2/results?term=ebselen&Search=Search.
- 134.Lu J, et al. Inhibition of bacterial thioredoxin reductase: an antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013;27:1394–1403. doi: 10.1096/fj.12-223305. [DOI] [PubMed] [Google Scholar]
- 135.Lieberman OJ, et al. High-throughput screening using the differential radial capillary action of ligand assay identifies ebselen as an inhibitor of diguanylate cyclases. ACS chemical biology. 2014;9:183–192. doi: 10.1021/cb400485k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Favrot L, et al. Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen. Nature communications. 2013;4:2748. doi: 10.1038/ncomms3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhabak KP, Mugesh G. Functional mimics of glutathione peroxidase: bioinspired synthetic antioxidants. Accounts of chemical research. 2010;43:1408–1419. doi: 10.1021/ar100059g. [DOI] [PubMed] [Google Scholar]
- 138.Beilhartz GL, et al. Comment on “A small-molecule antivirulence agent for treating Clostridium difficile infection”. Science translational medicine. 2016;8:370tc372. doi: 10.1126/scitranslmed.aad8926. [DOI] [PubMed] [Google Scholar]
- 139.Butler T. Plague into the 21st century. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2009;49:736–742. doi: 10.1086/604718. [DOI] [PubMed] [Google Scholar]
- 140.Pan NJ, et al. Targeting type III secretion in Yersinia pestis. Antimicrob Agents Chemother. 2009;53:385–392. doi: 10.1128/AAC.00670-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cornelis GR, et al. The virulence plasmid of Yersinia, an antihost genome. Microbiology and molecular biology reviews: MMBR. 1998;62:1315–1352. doi: 10.1128/mmbr.62.4.1315-1352.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bozue J, et al. A Yersinia pestis YscN ATPase mutant functions as a live attenuated vaccine against bubonic plague in mice. FEMS Microbiol Lett. 2012;332:113–121. doi: 10.1111/j.1574-6968.2012.02583.x. [DOI] [PubMed] [Google Scholar]
- 143.Ritchie JM, Waldor MK. The locus of enterocyte effacement-encoded effector proteins all promote enterohemorrhagic Escherichia coli pathogenicity in infant rabbits. Infect Immun. 2005;73:1466–1474. doi: 10.1128/IAI.73.3.1466-1474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pujol C, Bliska JB. Turning Yersinia pathogenesis outside in: subversion of macrophage function by intracellular yersiniae. Clinical immunology. 2005;114:216–226. doi: 10.1016/j.clim.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 145.Pujol C, Bliska JB. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun. 2003;71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Neyt C, Cornelis GR. Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol Microbiol. 1999;33:971–981. doi: 10.1046/j.1365-2958.1999.01537.x. [DOI] [PubMed] [Google Scholar]
- 147.Dacheux D, et al. Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol Microbiol. 2001;40:76–85. doi: 10.1046/j.1365-2958.2001.02368.x. [DOI] [PubMed] [Google Scholar]
- 148.Akeda Y, Galan JE. Chaperone release and unfolding of substrates in type III secretion. Nature. 2005;437:911–915. doi: 10.1038/nature03992. [DOI] [PubMed] [Google Scholar]
- 149.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Manzanillo PS, et al. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.De Leon J, et al. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J Biol Chem. 2012;287:44184–44191. doi: 10.1074/jbc.M112.420869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pym AS, et al. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 153.Rybniker J, et al. Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host Microbe. 2014;16:538–548. doi: 10.1016/j.chom.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 154.Takii T, et al. Simple fibroblast-based assay for screening of new antimicrobial drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46:2533–2539. doi: 10.1128/AAC.46.8.2533-2539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hsu T, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pang X, et al. Evidence for complex interactions of stress-associated regulons in an mprAB deletion mutant of Mycobacterium tuberculosis. Microbiology. 2007;153:1229–1242. doi: 10.1099/mic.0.29281-0. [DOI] [PubMed] [Google Scholar]
- 157.Pang X, et al. MprAB regulates the espA operon in Mycobacterium tuberculosis and modulates ESX-1 function and host cytokine response. J Bacteriol. 2013;195:66–75. doi: 10.1128/JB.01067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rohmer L, et al. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011;19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Olive AJ, Sassetti CM. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol. 2016;14:221–234. doi: 10.1038/nrmicro.2016.12. [DOI] [PubMed] [Google Scholar]
