

Abstract
Ancillary ligands, especially the tripodal ligands such as tris(triazolylmethyl)amines, have been widely used to accelerate the Cu-catalyzed azide-alkyne cycloaddition (CuAAC, a “click” reaction). However, the relationship between the activity of these Cu(I) complexes and their stability against air oxidation and ligand dissociation/exchange was seldom studied, which is critical for the applications of CuAAC in many biological systems. In this work, we synthesized twenty-one Cu(I) tripodal ligands varying in chelate arm length (five to seven atoms), donor groups (triazolyl, pyridyl and phenyl), and steric hindrance. The effects of these variables on the CuAAC reaction, air oxidation, and ligand dissociation were evaluated. Reducing the chelate arm length to five atoms, decreasing steric hindrance, or using a relatively weakly-binding ligand can significantly increase the CuAAC reactivity of the Cu(I) complexes, but the concomitant higher degree of oxidation cannot be avoided, which leads to rapid degradation of a histidine-containing peptide as a model of proteins. The oxidation of the peptide can be reduced by attaching oligo(ethylene glycol) chains to the ligands as sacrificing reagents. Using electrospray ionization mass spectrometry (ESI-MS), we directly observed the tri- and di-copper(I)-acetylide complexes in CuAAC reaction in the [5,5,5] ligand system and a small amount of di-Cu(I)-acetylide in the [5,5,6] ligand system. Only the mono-Cu(I) ligand adducts were observed in the [6,6,6] and [5,6,6] ligand systems.
Keywords: copper-catalyzed alkyne-azide cycloaddition, copper(I) ligands, dissociation constant, oxidation, ascorbic acid
Table of contents entry
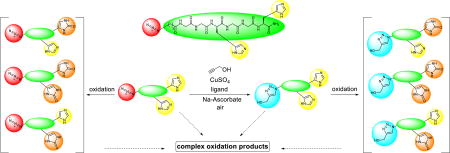
Oligo(ethylene glycol) tethered Cu(I) ligands sacrificially protect catalysts and biomolecules from being oxidized in the CuAAC reaction.
Introduction
Copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction1, 2 as a typical “click” reaction3 has been widely used in drug discovery,4–6 materials science,7, 8 bioconjugation,6, 9–15 and many other fields. Ancillary ligands such as nitrogen (amine, N(sp2)-Type), carbon (N-heterocyclic carbene), phosphorous and sulfur donors, have been extensively studied to accelerate the reaction, and to stabilize the Cu(I) against disproportionation and oxidation by O2.16 In bioconjugation reactions, the tripodal tris(triazolylmethyl)amine ligands, such as THPTA,17, 18 BTTAA,19 BTTES,20 BTTPS,21 BTTP,22 and 1 (Fig. 1 & 2),23 are the most efficient ligands that enable the reaction to be completed within minutes at tens of micromolar level of copper.24, 25 However, in the complex biological systems, the cytotoxicity of Cu(I)26–28 and the presence of strong-coordinating biological chelators that deactivate the copper catalyst24, 29 greatly hampered the application of CuAAC reaction in living systems.30
Fig. 1.

Structural formulas of tripodal tris(triazolylmethyl)amine ligands for bioconjugation reaction.
Fig. 2.

Structural formulas of Cu(I) ligands (1–21). The ligands are classified according to 1) donor types: [Tr,Tr,Tr] for 1–5, [Tr,Tr,Py] for 8, [Tr,Py,Py] for 9, [Py,Py,Py] for 10 and 11, [Py,Py,Ph] for 12–16, and [Tr,Tr,Ph] for 6 and 17–21, where Tr = triazoyl, Py = pyridyl, and Ph = phenyl; 2) chelate arm lengths: [6,6,6] for 2, 8–10, 12–14, 17 and 18, [5,5,5] for 1, 6, 11 and 21, [5,5,6] for 4 and 20, [5,6,6] for 5, 15, 16 and 19, and [7,7,7] for 3; and 3) methylation as in 13–16 and 18–21 to enhance the steric hindrance and electron donating property.
In most cases, a reducing agent (e.g., ascorbate) is needed to maintain the catalytically active Cu(I) oxidation state.2, 31 Nevertheless, the Cu(I)/Cu(II)/ascorbate/O2 redox cycle rapidly generates reactive oxygen species (ROS)32 that can degrade the biomolecules and the Cu(I) ligands,26–28 which is one of the major drawbacks of CuAAC reaction compared to the copper(I)-free bioconjugation methods.33 The oxidation mechanism of Cu(I) complexes has been extensive studied.34, 35 Increasing the denticity of ligands,36 expanding the size of the chelate ring,37 reducing the electron-donating ability of ligands,38 and adding sterically demanded substituents36 can improve the stability of Cu(I) complexes against oxidation. Tripodal ligands with a six-membered chelate ring bearing pyridyl and phenyl chelating arms exhibit a relatively high stability in air,37, 39–41 which are potential candidates for CuAAC reaction in bioconjugation.
On the other hand, the competition from biological Cu(I) ligands, such as glutathione (GSH, dissociation constants KD ≈ 9 ×10−12 M−1),29 an abundant copper-binding tripeptide presenting in micromolar concentration inside live cells,42, 43 also greatly reduce the catalytic activity of Cu(I) complexes in bioconjugation reactions.30 We performed CuAAC reaction inside live mammalian cells using tris(triazolylmethyl)amine ligands.30 Although an overall yield of ~18% was obtained on membrane proteins including those in the intracellular membranes, the yield of the reaction in cytosolic proteins was only 0.8%, which was attributed to the deactivation of the copper catalyst mainly by GSH. Therefore, to improve the efficiency of CuAAC reaction inside live cells, we need to develop efficient copper catalysts that are stable against dissociation by GSH.
Despite the above challenges for bioconjugations, the CuAAC reaction produces a small triazole linkage resembling a peptide linkage, which possibly imposes the least perturbation on the biological function of the conjugates.44 In addition, the small ethynyl and azido handles can be easily incorporated to synthetic-and bio-molecules. These unique advantages of CuAAC30 over the copper-free click reactions30, 45 have motivated us and other researchers to develop more efficient catalysts that overcome the above challenges.
In this work, we systematically evaluated the factors influencing the activity of Cu(I) complexes in CuAAC reaction and their stability against oxidation and dissociation, including the chelate arm length (atom number from the central tertiary amine to copper), donor types, and steric demands. A series of tripodal tertiary amine ligands bearing triazolyl, pyridyl, and phenyl arms with a chelate arm length of 5−7 atoms were analyzed. The apparent rate constant (kobs) and yield of a model CuAAC reaction, the initial rate (Vo) of the ascorbate oxidation catalyzed by the Cu(I) complexes, and the dissociation constant (KD) of the Cu(I) complexes were reported. The efficiency of these Cu(I) ligands was quantitatively analyzed by a factor representing the CuAAC activity vs. anti-oxidation capability. Electrospray ionization mass spectrometry (ESI-MS) was employed to analyze the active Cu(I)-acetylide intermediates in the CuAAC reaction. Finally, selected ligands were evaluated for their performance in the CuAAC reaction involving a histidine-containing peptide.
Results and discussion
Ligands and measurement methods
Twenty-one structurally-related tripodal amine Cu(I) ligands (1–21, Fig. 2) were synthesized to investigate the effect of ligand type, chelate arm lengths and steric demands on the CuAAC activity and stability against oxidation and dissociation. Ligand BTTAA19 reported as a highly efficient ligand for CuAAC in bioconjugation was used for comparison. The results were summarized in Fig. 3 and Table S1, ESI†.
Fig. 3.

