

SUMMARY
N6-methyladenosine (m6A), installed by the Mettl3/Mettl14 methyltransferase complex, is the most prevalent internal mRNA modification. Whether m6A regulates mammalian brain development is unknown. Here we show that m6A depletion by Mettl14 knockout in embryonic mouse brains prolongs cell cycle of radial glia cells and extends cortical neurogenesis into postnatal stages. m6A depletion by Mettl3 knockdown also leads to prolonged cell cycle and maintenance of radial glia cells. m6A-sequencing of embryonic mouse cortex reveals enrichment of mRNAs related to transcription factors, neurogenesis, cell cycle and neuronal differentiation, and m6A-tagging promotes their decay. Further analysis uncovers previously unappreciated transcriptional pre-patterning in cortical neural stem cells. m6A signaling also regulates human cortical neurogenesis in forebrain organoids. Comparison of m6A-mRNA landscapes between mouse and human cortical neurogenesis reveals enrichment of human-specific m6A-tagging of transcripts related to brain disorder risk genes. Our study identifies an epitranscriptomic mechanism in heightened transcriptional coordination during mammalian cortical neurogenesis.
In brief
m6A-dependent mRNA decay is critical for transcriptional pre-patterning in mammalian cortical neurogenesis.
INTRODUCTION
Proper development of the nervous system is critical for its function, and deficits in neural development have been implicated in many brain disorders, such as microcephaly, autistic spectrum disorders, and schizophrenia (Jamuar and Walsh, 2015; Taverna et al., 2014). In the embryonic mouse cortex, radial glia cells (RGCs) function as neural stem cells, sequentially giving rise to neurons residing in different cortical layers and then switching to glial production before their depletion during early postnatal stages (Taverna et al., 2014). Such a precise and predictable developmental schedule requires a highly coordinated genetic program (Okano and Temple, 2009). Indeed, previous studies have revealed transcriptional cascades that orchestrate the dynamics of mammalian cortical neurogenesis (Martynoga et al., 2012; Miller et al., 2014; Nord et al., 2015). Recent discoveries of widespread mRNA chemical modifications (Zhao et al., 2017a) raise the question of whether this mechanism plays any regulatory role in cortical neurogenesis.
Modified nucleotides in mRNAs were initially discovered over 40 years ago, but little was known about the extent, transcript identities, and potential functions of various reversible chemical modifications until very recently (Zhao et al., 2017a). High-throughput sequencing approaches have revealed a dynamic “epitranscriptome” landscape for many mRNA modifications in various organisms, including N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), 5-hydroxymethylcytosine (hm5C), and pseudouridine (ψ) (Li et al., 2016). Among these modifications, m6A is the most abundant internal modification in mRNAs of eukaryotic cells (Desrosiers et al., 1975). m6A profiling with cell lines has revealed m6A sites in over 25% of human transcripts, with enrichment in long exons, and near transcription start sites and stop codons (Meyer and Jaffrey, 2014). In mammals, m6A is installed by the methyltransferase complex consisting of Mettl3 (methyltransferase-like 3), Mettl14, Wtap (Wilms tumor 1-associated protein), KIAA1429, RBM15 (RNA-binding motif protein 15) and its paralogue (RBM15B) (Patil et al., 2016), whereas its removal is mediated by demethylases Fto (fat mass and obesity-associated) and Alkbh5 (alkB homolog 5) (Zhao et al., 2017a). Recent in vitro studies have identified multiple functions of m6A in mRNA metabolism, from processing in the nucleus to translation and decay in the cytoplasm (Zhao et al., 2017a). The field has just started to investigate physiological functions of m6A. For example, Mettl3 or Mettl14 knockdown reduces m6A levels and decreases self-renewal of primed mouse embryonic stem cells (mESCs) (Wang et al., 2014a), whereas Mettl3 knockout naïve mESCs exhibit improved self-renewal and impaired differentiation, due to dysregulated decay of m6A-tagged transcripts, such as Nanog (Batista et al., 2014; Geula et al., 2015).
Identification of the molecular machinery mediating m6A mRNA methylation provides an entry point to explore physiological functions of this pathway in vivo. Studies of Drosophila development showed that m6A methylation regulates sex determination and neuronal functions by modulating mRNA splicing (Haussmann et al., 2016; Lence et al., 2016). In Zebrafish embryos, m6A-tagging promotes clearance of maternal mRNAs and maternal-to-zygotic transition (Zhao et al., 2017e). In mice, germline Mettl3 deletion results in early embryonic lethality (Geula et al., 2015). Nothing is known about the role of m6A signaling during mammalian embryonic brain development in vivo. Here we used the Mettl14 conditional knockout mouse as a model to examine m6A function in embryonic cortical neurogenesis in vivo. We further investigated underlying cellular and molecular mechanisms. Finally, we extended our analysis to human embryonic cortical neurogenesis using induced pluripotent stem cell (iPSC)-derived forebrain organoids and compared m6A-mRNA landscapes between mouse and human cortical neurogenesis. Together, our results reveal critical epitranscriptomic control of mammalian cortical neurogenesis and provide insight into mechanisms underlying this highly coordinated developmental program.
RESULTS
Nervous system Mettl14 deletion extends cortical neurogenesis into postnatal stages
We first investigated the expression pattern of molecular players mediating m6A signaling during mouse embryonic cortical neurogenesis. Mining the recently published single-cell RNA-seq dataset of RGCs and their progeny (Telley et al., 2016) revealed that highest Mettl14 expression in RGCs and relatively constant levels of m6A methyltransferase components (Mettl3, Wtap), demethylases (Fto, Alkbh5) and m6A readers (Ythdf2, Ythdf3) during neurogenesis (Figure S1A). We next conditionally deleted Mettl14 in the developing mouse nervous system starting at E11.5 using the Nestin-Cre;Mettl14f/f conditional knockout (cKO) model (Figure S1B). We confirmed Mettl14 deletion at the protein level with Western blot analysis of E17.5 brains (Figure S1B). The cKO animals were smaller in size by P5 compared to wildtype (WT) littermates, and all cKO animals died before P25 (Figure S1C–D). Thus, the function of m6A molecular machinery in the nervous system is indispensable for life in the mammalian system.
We then examined cortical structures at P5. cKO mice exhibited enlarged ventricles with an adjacent dense layer of cells that resembled the embryonic germinal zone (Figure 1A). Immunohistological analysis showed the presence of Pax6+ and Nestin+ cells with radial fibers along the ventricle in cKO mice, but not in WT mice (Figure 1A–B). During mouse cortical development, Pax6+ RGCs are largely depleted by P5 (Dwyer et al., 2016). In contrast, a substantial number of Pax6+ cells were present in cKO mice at P5 (Figure 1C). Neurogenic Pax6+ RGCs give rise to intermediate progenitor cells (IPCs) expressing Tbr2/Eomes (Englund et al., 2005). The presence of Pax6+ cells in cKO mice was accompanied by Tbr2+ IPCs, which were absent in WT mice by P5 (Figure 1D–E). To confirm that cortical neurogenesis continued postnatally, we pulsed animals with EdU at P5 and analyzed 2 days later. Significant numbers of EdU+Pax6+ proliferating RGCs, EdU+Tbr2+ IPCs, and EdU+Tbr2+TuJ1+ neuroblasts were present in cKO mice, but very few in WT littermates (Figure 1F–G). These results indicate that cKO mice maintain neurogenic RGCs with extended cortical neurogenesis into postnatal stages.
Figure 1. Nervous system Mettl14 deletion results in residual radial glia cells and ongoing neurogenesis in the postnatal mouse cortex.

(A–C) Presence of neurogenic RGCs in P5 Nestin-Cre;Mettl14f/f cKO cortices. Shown are sample confocal images (A, B) and quantifications (C). Regions in white boxes are shown at a higher magnification. Scale bars: 500 μm (A, top panel), 50 μm (A, bottom panel), 100 μm (B). Values in (C) represent mean ± SEM (n = 4–7; ***: P < 0.001; *: P < 0.05; unpaired Student’s t-test).