- 160.Russell DG, et al. Mycobacterium tuberculosis Wears What It Eats. Cell Host Microbe. 2010;8:68–76. doi: 10.1016/j.chom.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Clemens DL, Horwitz MA. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J Exp Med. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tan S, Russell DG. Trans-species communication in the Mycobacterium tuberculosis-infected macrophage. Immunol Rev. 2015;264:233–248. doi: 10.1111/imr.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fratti RA, et al. Regulators of membrane trafficking and Mycobacterium tuberculosis phagosome maturation block. Electrophoresis. 2000;21:3378–3385. doi: 10.1002/1522-2683(20001001)21:16<3378::AID-ELPS3378>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 164.Schaible UE, et al. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J Immunol. 1998;160:1290–1296. [PubMed] [Google Scholar]
- 165.Van der Geize R, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rothchild AC, et al. iNKT cell production of GM-CSF controls Mycobacterium tuberculosis. PLoS Pathog. 2014;10:e1003805. doi: 10.1371/journal.ppat.1003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Griffin JE, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lancaster JR., Jr Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chemical research in toxicology. 2006;19:1160–1174. doi: 10.1021/tx060061w. [DOI] [PubMed] [Google Scholar]
- 170.Voskuil MI, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rohde K, et al. Mycobacterium tuberculosis and the environment within the phagosome. Immunol Rev. 2007;219:37–54. doi: 10.1111/j.1600-065X.2007.00547.x. [DOI] [PubMed] [Google Scholar]
- 172.McCune RM, et al. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J Exp Med. 1966;123:445–468. doi: 10.1084/jem.123.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tian J, et al. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of alpha-ketoglutarate decarboxylase. Proc Natl Acad Sci U S A. 2005;102:10670–10675. doi: 10.1073/pnas.0501605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bryk R, et al. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- 175.Bryk R, et al. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 176.Shi S, Ehrt S. Dihydrolipoamide acyltransferase is critical for Mycobacterium tuberculosis pathogenesis. Infect Immun. 2006;74:56–63. doi: 10.1128/IAI.74.1.56-63.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McMurray DN. Disease model: pulmonary tuberculosis. Trends in molecular medicine. 2001;7:135–137. doi: 10.1016/s1471-4914(00)01901-8. [DOI] [PubMed] [Google Scholar]
- 178.Bryk R, et al. Selective killing of nonreplicating mycobacteria. Cell Host Microbe. 2008;3:137–145. doi: 10.1016/j.chom.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.VanderVen BC, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 2015;11:e1004679. doi: 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Griffin JE, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol. 2012;19:218–227. doi: 10.1016/j.chembiol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gould TA, et al. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol. 2006;61:940–947. doi: 10.1111/j.1365-2958.2006.05297.x. [DOI] [PubMed] [Google Scholar]
- 182.Munoz-Elias EJ, et al. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- 183.Eoh H, Rhee KY. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proc Natl Acad Sci U S A. 2014;111:4976–4981. doi: 10.1073/pnas.1400390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Savvi S, et al. Functional characterization of a vitamin B-12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: Implications for propionate metabolism during growth on fatty acids. J Bacteriol. 2008;190:3886–3895. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dresen C, et al. A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J Biol Chem. 2010;285:22264–22275. doi: 10.1074/jbc.M109.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Evans ML, et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Molecular cell. 2015;57:445–455. doi: 10.1016/j.molcel.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cordero-Alba M, et al. SrfJ, a Salmonella type III secretion system effector regulated by PhoP, RcsB, and IolR. J Bacteriol. 2012;194:4226–4236. doi: 10.1128/JB.00173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Capyk JK, et al. Activity of 3-ketosteroid 9alpha-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem. 2011;286:40717–40724. doi: 10.1074/jbc.M111.289975. [DOI] [PMC free article] [PubMed] [Google Scholar]