Performance of the 21 ligands (1–21) of Cu+. (A) Apparent second-order rate constant (kobs) for the reaction of azido-coumarin AC (100 µM) with propargyl alcohol (50 µM) in the presence of the ligand (100 µM), CuSO4 (100 µM), and sodium ascorbate (5 mM) in phosphate buffered saline (PBS, pH = 7.4) in air at 24 ± 1 °C during 60 min.57–60 (B) The initial air-oxidation rate (Vo) of sodium ascorbate (200 µM) by diffused O2 in the presence of ligand/Cu(I) 1:1 complexes (100 µM) in PBS (pH = 7.4) at 24 ± 1 °C. (C) Dissociation constants of the Cu(I)-ligand complexes measured by competitive binding assay with Bca. (D) The trade-off between CuAAC over oxidation (kobs/Vo). (E) The trade-off between CuAAC over dissociation constants (kobs/KD). Error bars represent the standard deviation from three-repeated measurement. [m,n,o] represents the chelate arm length. Me in [m,n,o-Me] represents the methyl substitution on the methylene group between central amine and phenyl arm, for 13, 15, 18–21. Me2 in [m,n,o-Me2] represents the methyl substitutions on the 6-position of pyridyl arms, for 14 and 16.
The CuAAC reactivity of the ligand-Cu(I) complexes was evaluated with a fluorogenic reaction20, 46 between propargyl alcohol and azido-coumarin (AC) to yield the fluorescent coumarin-triazole product (CT, Scheme 1A). The yields were calculated based on a calibration curve of the fluorescence intensity of the CT (Fig. S4), and the rate constants (kobs) were fitted using second order kinetic model (Fig. 3A, and Fig. S6∓S7, ESI†).
Scheme 1.

Reactions for the measurement of (A) CuAAC activity; (B) initial rate of oxidation; and (C) KD of the Cu(I) complexes with the ligands listed in Fig. 2.
The stability of the Cu(I) complexes against oxidation in air was evaluated by the initial rate of oxidation of ascorbate acid (Asc), as the reducing agent for converting the oxidized Cu(II) back to Cu(I) (Scheme 1B).47 The initial rate (Vo) for the consumption of ascorbate in the presence of the Cu(I) complex (Fig. 3B) was monitored by the decrease of its maximum absorbance at 265 nm in aqueous solutions and plotted in Fig. S3, ESI†.
The dissociation constants (KD) of the ligand-Cu(I) complexes were determined using bicinchoninic anion (Bca) as a competing ligand (Scheme 1C),48, 49 which forms a 2:1 complex with Cu(I) (formation constant β2 = 1.58 × 1017 M−2) that is essentially pH-independent at pH > 7.0.48, 50 Lower affinity probes such as Ferene would give better accuracy for ligands with KD > 10−9 M,49 but all ligands in Fig. 2 possess sufficient binding affinity (Fig. 3C) that can compete with Bca at (Bca)/Cu(I) of 40 µM/16 µM. Excess of Bca was used to ensure the dominance of Cu(I)(Bca)2 over Cu(I)(Bca), which is also a prerequisite for use of the competitive equilibrium method to determine KD of the complexes.48, 50 The influence of anions and solvents on the CuAAC reaction has been investigated.51, 52 The anions (Cl−, SO42−, and PF6−) and solvent (H2O) in our system coordinate with Cu(I) much more weakly compared to the ligands, thus should have a limited influence on the reaction.
Effect of chelate arm length, chelate angle and Cu-N distance
In ligand/Cu(I) 1:1 complexes, the length of the three chelate arms (designated as [m,n,o], where m, n, o = 5–7 are the number of atoms from the central amine to copper, see Fig. 4) largely influenced the ability of the ligands to stabilize Cu(I) against O2 oxidation. Among all the ligands tested (Fig. 2), the tris(triazolylethyl)amine ligand 2 ([6,6,6]) offered the best protection of Cu(I), showing more than 9-fold lower oxidation rate compared to most of the other ligands tested (Fig. 3B & Table S1, ESI†), including the reported [6,6,6] tris(pyridylethyl)amine 10.53 Complex 18-Cu(I) with [6,6,6-Me] structure also showed high stability against oxidation. Note that the [6,6,6] 10-Cu(I)-Cl complex has been proved more stable against oxidation compared to the [5,5,5] 11-Cu(I)-Cl complex.35 The increased chelation arm length in the [6,6,6] 10-Cu(I)-Cl creates a more sterically demanding complex, forming a square-based pyramidal structure that offered more protection of Cu(I) against oxidation by O2 compared to the [5,5,5] 11-Cu(I)-Cl complex with a trigonal bipyramidal structure that promotes oxidation of Cu(I).37
Fig. 4.

DFT (B3LYP/LANL2DZ)-optimized geometries of the Cu(I) complexes bearing 1, 2, 3, 5, 10, 11 ligands (A–F, respectively), in which the EG4 group was replaced with a methyl group. Red: Cu, blue: N, gray: C. Hydrogen atoms were omitted for clarity. The average of the three bond angles between N(1)−Cu−N(2), oxidation rate Vo (10−8 M s−1), and CuAAC second-order rate constants kobs (M−1s−1) were labeled on each complex.
We performed density functional theory (DFT, B3LYP54/LANL2DZ55) calculations to obtain the optimized geometry of selected ligand-Cu(I) complexes, including the Cu(I) complexes bearing triazolyl (1, 2, 3 & 5) and pyridyl (10, 11) ligands. As shown in Fig. 4 and Table 1, the chelate arm length of Cu(I) complexes mainly influenced the angles between the aromatic nitrogen donor N(1), the Cu(I), and the central amino nitrogen N(2). For example, an average of 78.4° N(1)−Cu(I)−N(2) angle in 1-Cu(I) ([5,5,5]) and 95.1° in 2-Cu(I) ([6,6,6]) were calculated. A clear correlation between the chelate angle and the oxidation property was revealed, i.e., larger N(1)−Cu(I)−N(2) angle gives better protection of Cu(I) against oxidation. It can be elucidated by the proposed oxidation mechanism of Cu(I) complexes, involving the activation of O2 by formation of a Cu/O2 1:1 adduct, followed by coordination with another Cu(I).34 The smaller chelation angle in ligand 1 system significantly facilitates the formation of the Cu2O2 complex.37, 56 In comparison, the [6,6,6] complexes 2-Cu(I) and 10-Cu(I) adopt a pseudo-tetrahedral geometry on Cu(I), which offered better protection of the Cu(I) oxidation state.
Table 1.
DFT (B3LYP/LANL2DZ)-optimized bond angles and distances of Cu(I) complexes bearing ligands 1, 2, 3, 5
| Ligand | Chelate arm length | Bond angle ϑ [°] (N(1)−Cu−N(2)) | Distance [Å] Cu−N(1)a | Distance [Å] Cu-N(2)b |
|---|---|---|---|---|
| 1 | [5,5,5] | 78.33, 78.40, 78.44 | 2.027, 2.028, 2.032 | 2.474 |
| 2 | [6,6,6] | 95.02, 95.14, 95.25 | 2.017, 2.018, 2.019 | 2.393 |
| 3 | [7,7,7] | 75.52, 91.87, 91.51 | 2.016, 2.022, 2.032 | 4.028 |
| 5 | [5,6,6] | 79.05, 93.68, 94.34 | 1.986, 2.027, 2.031 | 2.472 |
| 10 | [6,6,6] | 99.31, 99.35, 99.43 | 2.058, 2.061, 2.063 | 2.226 |
| 11 | [5,5,5] | 82.25, 82.39, 82.51 | 2.056, 2.057, 2.063 | 2.252 |
N(1): the nitrogen on triazole or pyridine ring.
N(2): the central amine nitrogen.