(D–E) Presence of IPCs in P5 cKO cortices. Shown are sample confocal images (D; scale bars: 100 μm) and quantification (E). Values represent mean ± SEM (n = 6; ***: P < 0.001; **: P < 0.01; unpaired Student’s t-test).
(F–G) Ongoing neurogenesis in P5 cKO cortices. P5 pups were injected with EdU and analyzed 48 hr later. Shown in (F) are sample confocal images of the ventricular side of the primary somatosensory cortex. Arrows indicate Pax6+EdU+ cells (top) and Tbr2+TuJ1+EdU+ cells (bottom). Scale bars: 100 μm. Quantification of EdU+ cells with different markers is shown in (G). Values represent mean ± SEM (n = 6; ***: P < 0.001; unpaired Student’s t-test).
(H–K) Reduced production of upper-layer neurons and astrocytes in cKO cortices. Pregnant mice were injected with EdU at E15.5 and analyzed at P5. Shown are sample confocal images (H, J; scale bars: 100 μm) and quantification (I, K). Values represent mean ± SEM (n = 6; ***: P < 0.001; unpaired Student’s t-test).
See also Figure S1
To further characterize the impact of Mettl14 deletion on cortical development, we examined neuronal subtype and glia production. We pulsed animals with EdU at E15.5 and examined them at P5. Compared to WT littermates, cKO mice exhibited a significantly decreased number of EdU+Satb2+ neurons, suggesting a deficit in producing late-born upper-layer neurons (Figure 1H–I). Direct measurement of the number of different cortical neuron subtypes also showed a reduced number of Satb2+ upper-layer neurons, but comparable numbers of Tbr1+ and Ctip2/Bcl11b+ lower-layer early-born neurons in P5 cKO mice (Figure S1E–F). On the other hand, Ctip2+ neurons were reduced in number in the E17.5 cKO cortex, suggesting a delay in the production of neuron subtypes of different cortical layers, rather than differentiation deficits (Figure S1G–H). In addition, we observed a significant decrease in the number of s100β+ astrocytes in cKO mice at P5 (Figure 1J–K). Together, these results indicate that Mettl14 function is critical for proper temporal progression of neurogenesis and gliogenesis during mouse cortical development in vivo.
Mettl14 deletion in neural progenitor cells leads to protracted cell cycle progression
Given the well-defined temporal progression of cortical neurogenesis from RGCs (Okano and Temple, 2009), we suspected that there could be RGC deficits during embryonic stages in cKO mice. Interkinetic nuclear migration (INM), the periodic movement of the cell nucleus in phase with cell-cycle progression, is a common feature of developing neuroepithelial (Taverna et al., 2014). We pulsed animals with EdU at E17.5 to label cells in S-phase and followed positions of nuclei in EdU+Pax6+ RGCs (Figure 2A). While there was no difference at 0.5 hr after EdU labeling, nuclei of labeled RGCs were positioned further away from the ventricular surface at 6 hr in cKO mice compared to WT (Figure 2B), suggesting delayed INM and potential cell cycle deficits. To directly examine the S to M phase transition of the cell cycle, we analyzed expression of phospho-Histone 3 (pH3), an M phase marker, 2 hr after EdU labeling (Figure 2C). We found a significant decrease in the percentage of EdU+pH3+Pax6+ cells among all pH3+Pax6+ cells in cKO mice, suggesting a prolonged S to M phase transition of RGCs (Figure 2D). To examine cell cycle exit of proliferating neural progenitors, we analyzed expression of Ki67, a proliferation marker, 24 hr after EdU labeling (Figure 2E). We found a significant decrease in the percentage of Ki67 negative cells among EdU+ cells in cKO mice, indicating a delay in cell cycle exit (Figure 2F).
Figure 2. Mettl14−/− RGCs and NPCs exhibit prolonged cell cycle progression.

(A–B) Abnormal INM of RGCs in Mettl14 cKO cortices. Pregnant mice were injected with EdU at E17.5 and analyzed 0.5 or 6 hr later. Shown are sample confocal images (A; scale bars: 50 μm) and quantification of the distance from Pax6+EdU+ nuclei to the ventricular surface (B). Values for the percentages of nuclei in each 20 μm bin represent mean ± SEM (n = 4; ***: P < 0.001; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(C–D) Delayed S to M phase transition of RGCs in Mettl14 cKO mice. Pregnant mice were injected with EdU at E17.5 and analyzed 2 hr later. Shown in (C) are sample confocal images. Arrowheads point to Pax6+pH3+EdU+ cells and arrows point to Pax6+pH3+EdU− cells. Scale bar: 50 μm. Shown in (D) is the quantification of the percentage of Pax6+pH3+EdU+ cells, representing cells proceeded from S to M phase during the 2 hr chase, among total Pax6+pH3+ cells. Values represent mean ± SEM (n = 5 for WT and n = 8 for cKO; ***: P < 0.001; unpaired Student’s t-test).
(E–F) Delayed cell cycle exit of neural progenitors in Mettl14 cKO mice. Pregnant mice were injected with EdU at E17.5 and analyzed 24 hr later. Shown in (E) are sample confocal images. Arrowheads point to Ki67−EdU+ cells and arrows point to Ki67+EdU− cells. Scale bar: 50 μm. Shown in (F) is the quantification of the percentage of Ki67−EdU+ cells, representing cells exited from cell cycle, among total EdU+ cells. Values represent mean ± SEM (n = 6; ***: P < 0.001; unpaired Student’s t-test).
(G–J) Time-lapse imaging analysis of mouse NPCs showing prolonged S-G2-M phase length in the absence of Mettl14. WT and cKO mouse NPCs were electroporated with plasmid co-expressing a Cdk2 sensor (green) and the H2B-mCherry nuclear marker (red), cultured for 2 days, and imaged for 48 hr. Shown in (G) are sample time-lapse images with time stamps. Scale bars: 10 μm. Also shown are box plots of quantifications for the total cell cycle length (H; n = 38 for WT and n = 30 for cKO), G1 phase length (I; n = 20), and S-G2-M phase length (J; n = 20). Each dot represents data from one NPC (***: P < 0.001; unpaired Student’s t-test).
See also Figure S2.
To address the cell intrinsic effect of Mettl14 deletion on cell cycle progression, we performed time-lapse imaging of individual cortical neural progenitor cells (NPCs) cultured from E13.5 mouse cortex. We used a dual reporter system with nuclear localized H2B-mCherry and a GFP-tagged Cdk2 substrate, DNA Helicase B (DHB) (Spencer et al., 2013). Cdk2 becomes active during the G1-S transition and phosphorylates DHB-GFP, which is then translocated from nucleus to cytoplasm. Therefore, the presence of GFP in the mCherry+ nucleus indicates cells in G1 phase, whereas translocation to the cytoplasm indicates S phase initiation, and continual buildup of cytoplasmic GFP occurs until mitosis (Figure S2A). Quantification of the length between sequential mitoses showed an increase of the total cell cycle length in Mettl14 cKO NPCs (Figure 2G–H; Movie S1 and S2). Further analysis of different cell cycle phases revealed a specific increase of the S-G2-M phases in the absence of Mettl14, but no difference in the G1 phase (Figure 2I–J).
To quantify cell cycle characteristics at the population level, we pulsed NPCs with EdU for 30 min and performed flow cytometry analysis 0 or 5 hr later (Figure S2B). We found a significant decrease in the percentage of EdU+ cells that divided in Mettl14 cKO NPCs compared to WT at 5 hr, confirming a delay in cell cycle progression (Figure S2C–D).
