

Abstract
It is unknown how T cell antigen receptors from autoreactive CD8+ T cells recognize self. A new paper shows that an unorthodox, low-affinity way of interacting with targets may be at the basis of autoimmunity.
The human adaptive immune system relies heavily on casual encounters. Intravital microscopy has shown in tremendous detail how primed T cells survey their environment and bump into thousands of other cells in the process. Unless a foreign substance is recognized, these encounters are merely brushes, and the partners that meet soon continue their usual business. In patients suffering autoimmune disease, T cells somehow arrest in response to a presented self antigen and become inflammatory. How these autoreactive cells reach the periphery in the first place and why they recognize self remains a mystery. In this issue of Nature Immunology, Bulek et al. shed light on how the unusual structural interaction between a CD8+ T cell and its target may promote autoimmunity1.
From a structural perspective, productive confrontations between a T cell antigen receptor (TCR) on a CD8+ T cell and a peptide-loaded major histocompatibility complex (MHC) class I molecule (pMHCI) are generally a cordial affair. Studies of pathogen-specific CD8+ T cells have shown that their TCRs extensively contact both peptide and MHC class I domains. In doing so, TCRs and peptides show some structural plasticity to accommodate optimal interaction. Once contact is established, the binding partners clearly are not in a hurry to release their grip, showing high binding affinity. In essence, the adaptive immune system has evolved to recognize and eliminate a vast and ever-fluctuating array of foreign intruders as specifically and efficiently as possible. It is therefore thought that deviations from this structural interaction pattern may be at the very basis of autoimmune disease, leading the CD8+ T cells to ‘see’ pMHCI that they should instead ‘ignore’.
Until now, the subject of autoreactive TCRs has been addressed only through the use of MHC class II–restricted CD4+ T cells, for which several studies have indeed demonstrated structural abnormalities during TCR-pMHCII interactions. Such abnormalities include ‘sliding’ of the TCR toward the edge of the pMHCII complex, which results in less ability to contact the peptide moiety and unusual TCR docking angles2. Eccentric TCR binding and restricted peptide access seem to be common themes and have been hypothesized to underlie the degeneracy associated with autoreactive T cells3,4. However, the structural landscape of autoreactive TCR-pMHCI interactions is entirely uncharted. The inherently different architecture of pMHCI and pMHCII, notably the existence of an open peptide groove in pMHCII that allows the presentation of longer peptides, could potentially allow distinct alternative docking modes.
In their present study, Bulek et al. address this knowledge gap by presenting the protein structure of the well-documented autoreactive human 1E6 CD8+ T cell clone in contact with MHC class I displaying its cognate peptide1. There is little doubt that the 1E6 clone used here is a genuine player in human autoimmunity. This clone was isolated from a patient with type 1 diabetes and has been described before in an elegant study; it shows reactivity to a naturally processed epitope from the preproinsulin signal peptide5. Interestingly, high glucose concentrations stimulate the presentation of this epitope by beta cells of the pancreas, which in turn enter a downward spiral toward apoptosis because of enhanced killing by cytotoxic T lymphocytes. The fact that reactivity to the epitope is frequently found in blood samples from patients with type 1 diabetes and the finding that this clone exists in pancreata from such patients6 add robust support to the assumption that the structural data reported by Bulek et al.1 correlate with true autoreactive functionality.
A surprising finding of Bulek et al. is that the clone is able to directly lyse isolated human islets without the need for any exogenous cytokine stimulus to enhance the expression of MHC class I (ref. 1). The authors suggest that this may be an indication of potential primacy of this clone in disease etiology, allowing the responding T cells to initiate rather than just respond to local inflammation. It is of course impossible to compare the expression of MHC class I on isolated human beta cells with its endogenous expression, and the option that cellular stress in vitro leads to just enough MHC class I for limited targeting should be considered. Nonetheless, the considerable enhancement of the killing efficiency of 1E6 when inflammatory cytokines are added is perhaps more reflective of the local inflammatory conditions that are generally found shortly after diagnosis6.
Bulek et al. use a mutagenesis screen of the cognate peptide sequence to show that the centrally located residues determine reactivity1. In analogy with most pathogen-reactive clones, structural characterization shows a rather conventional, centrally oriented TCR docking mode. All other aspects of the interaction, however, demonstrate a less congenial liaison than that described above for pathogen-specific T cells. First, the degree of conformational plasticity during complex formation is minimal, designated by the authors as a rigid ‘lock-and-key’ docking. Second, the binding partners barely touch, and the MHC contact footprint is very narrow. The TCR contacts are highly peptide centric and dominated by hydrophobic ‘hot spots’. Finally, binding affinity is the weakest of any natural agonist antigen yet described and extends the range of natural agonist interactions far below what was thought functionally relevant.
The findings discussed above thus redefine the understanding of what constitutes a functionally relevant TCR-pMHC interaction (Fig. 1). The features described above suggest that a rather superficial recognition mechanism may suffice for T cell activation, as only a constrained portion of the peptide is scanned by the TCR. This binding mode hints at the possibility that this clone could potentially react with many different peptides. Indeed, another study has shown that the 1E6 TCR can recognize well over 1 × 106 different peptide sequences ten amino acids in length as well as (or, in many cases, much better than) the disease-relevant preproinsulin structure7. These findings serve to highlight the enormous potential of TCR degeneracy to be a causative factor in autoimmune disease. The search for environmental agents able to prime these cells is usually undertaken in hope of finding one, or a few, responsible antigen(s). The present data suggest that there could in fact be many. We propose that the establishment of a local ‘fertile field’, a proinflammatory microenvironment, by infectious agents may be required to make the low-avidity autoreactive cells become pathogenic. This could be achieved by a variety of mechanisms such as molecular mimicry and would probably be aided by enhanced ‘visibility’ of the beta cells as they begin to overexpress MHC class I (ref. 8).