The distance between Cu and N(1) does not vary a lot with the chelate arm length. The Cu(I)-N(2) interaction, on the other hand, reflects a plausible influence on the oxidation of Cu(I) complexes. The [6,6,6] systems that are most stable against oxidation did show the strongest Cu(I)-N(2) interaction among the calculated complex of the same donor type. However, the [7,7,7] system of no Cu(I)-N(2) interaction is better in protecting the Cu(I) against oxidation compared to the [5,5,5] system that exhibits weak Cu(I)-N(2) interaction (Table 1).
Although the [6,6,6] chelate structure is the best for stabilizing Cu(I) against oxidation, it also exhibits a poor CuAAC reactivity with >10-fold lower kobs values compared to the [5,5,5] system bearing the same ligand type. In general, the coordination geometry of Cu(I) complex generates the same effect on their activity in oxidation and CuAAC reaction: larger N(1)−Cu(I)−N(2) angle often gives lower CuAAC activity. An exception is the [7,7,7] 3-Cu(I) complex, which displayed much lower CuAAC activity compared to the [5,6,6] complex 5-Cu(I) with a similar N(1)−Cu(I)−N(2) angle (86.3° vs. 89.0°, respectively). The primary structural difference between these two complexes is the Cu(I)−N(2) interaction in the [5,6,6] complex (Cu(I)−N(2) 2.472 Å) that does not exist in the [7,7,7] complex. Therefore, we suggest that the central amine donor may also play a role in accelerating the CuAAC reaction.
Comparing all the Cu(I) complexes tested (Fig. 3), the influence of the chelate arm length (of the same ligand type) on Cu(I) oxidation follows the order of [5,5,5] > [7,7,7] ≈ [5,5,6] ≈ [5,6,6] >> [6,6,6], while on the CuAAC activity follows a general trend of [5,5,5] > [5,5,6] ≈ [5,6,6] >> [7,7,7] > [6,6,6] among all the tris(N-donor)amine ligands ([Tr,Tr,Tr] & [Py,Py,Py]) and the unsubstituted bis(N-donor)(phenyl)amine ligands ([Tr,Tr,Ph] & [Py,Py,Ph]). The methyl substitution(s) on the bis(N-donor)(phenyl)amine ligands (13−16 and 18−20) also showed influence on the ligand’s CuAAC activity (see discussions below).
The dissociation constants (KD) of the Cu(I) complexes reflect the extent of potential ligand exchange during the CuAAC reaction, e.g., with alkyne or azide. The KD of tested Cu(I) complexes (Fig. 3C) is in the range of 1 pM to 1 nM. Among the tris(triazolyl)amines, ligands 1 ([5,5,5]) and 4 ([5,5,6]) showed 10-folds weaker coordinating strength toward Cu(I) compared to the ligands 2, 3 and 5 that form [6,6,6], [7,7,7], and [5,6,6] chelate rings. The chelating strengths of the [7,7,7] 3-Cu(I) complex and the [5,6,6] 5-Cu(I) complex, the calculated chelate angles, and the oxidation rate of the two complexes are very close.
Increasing the chelate arm lengths in [Tr,Tr,Ph] ligands (6→17, 21→20→19→18) generally results in decreased CuAAC activity and oxidation rate (vide supra). However, the stability of these Cu(I) complexes (KD) follows the reversed trend: the [5,5,5] shows the strongest coordination, followed by [5,5,6], [5,6,6], and [6,6,6] structure.
Effect of ligand types
The phenyl ring offers weak π-coordination towards Cu(I). For the N-coordinating ligands, the π-accepting ability of triazolyl ligands is lower than that of the pyridyl ligands because of the presence of p lone pair on the alkylated nitrogen of triazole. The basicity of pyridine is also stronger than triazole.61 Therefore, the pyridine ligand can form a more stable complex with the d10-filled Cu(I) ion as compared to the triazole ligands.62 Indeed, as calculated (Table 2), the distances between Cu(I) and the central amino nitrogen N(2) in the triazolyl complexes are much longer than those in the pyridyl complexes of similar structure. It can be attributed to the weaker π-accepting ability of triazole rings compared to pyridines61 as a better acceptor of the back-donation of electron by Cu(I) thus strengthen the electron-donating interaction of the central amine toward the Cu(I).
Table 2.
Recovery (%) of the liganda during the CuAAC reaction
| Time (min) | 1 | 5 | BTTAA |
|---|---|---|---|
| 5 | 68 ± 7 | 99 ± 2 | 70 ± 4 |
| 10 | 46 ± 1 | 41 ± 1 | 45 ± 3 |
| 30 | 24 ± 1 | 20 ± 4 | 29 ± 2 |
| 60 | 19 ± 1 | 15 ± 1 | 26 ± 2 |
Derived from Eq. 27–29 (ESI†). Each data point was the mean of three replicates and the error bar represented the standard deviation (SD).
The measured KD for Cu(I) complexes bearing ligands 1–21 is in general agreement with this trend (Fig. 3C). For the [6,6,6] chelate structure, the KD increased in the order of [Py,Py,Py] (10) < [Tr,Py,Py] (9) < [Tr,Tr,Py] (8) ≈ [Tr,Tr,Tr] (2) << [Py,Py,Ph] (12) ≈ [Tr,Tr,Ph] (17), while for the [5,5,5] system: [Py,Py,Py] (11) << [Tr,Tr,Tr] (1) ≈ [Tr,Tr,Ph] (6). On the other hand, appending a triazolyl group to 1 as the tetra-triazole 7 further decreased the KD.
In the [6,6,6] system, although the tris(pyridyl) complex 10-Cu(I) possesses larger chelate angle and shorter Cu-N(2) distances than the tris(triazolyl) complex 2-Cu(I) (Fig. 4), the latter shows an order of magnitude lower Vo than most of the other ligands. This result indicates that Py ligand promotes oxidation due to stronger sigma donating ability and also stronger capacity for accepting back donation from Cu(I) than triazoyl ligand. Besides, the ligand 18 bearing [Tr,Tr,Ph]-[6,6,6-Me] structure also offers good stability. Introducing an extra N-donor to 1 (as ligand 7) has little effect on the oxidation rate. Therefore, as discussed above, the oxidation property of a Cu(I) complex can be affected by the chelating geometry and steric demands around the Cu(I) center, but the electronic environment53 and donor types may also work together to stabilize the complexes. In the [5,5,5] systems, triazole donors are more efficient than pyridines to enhance the CuAAC reactivity (Fig. 3A), with the reactivity (kobs) following the order [Tr,Tr,Tr] (1) > (BTTAA) > [Tr,Tr,Ph] (6) > [Py,Py,Py] (11). Replacement of a triazole donor in 1 with a weaker phenyl group (6) resulted in a decrease in the CuAAC reactivity while replacing a triazole with a stronger π-accepting pyridine (11) caused a significant decrease in the CuAAC reactivity.
Effect of steric demands
We have discussed the steric effect caused by the length of the chelate arms. In addition, to study the steric effect we introduced methyl groups either to the methylene groups between the central amine and the phenyl arms (as in 13, 15, 18–21, designated as [m,n,o-Me]), or to the 6-position of the pyridyl rings (as in 14 and 16, designated as [m,n,o-Me2]). By adding a methyl group at the benzylic CH2, the interaction between Cu(I) and the phenyl ring can be enhanced due to the reduced free rotation of phenyl ring. It was reported that such methyl substitution greatly reduced the reactivity with O2.39 In our systems, reduced rate of oxidation (Vo) was observed for 13 compared to 12, 18 to 17, and 21 to 6 (Fig. 3 and Table S1). However, the effect on KD was not consistent. The methyl substitution on the pyridine ring of ligand increased the stability of its Cu(I) complex against oxidation, i.e., 14 vs. 12 in Fig. 3. However, a larger drop in kobs than in Vo was observed. The generally higher influence of steric demands on CuAAC activity than oxidation can be first attributed to the smaller size of dioxygen compared to the azide and alkyne, which is facile to attack the ligand-Cu(I) complexes. In addition, during the oxidation reaction, only one dioxygen molecule is bridged by two Cu(I), while the CuAAC reaction need both azide and alkyne to interact with Cu(I), which is more susceptible to the steric hindrance of the ligand.