Mettl3 regulates embryonic cortical neurogenesis
Consistent with the finding that Mettl14 is a critical component of the m6A methyltransferase complex (Wang et al., 2017), Mettl14 deletion led to a significant reduction of m6A levels in mRNAs from both embryonic mouse cortex in vivo and cultured cortical NPCs (Figure 3A–B). To further assess our model that m6A methylation regulates cortical neurogenesis, we compared the phenotype of Mettl14 cKO to knockdown of Mettl3, another critical component of the m6A methyltransferase complex (Wang et al., 2017).
Figure 3. Mettl3 regulates cell cycle progression of NPCs and maintenance of embryonic cortical RGCs.

(A) Depletion of m6A-tagging on mRNAs purified from E15.5 and E17.5 Mettl14 cKO mouse forebrain. Shown in the left panels are sample images of m6A dot blot and methylene blue staining (for loading controls). Data were normalized to the averaged levels of WT samples and quantification is shown in the right panel. Values represent mean ± SEM (n = 3; **: P < 0.01; unpaired Student’s t-test).
(B) Depletion of m6A-tagging on mRNAs purified from Mettl14 cKO NPCs. Values represent mean ± SEM (n = 3; **: P < 0.01; unpaired Student’s t-test).
(C–D) Flow cytometry analysis of cell cycle status of mouse NPCs. NPCs were electroporated to co-express GFP and the control shRNA, or the shRNA against Mettl3. At day 4, NPCs were pulse-labeled with EdU (10 μM) for 30 min, cultured for 9 hr, followed by EdU and DNA content (DyeCycle Violet) staining and flow cytometry analysis. Shown are sample histograms of DNA content from GFP+EdU+ cells and the total cell population (as a reference; C) and quantification (D). Values in (D) represent mean ± SEM (n = 4; **: P < 0.01; unpaired Student’s t-test).
(E–G) Embryonic mouse cortices were electroporated in utero at E13.5 to co-express GFP and shRNA-control, or GFP and shRNA-Mettl3, and analyzed at E17.5. Shown in (E) are sample confocal images. Scale bars: 50 μm. The distribution of GFP+ cells in each zone (F) and the percentage of GFP+Pax6+ cells among total GFP+ cells (G) were quantified. VZ: ventricular zone; SVZ: subventricular zone; IZ: intermediate zone; CP: cortical plate. Values represent mean ± SEM (n = 4; ***: P < 0.001; **: P < 0.01; unpaired Student’s t-test).
See also Figure S3.
We first confirmed effective Mettl3 knockdown (KD) with Q-PCR and diminished m6A content in mRNAs from Mettl3 KD cells with dot blot analysis (Figure S3A–C and Table S1). We next performed population cell cycle analysis with EdU pulse-chase and flow cytometer quantification (Figure S3D). We found a significant reduction in the percentage of GFP+EdU+ NPCs that divided upon Mettl3 KD (Figure 3C–D), similar to the effect of Mettl14 cKO (Figure S2C–D).
To examine the impact of Mettl3 KD on RGC behavior in vivo, we electroporated plasmids co-expressing GFP and the shRNA against mouse Mettl3, or the control shRNA, in utero at E13.5 and analyzed GFP+ cells at E17.5. Newborn neurons normally migrate toward the cortical plate (CP) through the intermediate zone (IZ), whereas self-renewing RGCs remain in the ventricular zone (VZ) and subventricular zone (SVZ) (Taverna et al., 2014). Compared to the control group, GFP+ cells with Mettl3 KD were more abundant in the VZ and SVZ and less abundant in the CP (Figure 3E–F), similar to the result from EdU fate mapping in Mettl14 cKO mice (Figure 1H). There was also a significant increase in the percentage of GFP+Pax6+ cells among all GFP+ cells with Mettl3 KD compared to the control group (Figure 3G).
Together, these results indicate that decreasing m6A levels by either Mettl14 cKO or Mettl3 KD leads to consistent phenotypes of protracted cell cycle progression of cortical NPCs and reduced differentiation of RGCs during mouse embryonic cortical neurogenesis.
m6A tags transcripts related to transcription factors, cell cycle, and neurogenesis, and promotes their decay
To gain insight into the molecular mechanism underlying m6A regulation of cortical neurogenesis, we performed m6A-seq of mouse forebrain at E13.5, a stage enriched for neural stem cells. We identified 4,055 high confidence m6A peaks corresponding to 2,059 gene transcripts (Figure S4A and Table S2). Similar to previous findings from cell lines (Meyer and Jaffrey, 2014), our in vivo analysis showed enriched distribution of m6A sites near stop codons (Figure S4B). We found no correlation between transcript levels and m6A-tagging (Figure S4C). Notably, many transcripts encoding transcription factors were m6A-tagged, such as Pax6, Sox1, Sox2, Emx2, and Neurog2/Neurogenin 2 (Figure 4A–B). Gene ontology (GO) and Wikipathways analyses of m6A-tagged transcripts revealed enrichment of genes related to cell cycle, stem cell, and neuronal differentiation (Figure 4A–C and Table S3). We observed m6A-tagging of a similar group of transcripts in cortical NPCs derived from E13.5 mouse cortex (Figure S4D and Table S1).
Figure 4. m6A tags transcripts related to transcription factors, cell cycle, and neuronal differentiation in the embryonic mouse brain, and promotes their decay.

(A) Coverage plots from m6A-seq of E13.5 mouse forebrains showing representative examples of m6A-tagged (Sox1, Emx2, and Cdk9) and non m6A-tagged (Rad17) transcripts. Top and middle panels show read coverages normalized by library sizes from m6A pulled-down and input libraries, respectively, and bottom panels show gene structures (arrows point to the direction of transcription; S.C.: stop codon).
(B–C) GO analysis of m6A-tagged genes reveals enrichment for biological process terms related to transcription factors, neurogenesis, cell cycle, and stem cell differentiation. Also shown is Wikipathways gene set enrichment analysis. FDR: false discovery rate.
(D) Cumulative distribution of Log2(gene expression ratios) at time 5 hr post ActD over time 0 hr for m6A tagged genes (purple line) and non-m6A tagged genes (black line) for WT and Mettl14 cKO NPCs. D = value of Kolmogorov Smirnov test statistic corresponding to maximum difference between methylated and non-methylated distributions.
(E) Cumulative distribution of log2(fold change in ratios of gene expression) at 5 hr post ActD treatment over time 0 hr upon Mettl14 deletion. Top panel shows cumulative distribution for non-targets (black line) and transcripts with 1–1.9 m6A sites on average (bright red line), or 2 or more sites on average (dark red line). Bottom panel shows cumulative distribution for non-targets and m6A-tagged transcripts with 1–2, 2–3, 3–4, and 4 or more sites on average.
(F) Summary of half-life of a group of transcripts in WT and Mettl14 cKO NPCs. Values represent mean ± SEM (n = 4; ***: P < 0.001; **: P < 0.01; unpaired Student’s t-test
To determine the functional consequence of m6A-tagging, we explored whether Mettl14 deletion affects decay of m6A-tagged transcripts. Cortical NPCs derived from E13.5 WT and Mettl14 cKO mice were treated with Actinomycin D to halt de novo transcription, and RNA-seq was performed 0 and 5 hr later to obtain the ratio of mRNA levels for each gene in order to measure their stability (Figure S4E). Across the transcriptomes, m6A-tagged transcripts exhibited significantly lower ratios compared to non m6A-tagged transcripts in the WT NPCs, and this difference was reduced in cKO NPCs (Figure 4D and Table S4). Direct comparison of WT and Mettl14 cKO NPCs showed that m6A-tagged transcripts exhibited a larger increase in their ratio compared to non-tagged transcripts upon Mettl14 deletion; one m6A tag per transcript was sufficient to increase the ratio and there was a minimal additional effect of more tagging sites (Figure 4E). It should be noted that our m6A-seq method could not determine whether multiple sites are simultaneously methylated in the same transcript. We confirmed our result with the direct measurement of the half-life of a selected group of transcripts (Figure 4F and S4F; Table S1).