Figure 1.
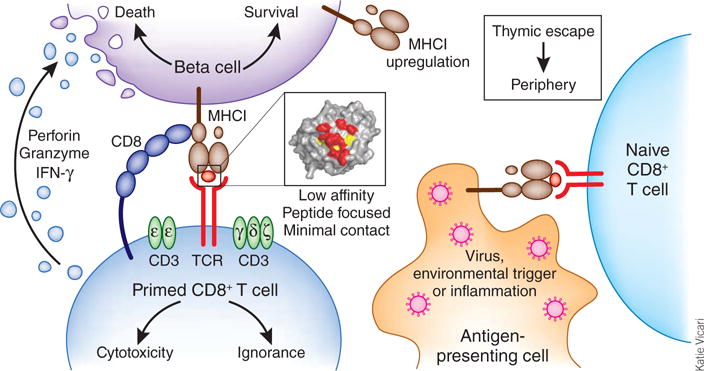
How autoreactive CD8+ T cells may induce autoimmune disease. CD8+ T cells specific for self somehow escape thymic negative selection and reach the periphery. These naive cells are subsequently primed by environmental triggers such as viruses, which are a source of high-affinity epitopes for optimal TCR ligation. Primed CD8+ T cells traffic to the pancreas, where they recognize endogenous target sequences, presented here by beta cells with hyper-expression of MHC class I. The data reported by Bulek et al. demonstrate that this interaction can be characterized by low-affinity and minimal, peptide-focused contacts1. Whereas the expected outcome of such a low-affinity TCR recognition would be ignorance, this subtle mode of interaction seems to suffice for the induction of cytotoxicity that results in beta-cell death. In patients with type 1 diabetes, high glucose concentrations lead to beta-cell stress and autophagy, which in turn could induce the presentation of neoantigens and enhanced killing of beta cells. IFN, interferon.
The repercussions of thymic escape are of importance equal to the issues discussed above. Is it reasonable to assume that CD8+ T cells encounter antigens differently in the thymus, or should the working hypothesis be that the antigenic repertoire differs from the periphery? The present work would favor the former theory, indicating involvement of degeneracy in recognition due to extremely limited TCR contact with the peptide sequence. As efficient as thymic selection may be, the process may simply fail to weed out all the problematic TCRs if they only have to bind as weakly, as has been found here, to trigger disease. The second scenario is supported by work showing that alternative peptide-loading conformations exist, depending on the loading pathway9. An alternative modification pathway could be post-translational modification, leading to ‘neoantigens’, as is seen after citrullination in rheumatoid arthritis. If it is assumed that such fundamental differences exist between the thymus and pancreas in peptide generation, it seems plausible that autoreactive T cells simply encounter the antigen for the first time in the pancreas.
Futures studies similar to those of Bulek et al.1 may aid in the rational design of altered peptide ligands. Although such ligands were once heralded as a successful avenue toward efficient antigen-specific tolerization, successful prediction of their tolerogenic potential has proven problematic. It is still largely unresolved how seemingly similar TCR-pMHC structures can lead to substantially divergent responses ranging from immunity to tolerance10. The present findings do not directly align with results obtained with nonobese diabetic mice showing that over time, high-avidity CD8+ T cells clones become dominant drivers of disease11. In those studies, depletion of low-affinity clones resulted in disease aggravation, with the corollary being that peptide tolerization should be targeted only to high-avidity clones11. However, cells of lower avidity might be potent disease drivers12, and the present study1 is well in line with that observation.
The controversies noted above could be resolved by more structural studies of functionally well-confirmed autoreactive clones that collectively could show how suboptimal peptide-MHC or TCR-pMHC interactions come about and affect downstream signaling. The difficulty here is of course that other contributors such as the coreceptors CD4 and CD8 and the various chains of the invariant signaling protein CD3 need to be mapped as well.
However casual they may seem, such autoreactive encounters are thus far more complex and ambiguous than previously assumed. Solving this particular puzzle will undoubtedly require a concerted effort, but the stakes are high, as here may lie the very key to understanding how autoimmunity develops.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Ken T Coppieters, Unit for Molecular Immunology and Inflammation, Department of Rheumatology, Ghent University Hospital, Ghent, Belgium.
Matthias G von Herrath, Type 1 Diabetes Center, The La Jolla Institute for Allergy and Immunology, La Jolla, California, USA.
References
- 1.Bulek M, et al. Nat Immunol. 2011;13:237–245. [Google Scholar]
- 2.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, et al. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sethi DK, et al. J Exp Med. 2011;208:91–102. doi: 10.1084/jem.20100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skowera A, et al. J Clin Invest. 2008;118:3390–3402. doi: 10.1172/JCI35449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coppieters KT, et al. J Exp Med. 2012;16:51–60. doi: 10.1084/jem.20111187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wooldridge L, et al. J Biol Chem. 2012;11:68–77. [Google Scholar]
- 8.von Herrath MG, Fujinami RS, Whitton JL. Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 9.Mohan JF, et al. Nat Immunol. 2010;11:350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding YH, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Immunity. 1999;11:45–56. doi: 10.1016/s1074-7613(00)80080-1. [DOI] [PubMed] [Google Scholar]
- 11.Han B, et al. Nat Med. 2005;11:645–652. doi: 10.1038/nm1250. [DOI] [PubMed] [Google Scholar]
- 12.von Herrath MG, Dockter J, Oldstone MB. Immunity. 1994;1:231–242. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]