Evaluation of data correlation and overall ligand efficiency
Collectively, the data for the 1:1 mixture of ligand 1–21 and BTTAA with Cu(I) (Fig. 3A–C and Table S1, ESI†) showed no direct correlation between the ligand-Cu(I) dissociation constant (KD) and the oxidation rate (Vo), with the correlation coefficient (r) being −0.16. In addition, no correlation between KD and the CuAAC reaction rate (kobs) was found (r = 0.08). However, there was a substantial correlation between the oxidation rate and CuAAC rate (r = 0.66). The statistical analysis on the present ligand systems indicate that: 1) neither Cu(I) oxidation nor CuAAC reactions is directly determined by the ligand-Cu(I) binding affinity; 2) in general, the Cu(I) catalysts showing higher CuAAC accelerating ability are more prone to be oxidized. Notably, current mechanistic studies suggest that both oxidation of Cu(I) by O2 and CuAAC reactions involve a bi-nuclear copper intermediate.35, 63, 64
Despite the positive correlation between the CuAAC activity and Cu(I) oxidation, there is room for optimizing the ligand efficiency. To evaluate the overall efficiency of tested ligands and to optimize the trade-off between the CuAAC activity and anti-oxidation ability, the kobs/Vo values of each ligand are plotted in Fig. 3D. In general, the tris(triazolyl) ligands are the most efficient ligands for CuAAC, having the best trade-off between CuAAC efficiency and anti-oxidation ability. The superior performance of the tris(triazolyl) ligands might be attributed to their relatively weak σ-donating ability. This type of ligand can be further optimized based on the kobs/Vo value. For example, the oligo(ethyleneglycol)-terminated tris(triazolyl) ligand 1 (kobs/Vo = 1.3) exhibits an enhanced anti-oxidation ability compared to the carboxylic acid-terminated BTTAA19 (kobs/Vo = 0.56, Table S1, ESI†) in PBS.
Faster oxidation of Cu(I) in air results in a drop of the CuAAC reaction yield (Table S1, ESI†) due to the rapid consumption of ascorbate reductant. Although ligand 1 exhibits the highest rate constant kobs (34 ± 7 M−1s−1), the reaction only achieved 89% yield during the 60 min reaction period. In comparison, the [5,6,6] ligand 5 with lower kobs (kobs = 11 ± 3 M−1s−1, kobs/Vo = 0.9) achieved the highest CuAAC yield (97%). The Cu(I) complex showed a 2-folds lower rate of ascorbate oxidation and 6-folds lower dissociation constant than the [5,5,5] ligand 1.
Note that both kobs and Vo may be substantially influenced by the solvent,17 and hence the optimization should be performed under conditions relevant to the specific application.
To evaluate the overall efficiency of tested ligands for their CuAAC activity over Cu(I) binding affinity, we plotted the kobs/KD values of each ligand in Fig. 3E. The tris(pyridylmethyl)amine ligand 11 showed much higher value than other ligands. This ligand not only accelerates the CuAAC reaction (kobs = 5.1 ± 0.5 M−1s−1 compared to ligand-free condition 0.6 ± 0.2 M−1s−1, Table S1 and Figure S7, ESI†), but also strongly binds the Cu(I) with a dissociation constant (KD = (1.5 ± 0.6) × 10−12 M−1) substantially lower than the cellular free thiol GSH (9 × 10−12 M−1).29 In addition, the 11-Cu(I) complex showed a reduced oxidation rate compared to the tris(triazolylmethyl)amine ligand 1.
Besides ligand 11, the tris(triazolyl)amine ligand 5 bearing one five-member ring and two six-member rings also exhibited a good CuAAC activity (kobs = 11 ± 3 M−1s−1), high Cu(I) binding affinity (KD = (18 ± 4) × 10−12 M−1, ten-fold smaller than ligand 1 and has the same order of magnitude as GSH), and slower oxidation compared to ligand 1. Therefore, ligand 11 and ligand 5 appear to be the best candidates among the ligands 1–21 for bioconjugation inside living mammalian cells.
Ligand capability of stabilizing di-Cu(I)-acetylide active species in CuAAC reaction
The mechanism of CuAAC reaction was proposed to go through a stepwise pathway involving two Cu(I) ions.64–70 The di-Cu(I)-acetylide has been proven as a much more active intermediate in the CuAAC reaction than the mono-Cu(I) species.63 Recently, results of our kinetic study of CuAAC reaction promoted by tris(triazoylmethl)amine ligands indicated the possibility that the trinuclear acelylide-Cu3-ligand complexes might be active intermediates in the reaction.71 Herein, we employed electrospray ionization mass spectrometry (ESI-MS) to detect Cu(I)-alkyne or Cu(I)-acetylide complexes with [Tr,Tr,Tr] ligands (1, 2, 4, 5) bearing different chelate ring size. By comparing the ability of these ligands in stabilizing the multinuclear Cu(I)-acetylide complexes, we aimed to find a possible correlation with the CuAAC reactivity.
ESI-MS has been widely employed for online monitoring of reactions, detecting elusive reaction intermediates,72 studying the metal-ligand solution equilibria,73, 74 and investigating organometallic reaction mechanisms.73, 75–77 In 2015, Iacobucci et al. reported the direct observation of CuAAC intermediates stabilized by the strong ligand triphenylphosphine using ESI-MS method.78 Our group also successfully detected multi-Cu(I) intermediates in the tris(triazolylmethyl)amine accelerated CuAAC reaction and quantitatively compared their activity.71
In the present study, we employed a water-soluble oligo(ethyleneglycol)-terminated alkyne (2-[2-(2-propyn-1-yloxyl)ethoxy]ethanol, PE, Fig. 5) to replace the volatile propargyl alcohol, while keeping the same Cu/ligand/alkyne ratio (1:1:0.5) as used in the kobs measurement, Cu(I) was generated in situ from reducing CuSO4 by sodium ascorbate.2, 31
Fig. 5.

Species corresponding to the assigned ESI-MS peaks obtained after mixing the alkyne with the ligand [Tr,Tr,Tr] 1, 2, 4 or 5, and CuSO4/Na-ascorbate. * and ** were formed by adventitious oxidation of the OEG side chain in the alkyne or the ligand during sample introduction to the ESI-MS. ESI-MS spectra of the Cu(I)-ligand-alkyne 1:1:0.5 mixture in water. CuSO4 (100 µM), ligand (100 µM), PE (50 µM), Na ascorbate (1 mM). (A) Ligand 1. (B) Ligand 2. (C) Ligand 4. (D) Ligand 5.
As shown in Fig. 5A, the [5,5,5] tris(triazolyl) amine ligand 1 formed a mixture of mono-Cu(I) ligand complex ([1+Cu]+), mono-Cu(I)-alkyne complex ([1+PE+Cu]+), a significant amount of di-Cu(I)-acetylide complex ([1+PE−H+2Cu]+), and a tiny amount of tri-Cu(I)-acetylide complex ([1+PE−H+3Cu]2+). The assignment is based on a good fitting of the theoretical and experimental m/z value and the corresponding isotope distribution patterns (Fig. S8, ESI†). In comparison, we did not detect any multi-Cu(I)-acetylides in the [6,6,6] (ligand 2) and [5,6,6] (ligand 5) ligand systems. Only the mono-Cu(I) ligand adducts were observed (Fig. 5B & 4D). Nevertheless, the ligand 4 with a shorter chelate arm length ([5,5,6]) generated a small amount of di-Cu(I)-acetylide ([4+PE−H+2Cu]+), showing low intensity in the spectrum (Fig. 5C). Adventitious oxidation of the OEG chain either in the alkyne or the ligand to vinyl or carboxylic acid derivatives (see below) was observed in the ESI-MS spectra of the mixtures, which can be attributed to the presence of a small amount of O2 during sample preparation and transfer to the mass spectrometer. The degree of oxidation increased with the rate of CuAAC reaction promoted by the ligands as 1 > 4 > 5 ≈ 2.