All together, these results support a model that m6A methylation of mRNAs related to cell cycle and neurogenesis confers their rapid turnover during the dynamic progress of cortical neurogenesis; a lack of m6A-tagging attenuates the decay of these mRNAs, resulting in deficits in temporal specification and cell cycle progression of NPCs.
Mettl14 deletion uncovers transcriptional pre-patterning for normal cortical neurogenesis
Among the 2,059 m6A-tagged genes in the E13.5 mouse cortex, two major GO terms were generation of neurons and neuronal differentiation (Figure 4C). For example, IPC marker Tbr2 and Neurog2, and neuronal markers Neurod1 and Neurod2 (Hevner et al., 2006), were m6A-tagged in E13.5 forebrain in vivo (Figure 5A) and in cultured cortical NPCs (Figure S4D). Q-PCR analysis of total mRNA showed increased levels of Tbr2, Neurog2, Neurod1, and Neurod2, but not non-tagged Rad17, in Mett14 cKO compared to WT NPCs (Figure 5B and Table S1). This result raised the possibility that neuronal lineage genes are already expressed in neural stem cells and their levels are actively suppressed post-transcriptionally by m6A-dependent decay; alternatively, Mettl14 deletion may transcriptionally upregulate these neuronal genes.
Figure 5. Post-transcriptional regulation of pre-patterning gene levels and protein production by m6A signaling in cortical neural stem cells.

(A) Coverage plots from m6A-seq of E13.5 mouse forebrains showing representative examples of m6A-tagged IPC (Tbr2 and Neurog2) and neuronal (Neurod1 and Neurod2) genes.
(B) Q-PCR analysis of total mRNA and 4sU-purified nascent mRNA from WT and Mettl14 cKO NPCs. All Ct values were first normalized to Gapdh control (not m6A-tagged), which were similar in both WT and cKO NPCs. The ratio (cKO over WT) was calculated for each experiment and values represent mean ± SEM (n = 3 cultures; ***: P < 0.001; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(C–F) Precocious expression of Tbr2 and Neurod1 proteins in RGCs in E17.5 Mettl14 cKO mice in vivo. Shown are sample confocal images (C, E; scale bars: 50 μm) and quantifications of the percentage of Tbr2+Pax6+ cells (D), or Neurod1+Pax6+ cells (F), among total Pax6+ cells (left panels, n = 6) and the density distribution of Tbr2+Pax6+ (D), or Neurod1+Pax6+ cells (F), from the ventricular surface (right panels, n = 4). Arrows indicate Tbr2+Pax6+ (C) or Neurod1+Pax6+ cells (E). Values in (D, F) represent mean ± SEM (***: P < 0.001; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
See also Figure S5.
To differentiate between these two possibilities, we quantified the levels of nascent mRNA using the 4-thiouridine (4sU) metabolic labeling approach (Duffy et al., 2015). We found comparable and even lower levels of nascent mRNA of neuronal lineage genes, such as Tbr2, Neurog2, and Neurod2, in Mettl14−/− NPCs in comparison to WT NPCs (Figure 5B and Table S1). The lower levels of nascent mRNA observed for some neuronal lineage genes in Mettl14 cKO NPCs could be explained by a negative feedback loop at the level of transcription, originating from elevated expression of stem cell genes, such as Emx2 and Sox1 (Figure 5B). Similarly, we found comparable levels of pre-mRNA for neuronal lineage genes in Mettl14 cKO compared to WT NPCs (Figure S5A and Table S1), suggesting that the increase in the total mRNA of neuronal lineage genes in Mettl14 cKO NPCs is not due to transcriptional upregulation. Together, these results support that neuronal lineage genes are already expressed in neural stem cells under normal cortical neurogenesis. Consistent with our result, mining the published single-cell RNA-seq dataset (Telley et al., 2016) revealed expression of neuronal lineage genes, such as Tbr2, Neurog2, Neurod6 and Tubb3/Tuj1, in individual RGCs in the embryonic mouse cortex in vivo (Figure S5B).
We next examined Tbr2 and Neurod1 protein levels in RGCs in vivo. Pax6+Tbr2+ cells were localized in the SVZ in WT at E17.5, but extended into the VZ in Mettl14 cKO mice (Figure 5C–D). Pax6+Neurod1+ cells were rare, but detectable just above the SVZ in WT cortices. In contrast, cKO mice exhibited a significantly increased number of Pax6+Neurod1+ cells with a much broader distribution, including in the SVZ and VZ (Figure 5E–F). To specifically examine expression in RGCs, we pulse-labeled juxtaventricular newborn cells by FlashTag (FT) (Telley et al., 2016). We found a significantly increased number of FT+Pax6+Tbr2+ and FT+Pax6+Neurod1+ cells in Mettl14 cKO cortex compared to those in WT 3 hr after labeling (Figure S5C–D). Given that FT+ cells at 3 hr upon labeling are exclusively undifferentiated RGCs (Telley et al., 2016), these results suggest that Mettl14 regulates neuronal lineage gene expression directly in RGCs.
To further assess our model that mRNA decay regulates neuronal lineage gene expression in RGCs, we performed in vivo knockdown experiments for the components of CCR4-NOT complex (Cnot7 and Cnot1), a major cytoplasmic mRNA deadenylase complex responsible for mRNA decay (Du et al., 2016; Schoenberg and Maquat, 2012). Both Cnot7 KD and Cnot1 KD led to increased numbers of Tbr2+Pax6+ and Neurod1+Pax6+ cells and location closer to the ventricular surface compared to the control shRNA (Figure S5E–F), phenotypes resembling Mettl14 cKO (Figure 5C–F).
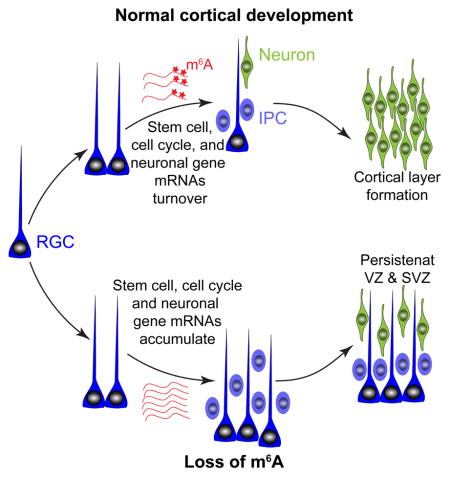
Taken together, our results suggest heightened transcriptional coordination and a previously unappreciated transcriptional pre-patterning mechanism for mammalian cortical neurogenesis, in which late IPC and neuronal genes are already transcribed in cortical neural stem cells and these transcripts are down regulated post-transcriptionally by m6A-dependent decay.
METTL14 regulates cell cycle progression of human cortical NPCs
We next examined whether m6A function is conserved in human cortical neurogenesis. Using a previously developed protocol (Yoon et al., 2014), we differentiated human iPSCs into a highly pure population of NESTIN+SOX2+ NPCs (hNPCs; 96.4 ± 1% among all cells; n = 5; Figure S6A). We co-expressed GFP and the validated shRNA against human METTL14 in these hNPCs (Figure S6B). After 4 days, we labeled cells with EdU for 30 min and performed cell cycle analysis with flow cytometer quantification 14 hr later (Figure S6C). Similar to results from mouse Mettl14 cKO NPCs (Figure S2C–D), we found a significant decrease in the percentage of GFP+EdU+ hNPCs that divided with METTL14 KD, indicating a delayed cell cycle progression (Figure 6A–B).
Figure 6. METTL14 regulates cell cycle progression of human NPCs.

(A–B) Flow cytometry analysis of cell cycle progression of hNPCs with METTL14 KD. Human NPCs were electroporated to co-express GFP and shRNA-control, or shRNA-METTL14. After 4 days, hNPCs were pulse-labeled with EdU (10 μM) for 30 min, incubated for 14 hr, followed by EdU and DNA content (DyeCycle Violet) staining and flow cytometry analysis, similarly as in Figure 3C–D. Values represent mean ± SEM (n = 4; **: P < 0.01; unpaired Student’s t-test).
(C–D) Flow cytometry analysis of cell cycle progression with METTL14 KD in human forebrain organoids. Day 45 forebrain organoids were electroporated to co-express GFP and shRNA-control, or shRNA-METTL14. After 7 days, forebrain organoids were pulse-labeled with EdU (10 μM) for 1 hr, cultured further for 14 hr, followed by dissociation and analysis similarly as in Figure 3C–D. Values represent mean ± SEM (n = 4; ***: P < 0.001; unpaired Student’s t-test).
See also Figure S6.
We recently developed a human iPSC-derived forebrain organoid model, which exhibits transcriptome profiles similar to fetal human cortex during development up to the second trimester (Qian et al., 2016). Around day 47, these forebrain organoids resemble mouse cortical neurogenesis at E13.5 (Figure S6D). We microinjected plasmids co-expressing GFP and the shRNA against human METTL14, or the control shRNA, into the lumen of forebrain organoids and performed electroporation to transfect RGCs (Figure S6E). After 7 days, we pulsed organoids with EdU for 1 hr and performed cell cycle analysis of GFP+ cells 14 hr later (Figure S6F). We observed a significant decrease in the percentage of GFP+EdU+ cells that divided with METTL14 KD (Figure 6C–D). Together, these results indicate that m6A mRNA methylation plays a conserved role in regulating cortical NPC cell cycle progression in both mouse and human.
m6A-seq of human forebrain brain organoids and fetal brain reveals conserved and unique m6A landscape features compared to embryonic mouse forebrain
Finally, we performed m6A-seq of day 47 human forebrain organoids. We detected 11,994 high confidence m6A peaks associated with 4,702 transcripts (Figure S7A and Table S2). Our previous systematic RNA-seq analyses of human forebrain organoids at different stages revealed that transcriptomes of organoids around day 47 were similar to human fetal cortex at 8–12 post-conception weeks (PCW) (Qian et al., 2016). We further performed m6A-seq of PCW11 fetal human brain and identified 10,980 high confidence peaks associated with 5,049 transcripts (Figure S7B and Table S2). m6A sites were enriched near transcription start sites and stop codons for both human samples (Figure S7C–D). Furthermore, m6A profiles from both samples showed significant overlap (Figure 7B). GO analysis of m6A-tagged transcripts shared in both samples showed enrichment of genes related to neurogenesis, neuronal differentiation and development (Figure 7C and Table S5). Many recently identified risk genes for schizophrenia and autistic spectrum disorders have been shown to be dynamically expressed and play critical roles during mammalian embryonic brain development (Ohi et al., 2016; Tebbenkamp et al., 2014). Notably, disease ontology analysis of these m6A-tagged genes shared in both human samples showed enrichment related to mental disorders, mental retardation, schizophrenia and bipolar disorder (Figure 7C and Table S5).
Figure 7. Conserved and unique features of m6A mRNA methylation in human forebrain organoids, human fetal brain and embryonic mouse forebrain.

(A) Representative plots of two m6A-tagged transcripts in day 47 human forebrain organoids and PCW11 human fetal brain, but not in mouse E13.5 forebrain.
(B) Venn diagram showing shared m6A-tagged transcripts between day 47 human forebrain organoids and PCW11 fetal human brain.
(C) GO and disease ontology analyses of shared m6A-tagged genes in day 47 human forebrain organoids and PCW11 human fetal brain.
(D) Venn diagram showing shared and unique m6A-tagged transcripts among mouse E13.5 forebrain, day 47 human forebrain cortex, and PCW11 fetal human brain. Only ortholog genes expressed in all three samples were used for analysis.
(E–F) Disease ontology analysis of transcripts uniquely m6A-tagged in human shows enrichment for neurodevelopmental diseases, whereas disease ontology analysis of commonly m6A-tagged transcripts showed enrichment for oncogenic processes.
We further performed comparison among m6A landscapes during mouse and human cortical neurogenesis. About 19.3%, 34.7% and 31.4% of detected transcripts exhibited m6A-tagging in E13.5 mouse brain, day 47 human forebrain organoids, and PCW11 human fetal brain, respectively (Figure S7E). Therefore, m6A mRNA methylation appears to be more prevalent in human. Among transcripts expressed in all three samples, 856 genes were commonly m6A-tagged (Figure 7D). These commonly m6A-tagged transcripts are enriched for genes related to neurogenesis and neuronal differentiation (Figure S7F and Table S5). Notably, 1,173 transcripts were expressed in both species, but only m6A-tagged in both human samples (Figure 7D). Ontology analysis of these human-specific m6A-tagged transcripts showed enrichment of genes related to mental disorders and mental retardation (Figure 7E–F and Table S5). Among genes associated with the 108 loci recently identified for genetic risk of schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics, 2014), 60 genes were m6A-tagged in human and 21 genes were uniquely tagged in both human forebrain organoids and fetal brain, but not in mouse E13.5 forebrain. On the other hand, the gene set of m6A-tagged transcripts shared between mouse and human is enriched for oncogenic processes (Figure 7E).
DISCUSSION
From flies to mammals, neurogenesis is a highly coordinated process with sequential waves of gene expression (Kohwi and Doe, 2013). Here we revealed a critical role of m6A mRNA methylation in this process in the mammalian system in vivo. Our results suggest a model that m6A-tagging of transcripts related to neural stem cells, cell cycle, and neuronal differentiation confers their rapid turnover to control the transcriptome composition at different phases of the dynamic cortical neurogenesis process. The observation of RGCs expressing markers thought to be expressed only in late IPCs and post-mitotic neurons in Mettl14 cKO mice led to the discovery of transcriptional pre-patterning in normal cortical neurogenesis and identifies m6A mRNA methylation as a key mechanism to prevent precocious expression of genes of later lineage status at the protein level in stem cells. We also provide the emerging “epitranscriptomic” field with databases of m6A mRNA landscapes of mouse and human cortical neurogenesis and identify intriguing human-specific features.
Transcriptional pre-patterning for cortical neurogenesis
The concept of pre-patterning initially came from analysis of chromatin states within multipotent progenitors to regulate the fate choice for liver and pancreas (Xu et al., 2011). Recent genome-wide mapping studies have suggested that epigenetic pre-patterning is important for spatio-temporal regulation of gene expression and may be a widespread phenomenon in cell fate decision (Chen and Dent, 2014). Our study reveals transcriptional pre-patterning in normal cortical neural stem cells in vivo. Consistent with our model, Pax6 has been shown to bind and activate both Tbr2 and Neurod1 promoters (Sansom et al., 2009). We showed that pre-patterning transcripts are tagged with m6A and subjected to rapid decay, therefore most of them are present in low levels among the bulk mRNA preparation and little protein under normal conditions – a likely reason why such a mechanism has escaped detection in previous studies. While epigenetic mechanisms play a key role in transcriptional regulation during neurogenesis (Yao et al., 2016), epitranscriptomic regulation as a post-transcriptional mechanism could provide the speed and additional specificity, while maintaining plasticity of gene expression. By working in concert, the epigenetic landscape can permit transcription of certain genes, such as genes defining late lineage states, while the epitranscriptome prevents aberrant protein production. Future studies are needed to investigate whether transcriptional pre-patterning is a general mechanism in fate specification of other stem cells during development.
Heightened transcriptional coordination of mammalian cortical neurogenesis by m6A
Our study provides the in vivo evidence in the mammalian system to support the emerging notion that m6A methylation plays a critical role in developmental fate transition. The precise and predictable developmental schedule of cortical neurogenesis requires rapid, tightly controlled changes in gene expression (Okano and Temple, 2009). Our results suggest that epitranscriptomic m6A-tagging, via regulation of mRNA decay, provides a key mechanism for temporal control of dynamic gene expression, which in turn regulates cell cycle progression of cortical neural stem cells in both mouse and human.