The presence of a larger portion of di-Cu(I)-acetylide active species in the Cu(I)/1/alkyne 1:1:0.5 system was consistent with the superior activity of ligand 1 in CuAAC reaction compared to other three ligands (Fig. 3A, kobs: 1 >> 4 ≈ 5 >> 2).
Furthermore, by increasing Cu(I)/ligand ratio from 1:1 to 2:1, there are 1.3-fold more tri-Cu(I) species, 15% less di-Cu(I) species, 22% more mono-Cu(I) species, and 0.8-fold less free Cu(I) species generated (Fig. S9 & Table S3, ESI†).
Peptide oxidation assay
To evaluate the performance of the lead ligands 1 and 5 in bioconjugation with CuAAC reaction, we incorporated an azido group to the N-terminal of a histidine-containing peptide Gly-Gly-His-Gly-Gly-His, the 15–20 fragment of the antimicrobial peptide Shepherin I (Shep I).79, 80 The reported ligand BTTAA was also tested for comparison. As shown in Scheme 2, the azido-peptide AP was subjected to the common CuAAC reaction conditions for bioconjugation, using propargyl alcohol (1 equiv., 50 µM) in the presence of ligand (2 equiv.), CuSO4 (1 equiv.) and sodium ascorbate (10 equiv.). The product triazolyl-peptide TP yield and extent of oxidation during the CuAAC reaction were monitored by liquid chromatography-mass spectrometry (LC-MS), using samples taken at 5, 10, 30, and 60 min reaction time. The result is depicted in Fig. 6 where the red and blue bars representing the remaining azidopeptide AP and product triazolyl-peptide TP, respectively.
Scheme 2.

The CuAAC reaction of the peptide AP (50 µM) with propargyl alcohol (50 µM), ligand (100 µM), CuSO4 (50 µM), and sodium ascorbate (500 µM) in Milli-Q water at 24 ± 1 °C in air to form the product TP and the oxidation byproducts (2-imidazolidone derivatives) API/II/III, TPI/II/III as well as a complex mixture of other oxidation byproducts.
Fig. 6.

Recovery (%) of the azido-peptide peptides AP (red) and the triazolyl-peptide TP (blue) during CuAAC reaction of AP in air with propargyl alcohol. Reaction conditions: the azido-peptide AP (50 µM), propargyl alcohol (50 µM), ligand (100 µM), CuSO4 (50 µM), and sodium ascorbate (500 µM) in Milli-Q water at 24 ± 1 °C. There was no ligand added in the (−) control. The red and blue bars represent the % amount of AP and TP relative to the amount of AP starting material, respectively, measured by LC-MS. The total represents the % peptide starting material and product withstanding the oxidation under the CuAAC conditions. Each data point was the mean of three replicates and the error bar represented the standard deviation.
The results show that without the ligands, a large extent of oxidative side reaction also took place and both the azidopeptide and triazolyl-peptide were rapidly oxidized. Only <4% of the peptides AP and TP remained intact after reacting for 30 min. The resultant by-products mainly include the oxidized histidine residue, showing an increase of mass by 16 amu or 32 amu, consistent with the oxidation of the imidazole ring in the histidine residues to the 2-imidazolidone derivatives API/II/III, TPI/II/III (Scheme 2),81 which were observed by LC-MS (Fig. S10, ESI†).
On the contrary, the presence of the ligands, especially the OEG-tethered [5,5,5] ligand 1 and [5,6,6] ligand 5, significantly reduced the peptide oxidation during CuAAC reaction. These two ligands offered better protection to peptide compared to the ligand BTTAA. As shown in Fig. 6, 44%, 61% and 24% of the peptides AP and TP remained intact in the presence of ligands 1, 5, and BTTAA, respectively. The tendency of the peptide oxidation follows the order of 5 < 1 < BTTAA, which is in accordance with the aforementioned ascorbate oxidation rate Vo (Fig. 3B). Thus, the peptide oxidation is correlated to the amount of reactive oxygen species (ROS) generated by Cu(I)/Cu(II)/ascorbate/O2 redox cycle.26–28, 32
As recommended by Finn et al.,17 a typical bioconjugation CuAAC reaction can be conducted with 5−6 equivalents of ligand in order to maintain the Cu(I) oxidation state in air. However, due to the triazoles-Cu(I) coordination that competes with the alkyne, the overall reaction rate was decreased when increasing the ligand to Cu(I) ratio from 1:1 to 6:1. Therefore, we employed the ligand/Cu(I) 2:1 ratio as the standard condition in our bioconjugation study. The ligand 1 was most efficient in CuAAC reaction, resulting in 32% product yield compared to 20% achieved by BTTAA system. During the 60 min reaction period, the CuAAC efficiency correlated with the oxidation rate of Cu(I) and peptides AP and TP. For ligand 1, the maximum yield of TP reached around 30 min, beyond which the yield started to decline due to oxidation. In comparison, for BTTAA, the maximum yield was achieved around 10 min.
As shown in Table 2, the % recovery of ligand 1 was almost the same as that of BTTAA (LC-MS quantification at 5, 10, and 30 min). However, the CuAAC reaction with BTTAA almost ceased at 10 min due to complete consumption of ascorbate or oxidation of the ligand. To rationalize the better performance of the ligand 1, we hypothesized that the OEG chain tethered on 1 sacrificially protected the tris(triazolylalkyl)amine core structure against oxidization by ROS generated by the Cu(I)/Cu(II)/ascorbate/O2 redox cycle, while the BTTAA ligand was oxidized on the tris(triazolylalkyl)amine core structure. Therefore, although the OEG degradation was prevalent, the CuAAC catalytic core [tris(triazolylalkyl)amine] of ligand 1 was intact. Indeed, a series of the OEG degradation products were observed by LC-MS (Fig. 7), including aldehydes and carboxylic acids and shortened OEG chains.
Fig. 7.

Total ion mass spectrum of ligand 1 fragments (total ion). The spectrum was obtained from the 60 min CuAAC reaction mixture containing the peptides by zoom in the region of m/z = 520–840 and retention time from 5–7 min. The experimental condition is given in ESI†. The oligo ethylene glycol (OEG) unit is designated as the [m,n,l] system where m, n, l = 1–4. The labeled peaks were summarized in Table S4, ESI† (Red: alcohol derivatives; Yellow: aldehyde derivatives; Blue: acid derivatives; Green: ether derivatives).82, 83 The single and multiple labeling represents the single and multiple components of the ligand fragments, respectively.
In the case of [5,6,6] ligand 5, although the system offered the best protection of Cu(I), the rise of ligand/Cu(I) ratio from 1:1 to 2:1 resulted in a drop of the CuAAC reaction rate. The excessive ligands reduced the accessibility to alkynes and azides for CuAAC reaction. Hence, while ligand 5 gave 97% yield in the fluorogenic CuAAC reaction (Cu/5 1:1, Fig. 3A), but <10% yield in the peptide CuAAC reaction (Cu/5 2:1, Fig. 6).