There are three major categories of m6A-tagged transcripts in the embryonic mouse brain. First, many classic transcription factors involved in neural stem cell maintenance and neurogenesis, such as Pax6, Sox2, Emx2, and Tbr2, are m6A-tagged and subject to rapid decay. Second, cell cycle-related transcripts, such as Cdk9, Ccnh/Cyclin H, and Cdkn1C/p57, are m6A-tagged. Functionally, the loss of m6A-tagging leads to prolonged cell cycle progression of cortical NPCs, resulting in delayed generation of different neuronal subtypes, extension of cortical neurogenesis into postnatal stages and deficits in astrocyte generation in vivo. Third, transcripts that were generally thought to be expressed only in later IPCs and post-mitotic neurons, such as Neurod1 and Neurod2, are m6A-tagged and expressed in neural stem cells. While expression of transcription factors is known to overlap during different stages of mammalian cortical neurogenesis (Hevner et al., 2006), our finding suggests a greater degree of transcriptional coordination than previously thought. On the other hand, expression of detectable neuronal proteins in a significant number of RGCs located in the SVZ in the absence of Mettl14 highlights the critical role of the epitranscriptomic mechanism in preventing precocious gene expression during the normal process of mammalian cortical neurogenesis.
Conserved and unique features of human m6A landscape during cortical neurogenesis
Our study provides databases of m6A mRNA landscapes during mouse and human cortical neurogenesis. Consistent with a similar role for m6A mRNA methylation in regulating cell cycle progression of cultured human NPCs and mouse NPCs in vitro and in vivo, the shared m6A-tagged transcripts in our mouse and human samples are enriched with genes related to neural stem cells, cell cycle, and neurogenesis. Notably, many genes associated with genetic risk for mental disorders, such as schizophrenia and autistic spectrum disorders, are only m6A-tagged in humans, but not in mice, raising the possibility that epitranscriptomic dysregulation may contribute to these human brain disorders. So far, one association study found evidence of ALKBH5 in conferring genetic risk for major depression disorder (Du et al., 2015), and two studies identified association of FTO mutations with growth retardation and developmental delay (Boissel et al., 2009; Daoud et al., 2016).
In summary, our study identifies a critical and conserved role of an m6A epitranscriptomic mechanism in the temporal control of mammalian cortical neurogenesis via promotion of mRNA decay of transcripts related to transcription factors, neural stem cells, cell cycle, and neuronal differentiation. Future studies will address how this epitranscriptomic mechanism interacts with various epigenetic mechanisms to coordinate dynamic transcriptomes during brain development, and how dysregulation of epitranscriptomic mechanisms may contribute to brain disorders.
STAR METHODS
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Pax6 | BioLegend | PRB-278P RRID: AB_2313780 |
| Mouse anti-Pax6 | BD Bioscience | 561664 RRID: AB_10895587 |
| Rabbit anti-Tbr2 | Abcam | ab23345 RRID: AB_778267 |
| Chicken anti-GFP | Aveslab | GFP-1020 RRID: AB_10000240 |
| Rabbit anti-Ki67 | Thermo Fisher | RM-9106 RRID: AB_2335745 |
| Goat anti-Sox2 | Santa Cruz | sc-17320 RRID: AB_2286684 |
| Chicken anti-Nestin | Aveslab | NES RRID: AB_2314882 |
| Rabbit anti-m6A | Synaptic Systems | 202 003 RRID: AB_2279214 |
| Mouse anti-m6A | Synaptic Systems | 202 111 RRID: AB_2619891 |
| Rabbit anti-Actin | Cytoskeleton | AAN01 RRID: AB_10708070 |
| Rabbit anti-GFAP | Dako | Z0334 RRID: AB_10013382 |
| Mouse anti-βIIITubulin (TuJ1) | Sigma-Aldrich | T3952 RRID: AB_1841226 |
| Rabbit anti-S100β | Abcam | ab52642 RRID: AB_882426 |
| Mouse anti-SATB2 | Abcam | ab92446 RRID: AB_10563678 |
| Rabbit anti-Tbr1 | Abcam | ab31940 RRID: AB_2200219 |
| Rat anti-CTIP2 | Abcam | ab18465 RRID: AB_2064130 |
| Rabbit anti-Phospho-Histone H3 | Cell Signaling | 9701S RRID: AB_331534 |
| Rabbit anti-Mettl14 | ABclonal | AB8530 |
| Mouse anti-Neurod1 | Abcam | ab60704 RRID:AB_943491 |
| Goat anti-rabbit IgG-HRP | Santa Cruz | sc-2004 RRID: AB_631746 |
| Goat anti-mouse IgG-HRP | Santa Cruz | sc-2005 RRID: AB_631736 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma-Aldrich | A1410 |
| DAPI | Thermo Fisher Scientific | D1306 RRID: AB_2629482 |
| StemPro Accutase Cell Dissociation Reagent | Thermo Fisher Scientific | A1110501 |
| Matrigel Matrix | Corning | 354277 |
| 7-AAD | Thermo Fisher Scientific | A1310 |
| Vybrant DyeCycle Violet | Thermo Fisher Scientific | V35003 |
| Y-27632 | Cellagen Technology | C9127-2s |
| EdU | Thermo Fisher Scientific | A10044 |
| Phosphatase Inhibitor Cocktail | Cell Signaling | 5870 |
| Protease Inhibitor Cocktail | Sigma | P8340 |
| 4x Laemmli Sample Buffer | Bio-Rad | 1610747 |
| Methylene blue | Sigma-Aldrich | M9140 |
| A83-01 | Stemcell Technologies | 72022 |
| Dorsomorphin | Stemcell Technologies | 72102 |
| SB431542 | Stemcell Technologies | 72232 |
| CHIR99021 | Stemcell Technologies | 72052 |
| Dulbecco’s Phosphate-Buffered Saline (DPBS) | Corning | 21-031 |
| Dulbecco’s Modification of Eagle’s Medium (DMEM) | Corning | 10-013 |
| DMEM/F-12, HEPES | Gibco | 11330-032 |
| Neurobasal® Medium | Gibco | 21103049 |
| KnockOut™ Serum Replacement | Gibco | 10828028 |
| GlutaMAX™ Supplement | Gibco | 35050061 |
| MEM Non-Essential Amino Acids Solution | Gibco | 11140050 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140122 |
| 2-Mercaptoethanol | Gibco | 21985023 |
| N-2 Supplement | Gibco | 17502048 |
| B-27® Supplement | Gibco | 17504044 |
| Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | 354230 |
| Insulin solution | Sigma-Aldrich | I0516 |
| Fetal Bovine Serum (FBS) | Corning | 35-010 |
| 0.1% Gelatin in Water | Stemcell Technologies | 7903 |
| Advanced DMEM/F-12 | Gibco | 12634010 |
| Human recombinant LIF | Stemcell Technologies | 78055 |
| Compound E | Stemcell Technologies | 73952 |
| Costar® 6 Well Clear Flat Bottom Ultra Low Attachment plate | Sigma-Aldrich | CLS3471 |
| Recombinant Human FGF-basic | Peprotech | 100-18B |
| SUPERase In RNase Inhibitor | Thermo Fisher Scientific | AM2694 |
| TURBO DNase (2 U/μL) | Thermo Fisher Scientific | AM2238 |
| N6-Methyladenosine monophosphate sodium salt | Sigma-Aldrich | M2780 |
| Agencourt AMPure XP | Beckman | A63880 |
| Dynabeads® Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D |
| CellTrace CFSE | Thermo Fisher Scientific | C34554 |
| luciferase control RNA | Promega | L4561 |
| TRIzol Reagent | Thermo Fisher Scientific | 15596026 |
| 4-thiouridine | Carbosynth | T4509 |
| MTSEA biotin- XX | Biotium | 90066 |
| Dimethylformamide (DMF) | Sigma-Aldrich | D4551 |
| Pierce Streptavidin Magnetic Bead | Thermo Fisher Scientific | 88816 |
| Critical Commercial Assays | ||
| Click-iT EdU Assay Kits for Flow Cytometry | Thermo Fisher Scientific | C10635 |
| Click-iT EdU Alexa Fluor 647 Imaging Kit | Thermo Fisher Scientific | C10340 |
| Dynabeads mRNA Purification Kit | Thermo Fisher Scientific | 61006 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | 18080051 |
| Fast SYBR Green Master Mix | Thermo Fisher Scientific | 4385610 |
| NEBNext Ultra RNA Library Prep Kit for Illumina | New England Biolabs | E7530 |
| Dynabeads mRNA DIRECT Purification Kit | Invitrogen | 61011 |
| RNeasy Mini Kit | Qiagen | 74106 |
| Quant-IT PicoGreen dsDNA Assay Kit | Thermo Fisher Scientific | P7589 |
| RNeasy MinElute Kit | Qiagen | 74204 |
| Deposited Data | ||
| Raw and analyzed data | This Paper | GSE99017 |
| Experimental Models: Cell Lines | ||
| B16-F10 | ATCC | CRL-6475 RRID: CVCL_0159 |
| C12 (iPSC from normal human foreskin fibroblasts) | Wen et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Cg-Tg(Nes-cre)1Kln/J | Jackson Laboratory | 003771 RRID: IMSR_JAX:003771 |
| Mouse: Mettl14floxed;floxed | From Dr. Chuan He | N/A |
| Mouse: Crl:CD1(ICR) | Charles River Laboratory | RRID: IMSR_CRL:22 |
| Mouse: Crl:CF188 | Charles River Laboratory | RRID:IMSR_CRL:23 |
| Oligonucleotides | ||
| shRNA control sequence for mouse and human: UUCUCCGAACGUGUCACGU | Qiagen | SI03650318 |
| shRNA targeting sequence: mouse Mettl3: GCACACUGAUGAATCUUUAGG | Wang et al., 2014 | N/A |
| shRNA targeting sequence: human Mettl14: CCUGAAAUUGGCAAUAUAGAA | Wang et al., 2014 | N/A |
| shRNA targeting sequence: mouse Cnot1: GUGGACAAUUUAACCAAGA | Du et al., 2016 | N/A |
| shRNA targeting sequence: mouse Cnot7: AACAAGUCUACAUUACACCGC | Du et al., 2016 | N/A |
| Primers for Q-PCR analysis | See Table S1 | N/A |
| Recombinant DNA | ||
| pUEG | Ge et al., 2006 | N/A |
| cFUGW | Lois et al. 2002 | Addgene plasmid 14883 |
| pCAG-GFP | Matsuda and Cepko, 2004 | Addgene plasmid: 11150 |
| pPGK-H2B-mCherry-2A-DHB(aa994-1087)-GFP | From Dr. Sergi Regot (Spencer et al., 2013) | N/A |
| Software and Algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Imaris | Bitplane | http://www.bitplane.com/imaris/imaris |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Tophat2 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Fastx Toolkit | http://hannonlab.cshl.edu/fastx_toolkit | http://hannonlab.cshl.edu/fastx_toolkit/download.html |
| MACS2 | Zhang et al. 2008 | https://github.com/taoliu/MACS/wiki/Install-macs2 |
| BedTools | Quimlan and Hall, 2010 | Bedtools.readthedocs.io/en/latest/content/instalation.html |
| Metagene R Package | Beauparlant et al., 2014 | http://bioconductor.org/packages/release/bioc/tml/metagene.html |
| HTSeq | Anders et al., 2014 | http://www-huber.embl.de/HTSeq/doc/install.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RSEM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM |
| EBSeq | Leng et al., 2013 | https://github.com/lengning/EBSeq |
| Toppgene | Chen et al., 2009 | https://toppgene.cchmc.org |
| ConsensusPathDB | Herwig, 2016 | http://cpdb.molgen.mpg.de |
| Webgestalt | Zhang et al., 2005 | http://www.webgestalt.org/option.php |
| Cytoscape | Shannon et al., 2003 | http://www.cytoscape.org/download.php |
| Single-cell transcriptomic atlas of the developing neocortex | Telley et al., 2016 | http://genebrowser.unige.ch/science2016/ |
| Other | ||
| SuperSignal West Dura Extended Duration Substrate | Thermo Fisher Scientific | 34075 |
| Nucleofector Kits for Mouse Neural Stem Cells | Lonza | VAPG-1004 |
| Nucleofector 2b Device | Lonza | AAB-1001 |
| Square wave electroporator | Nepa Gene | CUY21SC |
| Tweezers with platinum disk electrode | Nepa Gene | CUY650-5 |
| Immun-Blot PVDF Membrane | Bio-Rad | 1620177 |
| Amersham Hybond-N+ membrane | GE Healthcare | RPN119B |
| Stratagene Stratalinker 2400 UV Crosslinker | Agilent Genomics | 53274-1 |
| 12-well Spinning Bioreactor | Qian et al. 2016 | N/A |
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
-
IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. You do not need to delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Hongjun Song (shongjun@mail.med.upenn.edu). There are no restrictions on any data or materials presented in this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Exons 7, 8, and 9 of mouse Mettl14 were targeted by inserting a single loxP site in intron 6 and an FRT-flanked neomycin resistance gene coupled with a loxP site in intron 9, with the consideration that they contain the DPWW active motif (Figure S1B). The targeting construct was electroporated into 129 mESCs, selected for neomycin resistance, screened for homologous recombination by Southern blotting, and selected mESC clones were used to generate chimeric mice by injection into C57BL/6J mouse blastocysts. Chimeric mice were bred to wild type C57BL/6J mice to test for germline transmission of the mutant allele, which was identified by PCR. The PCR-positive lines were crossed with a β-actin promoter-driven Flp recombinase to remove the neomycin resistance gene via FRT site recombination. The neomycin cassette-deleted mice were identified by PCR, and the resultant Mettl14f/f a llele and Nestin-Cre+/Tg mice (Jackson Laboratory stock: 003771) were used to generate Nestin-Cre+/Tg; M ettl14 +/f mice and Nestin-Cre+/+; M ettl14 f/f mice. WT and cKO mice were generated by crossing Nestin-Cre+/Tg; M ettl14 +/f males and Nestin-Cre+/+; M ettl14 f/f females.
For in utero electroporation analysis, timed-pregnant CD1 mice (Charles River Laboratory) at E13.5 were used as previously described (Yoon et al., 2014). Timed pregnant mice were euthanized by cervical dislocation, and embryos were euthanized by decapitation before the dissection step. All animal procedures used in this study were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee of Johns Hopkins University School of Medicine.
Primary mouse NPCs
Mouse NPCs were isolated from Mettl14 WT and cKO mouse embryonic cortices and cultured in Neurobasal medium (Gibco BRL) containing 20 ng/ml FGF2, 20 ng/ml EGF, 5 mg/ml heparin, 2% B27 (v/v, Gibco BRL), Glutamax (Invitrogen), Penicillin/Streptomycin (Invitrogen) on culture dishes pre-coated with Matrigel matrix (2%, Corning).