Proposed mechanism of CuAAC and Cu(I) oxidation
On the basis of the data from ESI-MS, Cu(I) oxidation,36, 84–86 and our previously proposed mechanism involving a di- or tri-Cu(I)-acetylide stabilized by tris(triazoylmethyl)amine ligands,87 we proposed the mechanism of CuAAC and O2 oxidation for Cu(I)-ligand I (Scheme 3). Due to the positive correlation between the CuAAC activity and Cu(I) oxidation (Fig. 3 and Table S1, ESI†), the alkyne and O2 coordination are the competing processes.87 In the CuAAC pathway, the alkyne interacts with I in both σ- and π-coordination modes, resulting in IIc1 or IIc2 (see ESI-MS of ligand 1 with the tri-Cu(I) complex in Fig. 5). The di- and trinuclear Cu(I) complex IIc1 and IIc2 coordinates with an azide to form the intermediate IIIc1 and IIIc2.84, 85 IIIc2 coordinates with another ligand or alkyne to form the intermediate IVc2. IIIc1 and IVc2 goes through the transition state metallacycle TSc1 and TSc2, leading to the triazolide IVc1 and Vc2.87 Protonation of the triazolide IVc1 and Vc2 by proximal alkyne or protonic solvent generates the triazole product and regenerates the catalyst IIc1 and IIc2.87 On the other hand, in the Cu(I) oxidation pathway, dioxygen is competing with alkyne in coordination with I. The oxygen coordinates with I in a σ-coordination mode to form intermediate IIo.81–83 The active mono-Cu(II) complex IIo interacts with another mono-Cu(I) complex I, and generates di-copper complex IIIo.81–83 The Cu(I) ion in IIIo rapidly oxidizes to Cu(II) ion in IVo, through the inner-sphere electron transfer pathway.84 Finally, the overall reactions form the peroxodicopper(II) complex Vo.81–83 Sodium ascorbate reduces Cu(II) ion back to Cu(I) ion, accompanied with the formation of reactive oxygen species (ROS).32 As discussed above, the oligo(ethylene glycol) (OEG) side arms from tris-triazole amine ligands can be sacrificially oxidized to partially protect the active Cu(I) catalyst in the CuAAC reaction.
Scheme 3.

Proposed mechanism of CuAAC and Cu(I) oxidation. L: ligands; L’: ligands or alkyne; Aky: alkyne; ROS: reactive oxygen species; OEG: oligo ethylene glycol side arms from ligands.
Conclusions
In summary, we have systematically investigated the structural effects of Cu(I)-ligand complexes including chelate arm length, donor type (triazoyl, pyridyl and phenyl) and steric demands on the CuAAC reactivity, stability against oxidation, and ligand dissociation/exchange. The tris(triazolylethyl)amine ligand 2 offered the best protection on Cu(I) against oxidation, the tris(pyridylethyl)amine ligand exhibited the best Cu(I) binding affinity, and the tris(triazolylmethyl)amine ligand 1 offered the best CuAAC activity.
Chelate arm length strongly affects both the O2 and CuAAC reactivity of Cu(I)-ligand complexes. In general, larger N(1)−Cu(I)−N(2) angle gives better protection of Cu(I) against oxidation, but also lower the CuAAC activity. The influence of chelate arm length (of the same ligand type) on Cu(I) oxidation follows the order of [5,5,5] > [7,7,7] ≈ [5,5,6] ≈ [5,6,6] >> [6,6,6], and in CuAAC reaction follows [5,5,5] > [5,5,6] ≈ [5,6,6] >> [7,7,7] > [6,6,6] for the tris(N-donor)amine ligands ([Tr,Tr,Tr] & [Py,Py,Py]). The central amine donor could play a role in accelerating the CuAAC reaction, but has no evident influence on the oxidation process. Optimization of the ligands with mixed chelate arm lengths ([5,6,6] and [5,5,6]) may achieve better trade-off between the CuAAC reactivity and stability of the catalyst.
Ligand donor type is the most important factor in ligand stability against dissociation/exchange. The phenyl ligands can offer a weak protection on Cu(I), while the pyridyl group is the strongest chelator. The pyridine ring offered the best π-accepting ability that strengthened the electron-donating interaction of central amine toward the Cu(I). No clear correlation between the ligand types and the stability of Cu(I) complexes against oxidation was observed, but in the [5,5,5] systems, triazole donors showed better activity than pyridines in CuAAC reaction.
For a given chelate arm length and donor type, incremental steric demands within a family of ligands tend to retard all the processes (oxidation, dissociation and CuAAC). In general, the influence of steric hindrance on the CuAAC activity is higher than the oxidation process.
The CuAAC activity of Cu(I) complexes followed the ranking of 1 ≥ BTTAA > 7 > 6 > 20 > 5 > 4 > 21 > 11, 12, 15, 16, 19 > 2, 3, 8, 9, 10, 13, 14, 17, 18. It can be correlated with their ability of forming the active tri- and di-Cu(I) acetylide species, which was observed by ESI-MS. The ligand 1 showed a much higher portion of di-Cu(I) acetylide than other ligands exhibiting lower CuAAC activity.
In the demanding bioconjugation applications, the ligand protection is compulsory. The OEG-tethered tris(triazolyl)amine ligands are recommended for their good performance in protecting the oxidative damage to biomolecules while maintaining the active tris(triazolyl)amine catalytic structure, which was achieved by the sacrificing oxidation of OEG chain.
Our result indicated that tris(pyridylmethyl)amine ligand 11 and the tris(triazolyl)amine-[5,6,6] ligand 5 might be good ligands for intracellular CuAAC reaction since both ligands exhibited high Cu(I) affinity, good stability against oxidation, and a considerable CuAAC activity.
Experimental
Determination of Cu(I)-ligand dissociation constant (KD)
Following the method described by Xiao et al.,48, 50 the dissociation constants (KD) of Cu(I) complexes with the ligand BTTAA and 1–21 were determined using Bca as the probe (Scheme 1). The concentration of CuI(Bca)2 was measured with a Varian Cary 50 Bio UV-visible spectrophotometer. For details, see ESI†.
Measurement of ascorbic acid oxidation rate (Vo)
The ascorbate decomposition (Scheme 1) in the presence of [Cu(I)-ligand] = 100 µM and initial [AA] = 200 µM was monitored by the decrease of ascorbate absorbance at 265 nm (ε = 1.5 × 104 M−1 cm−1) recorded with a Varian Cary 50 Bio UV-visible spectrophotometer. For details, see ESI†.
Measurement of CuAAC apparent rate constant (kobs)
A modified fluorogenic assay was employed in a 96 well plate to evaluate the relative activity and kinetic properties of each ligand for the model CuAAC reaction of non-fluorescent 3-azido-7-hydroxycoumarin (100 µM) and propargyl alcohol (50 µM) in the presence of ascorbate (5 mM) and CuSO4 (100 µM) in 0.01 M PBS (Scheme 1).46 Formation of the fluorescent triazole product was monitored using a Perkin Elmer HTS 7000 BioAssay Reader to estimate the yields and kinetic constants. For details, see ESI†.
Characterization of Cu(I)-ligand-alkyne complexes
Deoxygenated water was used as the solvent for the reaction. Positive ion ESI-MS data were acquired using a Thermo Finnigan LCQ Deca XP ion trap mass spectrometer operated with Xcalibur® software package (Thermo Fisher Scientific Inc.). The spectra were scanned in the m/z range from 400 to 1200. An optimization procedure carried out at the beginning of this work was conducted to achieve a good signal intensity. The ESI spray condition was: flow rate 10 µL/min; electrospray capillary voltage: 4.49 kV; source temperature: 40 °C. Typically, all MS measurements were carried out 3 min after mixing the reactants and the accumulated ESI-MS time is 5 min.
The preparation of the Cu(I)-ligand-alkyne complexes was conducted in the anaerobic chamber. 100 µL ligand (1 mM), 50 µL PE (1 mM), 100 µL CuSO4 (1 mM), and 100 µL sodium ascorbate (10 mM) in Milli-Q water (650 µL) were mixed together in a 1.5 mL Eppendorf tube and vortexed for 30 seconds. A total sample amount was 250 µL for ESI-MS injection. For details, see ESI†.
Peptide oxidation Assay
The biomolecule (peptide) oxidation was evaluated by the % peptide remaining during the model CuAAC reaction of azidopeptide (50 µM), propargyl alcohol (50 µM), ligand (100 µM), CuSO4 (50 µM), and sodium ascorbate (500 µM) in Milli-Q water. The % peptide remaining was quantified by LC MS/MS using a Thermo Finnigan LCQ Deca XP Plus with a gradient elution (2–70%, 0.1% formic acid in acetonitrile/0.1% formic acid in H2O, 10 min) through a C18 column (Kinetex™ 5 µm XB-C18 100 Å, LC Column 50 × 4.6 mm, Phenomenex Inc.). For details, see ESI†.
Computational method
The DFT calculations were performed using Q-Chem 4.1 on [CuI(1)]+, [CuI(2)]+, [CuI(3)]+, [CuI(5)]+, [CuI(10)]+ and [CuI(11)]+ complexes, in which anion of each Cu(I) complex has been omitted and each oligo(ethylene glycol) chain was replaced with a methyl group for simplicity.88 Optimizations were started from reported crystallographically derived parameters using the B3LYP/LANL2DZ basis set.37, 56, 89 For details, see ESI†.
Synthesis of tripodal amine ligands
The detailed synthesis, purification, and chemical characterization were given in ESI†.
Evaluation of data correlation
Correlation coefficient (r) was calculated by the CORREL function in Excel to find the correlation coefficient between each two parameters.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grants (R21HD058985 and 5R01EY013175), the National Science Foundation grant (DMR-1508722), The Alliance for NanoHealth grant (W81XWH-11-02-0168) and the GEAR grant from the University of Houston.
Footnotes
Electronic supplementary information (ESI) available: Detailed experimental procedures, characterizations and supporting data. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 2.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HC, Finn M, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med. Res. Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- 5.Hein CD, Liu X-M, Wang D. Pharm. Res. 2008;25:2216–2230. doi: 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thirumurugan P, Matosiuk D, Jozwiak K. Chem. Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 7.Xi W, Scott TF, Kloxin CJ, Bowman CN. Adv. Func. Mat. 2014;24:2572–2590. [Google Scholar]
- 8.Golas PL, Matyjaszewski K. Chem. Soc. Rev. 2010;39:1338–1354. doi: 10.1039/b901978m. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad Fuaad AA, Azmi F, Skwarczynski M, Toth I. Molecules. 2013;18:13148–13174. doi: 10.3390/molecules181113148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang W, Becker ML. Chem. Soc. Rev. 2014;43:7013–7039. doi: 10.1039/c4cs00139g. [DOI] [PubMed] [Google Scholar]
- 11.Yang M, Li J, Chen PR. Chem. Soc. Rev. 2014;43:6511–6526. doi: 10.1039/c4cs00117f. [DOI] [PubMed] [Google Scholar]
- 12.Hu Q-Y, Allan M, Adamo R, Quinn D, Zhai H, Wu G, Clark K, Zhou J, Ortiz S, Wang B. Chem. Sci. 2013;4:3827–3832. [Google Scholar]
- 13.Jiang H, Zheng T, Lopez-Aguilar A, Feng L, Kopp F, Marlow FL, Wu P. Bioconjugate Chem. 2014;25:698–706. doi: 10.1021/bc400502d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin W, Du Y, Zhu Y, Chen X. J. Am. Chem. Soc. 2014;136:679–687. doi: 10.1021/ja410086d. [DOI] [PubMed] [Google Scholar]
- 15.El-Sagheer AH, Brown T. Chem. Soc. Rev. 2010;39:1388–1405. doi: 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]
- 16.Díez-González S. Catal. Sci. Technol. 2011;1:166–178. [Google Scholar]
- 17.Hong V, Presolski SI, Ma C, Finn M. Angew. Chem. Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong V, Steinmetz NF, Manchester M, Finn M. Bioconjugate Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angew. Chem. Int. Ed. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soriano del Amo D, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P. J. Am. Chem. Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Hong S, Tran A, Jiang H, Triano R, Liu Y, Chen X, Wu P. Chem. Asian J. 2011;6:2796–2802. doi: 10.1002/asia.201100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang M, Jalloh AS, Wei W, Zhao J, Wu P, Chen PR. Nat. Commun. 2014:5. doi: 10.1038/ncomms5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Li K, Cai C. Chem. Commun. 2011;47:3186–3188. doi: 10.1039/c0cc05376g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 25.Gupte A, Mumper RJ. Cancer Treat. Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy DC, McKay CS, Legault MC, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z, Pezacki JP. J. Am. Chem. Soc. 2011;133:17993–18001. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- 27.Abel GR, Jr, Calabrese ZA, Ayco J, Hein JE, Ye T. Bioconjugate Chem. 2016;27:698–704. doi: 10.1021/acs.bioconjchem.5b00665. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Cai H, He J, Chen H, Lam S, Cai T, Zhu Z, Bark SJ, Cai C. Bioconjugate Chem. 2016;27:2315–2322. doi: 10.1021/acs.bioconjchem.6b00267. [DOI] [PubMed] [Google Scholar]
- 29.Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Nature. 2010;465:645–648. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Wang L, Yu F, Zhu Z, Shobaki D, Chen H, Wang M, Wang J, Qin G, Erasquin UJ, Ren L, Wang Y, Cai C. Chem. Sci. 2017;8:2107–2114. doi: 10.1039/c6sc02297a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis WG, Magallon FG, Fokin VV, Finn M. J. Am. Chem. Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 32.Pham AN, Xing G, Miller CJ, Waite TD. J. Catal. 2013;301:54–64. [Google Scholar]
- 33.Becer CR, Hoogenboom R, Schubert US. Angew. Chem. Int. Ed. 2009;48:4900–4908. doi: 10.1002/anie.200900755. [DOI] [PubMed] [Google Scholar]
- 34.Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 35.Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 36.Hatcher LQ, Karlin KD. Adv. Inorg. Chem. 2006;58:131–184. [Google Scholar]
- 37.Schatz M, Becker M, Thaler F, Hampel F, Schindler S, Jacobson RR, Tyeklár Z, Murthy NN, Ghosh P, Chen Q. Inorg. Chem. 2001;40:2312–2322. doi: 10.1021/ic000924n. [DOI] [PubMed] [Google Scholar]
- 38.Zhang CX, Kaderli S, Costas M, Kim E-i, Neuhold Y-M, Karlin KD, Zuberbühler AD. Inorg. Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- 39.Osako T, Tachi Y, Taki M, Fukuzumi S, Itoh S. Inorg. Chem. 2001;40:6604–6609. doi: 10.1021/ic010625d. [DOI] [PubMed] [Google Scholar]
- 40.Osako T, Tachi Y, Doe M, Shiro M, Ohkubo K, Fukuzumi S, Itoh S. Chem. Eur J. 2004;10:237–246. doi: 10.1002/chem.200305263. [DOI] [PubMed] [Google Scholar]
- 41.Itoh S, Tachi Y. Dalton Transactions. 2006:4531–4538. doi: 10.1039/b607964d. [DOI] [PubMed] [Google Scholar]
- 42.Xiao Z, Brose J, Schimo S, Ackland SM, La Fontaine S, Wedd AG. J. Biol. Chem. 2011;286:11047–11055. doi: 10.1074/jbc.M110.213074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maryon EB, Molloy SA, Kaplan JH. Am. J. Physiol. Cell Physiol. 2013;304:C768–C779. doi: 10.1152/ajpcell.00417.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valverde IE, Bauman A, Kluba CA, Vomstein S, Walter MA, Mindt TL. Angew. Chem. Int. Ed. 2013;52:8957–8960. doi: 10.1002/anie.201303108. [DOI] [PubMed] [Google Scholar]
- 45.Prescher JA, Bertozzi CR. Nat. Chem. Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 46.Sivakumar K, Xie F, Cash BM, Long S, Barnhill HN, Wang Q. Org. Lett. 2004;6:4603–4606. doi: 10.1021/ol047955x. [DOI] [PubMed] [Google Scholar]
- 47.Scarpa M, Vianello F, Signor L, Zennaro L, Rigo A. Inorg. Chem. 1996;35:5201–5206. [Google Scholar]
- 48.Xiao Z, Wedd AG. Nat. Prod. Rep. 2010;27:768–789. doi: 10.1039/b906690j. [DOI] [PubMed] [Google Scholar]
- 49.Xiao Z, Gottschlich L, van der Meulen R, Udagedara SR, Wedd AG. Metallomics. 2013;5:501–513. doi: 10.1039/c3mt00032j. [DOI] [PubMed] [Google Scholar]
- 50.Djoko KY, Xiao Z, Huffman DL, Wedd AG. Inorg. Chem. 2007;46:4560–4568. doi: 10.1021/ic070107o. [DOI] [PubMed] [Google Scholar]
- 51.Jin L, Romero EA, Melaimi M, Bertrand G. J. Am. Chem. Soc. 2015;137:15696–15698. doi: 10.1021/jacs.5b11028. [DOI] [PubMed] [Google Scholar]
- 52.Presolski SI, Hong V, Cho S-H, Finn MG. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei N, Murthy NN, Tyeklar Z, Karlin KD. Inorg. Chem. 1994;33:1177–1183. [Google Scholar]
- 54.Lee C, Yang W, Parr RG. Physical review B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 55.Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270–283. [Google Scholar]
- 56.Karlin KD, Hayes JC, Juen S, Hutchinson JP, Zubieta J. Inorg. Chem. 1982;21:4106–4108. [Google Scholar]
- 57.Rodionov VO, Fokin VV, Finn M. Angew. Chem. Int. Ed. 2005;117:2250–2255. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 58.Presolski SI, Hong V, Cho S-H, Finn M. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodionov VO, Presolski SI, Díaz Díaz D, Fokin VV, Finn M. J. Am. Chem. Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 60.Rodionov VO, Presolski SI, Gardinier S, Lim Y-H, Finn M. J. Am. Chem. Soc. 2007;129:12696–12704. doi: 10.1021/ja072678l. [DOI] [PubMed] [Google Scholar]
- 61.Schulze B, Schubert US. Chem. Soc. Rev. 2014;43:2522–2571. doi: 10.1039/c3cs60386e. [DOI] [PubMed] [Google Scholar]
- 62.Colasson B, Le Poul N, Le Mest Y, Reinaud O. Inorg. Chem. 2011;50:10985–10993. doi: 10.1021/ic201540x. [DOI] [PubMed] [Google Scholar]
- 63.Worrell BT, Malik JA, Fokin VV. Science. 2013;340:457–460. doi: 10.1126/science.1229506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu L, Brassard CJ, Zhang X, Guha PM, Clark RJ. Chem. Rec. 2016;16:1501–1517. doi: 10.1002/tcr.201600002. [DOI] [PubMed] [Google Scholar]
- 65.Rodionov VO, Fokin VV, Finn M. Angew. Chem. Int. Ed. 2005;44:2210–2215. doi: 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]
- 66.Berg R, Straub BF. Beilstein J. Org. Chem. 2013;9:2715–2750. doi: 10.3762/bjoc.9.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahlquist M, Fokin VV. Organometallics. 2007;26:4389–4391. [Google Scholar]
- 68.Straub BF. Chem. Commun. 2007:3868–3870. doi: 10.1039/B706926J. [DOI] [PubMed] [Google Scholar]
- 69.Cantillo D, Avalos M, Babiano R, Cintas P, Jimenez JL, Palacios JC. Org. Biomol. Chem. 2011;9:2952–2958. doi: 10.1039/c0ob01001d. [DOI] [PubMed] [Google Scholar]
- 70.Kalvet I, Tammiku-Taul J, Mäeorg U, Tämm K, Burk P, Sikk L. ChemCatChem. 2016;8:1804–1808. [Google Scholar]
- 71.Chen H, Cai C, Li S, Ma Y, Luozhong S, Zhu Z. Chem. Eur. J. 2017;23:4730–4735. doi: 10.1002/chem.201700555. [DOI] [PubMed] [Google Scholar]
- 72.Iacobucci C, Reale S, De Angelis F. Angew. Chem. Int. Ed. 2016;55:2980–2993. doi: 10.1002/anie.201507088. [DOI] [PubMed] [Google Scholar]
- 73.Di Marco VB, Bombi GG. Mass Spectrom. Rev. 2006;25:347–379. doi: 10.1002/mas.20070. [DOI] [PubMed] [Google Scholar]
- 74.Keith-Roach MJ. Anal. Chim. Acta. 2010;678:140–148. doi: 10.1016/j.aca.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 75.Schröder D. Acc. Chem. Res. 2012;45:1521–1532. doi: 10.1021/ar3000426. [DOI] [PubMed] [Google Scholar]
- 76.Hinderling C, Adlhart C, Chen P. Angew. Chem. Int. Ed. 1998;37:2685–2689. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2685::AID-ANIE2685>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 77.Santos LS. Eur. J. Org. Chem. 2008;2008:235–253. [Google Scholar]
- 78.Iacobucci C, Reale S, Gal J-F, De Angelis F. Angew. Chem. Int. Ed. 2015;54:3065–3068. doi: 10.1002/anie.201410301. [DOI] [PubMed] [Google Scholar]
- 79.Remuzgo C, Oewel T, Daffre S, Lopes TS, Dyszy F, Schreier S, Machado-Santelli G, Teresa Machini M. Amino Acids. 2014;46:2573–2586. doi: 10.1007/s00726-014-1811-2. [DOI] [PubMed] [Google Scholar]
- 80.Park CJ, Park CB, Hong S-S, Lee H-S, Lee SY, Kim SC. Plant Mol. Biol. 2000;44:187–197. doi: 10.1023/a:1006431320677. [DOI] [PubMed] [Google Scholar]
- 81.Uchida K, Kawakishi S. Bioorg. Chem. 1989;17:330–343. [Google Scholar]
- 82.Gallet G, Carroccio S, Rizzarelli P, Karlsson S. Polymer. 2002;43:1081–1094. [Google Scholar]
- 83.Qin G, Cai C. Chem. Commun. 2009:5112–5114. doi: 10.1039/B911155G. [DOI] [PubMed] [Google Scholar]
- 84.Fukuzumi S, Karlin KD. Coord. Chem. Rev. 2013;257:187–195. doi: 10.1016/j.ccr.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 86.Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 87.Zhu L, Brassard CJ, Zhang X, Guha P, Clark RJ. Chem. Rec. 2016 doi: 10.1002/tcr.201600002. [DOI] [PubMed] [Google Scholar]
- 88.Krylov AI, Gill PMW. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013;3:317–326. [Google Scholar]
- 89.Donnelly PS, Zanatta SD, Zammit SC, White JM, Williams SJ. Chem. Commun. 2008:2459–2461. doi: 10.1039/B719724A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.