Human iPSC cultures and fetal brain sample
The human iPSC line used in the current study (C1) was fully characterized (Wen et al., 2014; Yoon et al., 2014). iPSCs were cultured in stem cell medium, consisting of DMEM:F12 (Invitrogen) supplemented with 20% Knockout Serum Replacer (Gibco), 1X Non-essential Amino Acids (Invitrogen), 1X Penicillin/Streptomycin (Invitrogen), 1X 2-Mercaptoenthanol (Millipore), 1X Glutamax (Invitrogen), and 10 ng/ml FGF-2 (Peprotech). Culture medium was changed every day. Human iPSCs were passaged every week onto a new plate pre-seeded with irradiated CF1 mouse embryonic fibroblasts (Charles River Laboratory). Human iPSCs were detached from the plate by treatment of 1 mg/ml Collagenase Type IV (Invitrogen) for 1 hr. iPSC colonies were further dissociated into smaller pieces by manual pipetting. All studies were performed under approved protocols of Johns Hopkins University School of Medicine. Human iPSCs were differentiated into primitive hNPCs according to a previously published protocol (Li et al., 2011). Briefly, iPSCs were passaged onto MEF feeders, and after 3 days, induction medium containing Advanced DMEM:F12 (50%) and Neurobasal medium (50%), CHIR99201 (4 μM, Cellagentech), SB431542 (3 μM, Cellagentech), Bovine serum albumin (5 μg/ml, Sigma), hLIF (10 ng/ml, Millipore), Compound E (0.1 μM, EMD Millipore), Glutamax (Invitrogen), Pen/Strep, supplemented with N2 and B27 (Invitrogen), was added to the culture. After 6 days of differentiation, hNPCs were dissociated with Accutase (Invitrogen) and plated, with the aid of a ROCK inhibitor (Y-27632, 3 μM, Cellagentech), onto culture dishes pre-coated with Matrigel matrix (2%, Corning).
The PCW11 fetal human cortical tissue was used for m6A-seq. All procedures used in this study were performed in accordance with the protocol approved by the Institutional Stem Cell Research Oversight Committee of Johns Hopkins University School of Medicine and Lieber Institute for Brain Development.
METHOD DETAILS
DNA constructs
For knockdown experiments for mouse genes, short hairpin RNA sequences (see KEY RESOURCE TABLE) were cloned into the retroviral vector expressing GFP under the control of the EF1α promoter and a specific shRNA under the control of human U6 promoter (pUEG) as previously described (Ge et al., 2006). For knockdown experiments for human METTL14, a short hairpin RNA sequence was cloned into the lentiviral vector expressing GFP under the control of the human ubiquitin C promoter and the specific shRNA under the control of human U6 promoter (cFUGW: Addgene plasmid 14883) as previously described (Yoon et al., 2014). The efficacy of each shRNA was confirmed in mouse B16-F10 cells (ATCC), or hNPCs derived from the C1 iPSC line.
In utero electroporation and FlashTag
In utero electroporation was performed as described previously (Yoon et al., 2014). In brief, timed-pregnant CD1 mice (Charles River Laboratory) at E13.5 or E14.5 were anesthetized and the uterine horns were exposed and approximately 1 to 2 μl of plasmid DNA, 0.5 μg/μl pCAG-GFP (Addgene plasmid: 11150) and 2.5 μg/μl cFUGW plasmid with the control shRNA, or the shRNA against mouse Mettl3, Cnot1 and Cnot7, was injected manually into the lateral ventricles of embryos using a calibrated micropipette. Five pulses (40 V, 50 ms in duration with a 950 ms interval) were delivered across the uterus with two 5-mm electrode paddles (CUY650-5, Nepa Gene) positioned on either side of the head by a square wave electroporator (CUY21SC, Nepa Gene). After electroporation, the uterus was placed back in the abdominal cavity and the wound was sutured. Mouse embryos were analyzed at E17.5. For FlashTag of RGCs, 1 μl of 10 μM of a carboxyfluorescein succinimidyl ester (CellTrace CFSE, ThermoFisher) was injected into the lateral ventricle of the E17.5 embryos using a calibrated micropipette. Mouse embryos were collected 3 hr later, fixed with with 4% paraformaldehyde in PBS overnight at 4°C for analysis. All animal procedures were performed in accordance with the protocol approved by the Johns Hopkins Institutional Animal Care and Use Committee.
Immunohistology and confocal imaging
For EdU labeling, timed pregnant mice were injected with EdU (150 mg/kg bodyweight, Invitrogen) at defined time points before euthanasia. For immunostaining of tissue sections, brains were fixed with 4% paraformaldehyde in PBS overnight at 4°C as previously described (Yoon et al., 2014). Samples were cryoprotected in 30% sucrose in PBS, embedded in OCT compound, and sectioned coronally (20 μm-thickness) on a Leica CM3050S cryostat. Brain sections were blocked and permeabilized with the blocking solution (5% normal donkey serum, 3% Bovine serum albumin, and 0.1% Triton X-100 in PBS) for 1 hr at room temperature, followed by incubation with primary antibodies diluted in the blocking solution at 4°C overnight. After washing, secondary antibodies diluted in blocking solution were applied to the sections for 1 hr at room temperature. Nuclei were visualized by incubating for 10 min with 0.1 μg/ml 4,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific) in PBS. For EdU labeling, Click-iT EdU Alexa Fluor 647 Imaging Kit (Thermo Fisher Scientific) was used following the manufacturer’s protocol (Qian et al., 2016). Stained sections were mounted with ProLong Gold anti-fade reagents (Thermo Fisher Scientific) and analyzed. All the antibodies used are listed in KEY RESOURCE TABLE.
Mouse and human NPC electroporation
Approximately 1.0 × 106 mouse or human NPCs were resuspended in 100 μL Mouse Neural Stem Cell Nucleofector Solution from the Lonza Nucleofector Kit for Mouse Neural Stem Cells (Lonza, VAPG-1004). Additionally, 10 μg of the appropriate plasmid was added to the cell solution. The solution was then placed in a cuvette provided in the Nucleofector Kit and electroporated using a Lonza Nucleofector 2b device (LONZA) (Ma et al., 2008). Next, the cells were resuspended in NPC media as described above with Rock Inhibitor (Y-27632, 3 μM, Cellagentech) to reduce cell death. Cells were allowed to grow for at least 3 days before analysis.
Human forebrain organoid culture
Protocols for generation of forebrain organoids were detailed previously (Qian et al., 2016; Xu et al., 2016; Yoon et al., 2017). Briefly, human iPSCs were cultured in stem cell medium, consisting of DMEM:F12 (Invitrogen) supplemented with 20% Knockout Serum Replacer (Gibco), 1X Non-essential Amino Acids (Invitrogen), 1X Penicillin/Streptomycin (Invitrogen), 1X 2-Mercaptoenthanol (Millipore), 1X Glutamax (Invitrogen), and 10 ng/ml FGF-2 (Peprotech) on irradiated CF1 mouse embryonic fibroblasts (Charles River). On day 1, iPSC colonies were detached by treatment of 1 mg/ml Collagenase Type IV (Invitrogen) for 1 hr and transferred to an Ultra-Low attachment 6-well plate (Corning Costar), containing 3 ml of stem cell medium (without FGF-2), plus 2 μM Dorsomorphine (Sigma) and 2 μM A83-01 (Tocris). On days 5–6, half of the medium was replaced with induction medium consisting of DMEM:F12, 1X N2 Supplement (Invitrogen), 1X Penicillin/Streptomycin, 1X Non-essential Amino Acids, 1X Glutamax, 1 μM CHIR99021 (Cellagentech), and 1 μM SB-431542 (Cellagentech). On day 7, organoids were embedded in Matrigel (Corning) and continued to grow in induction medium for 6 more days. On day 14, embedded organoids were mechanically dissociated from Matrigel and transferred to each well of a 12-well spinning bioreactor (SpinΩ) containing differentiation medium, consisting of DMEM:F12, 1X N2 and B27 Supplements (Invitrogen), 1X Penicillin/Streptomycin, 1X 2-Mercaptoenthanol, 1X Non-essential Amino Acids, 2.5 μg/ml Insulin (Sigma).
Forebrain organoid electroporation and analysis
Day 45 forebrain organoids were transferred into PBS solution in a 10 cm petri dish for electroporation. A mixture of 0.5 μl of plasmid DNA and 0.05% Fast green was injected into the lumen of neural tube structures in forebrain organoids using a calibrated micropipette (Yoon et al., 2017). About 3–4 locations on one side of each forebrain organoid were targeted by the injection. The DNA-injected side of the organoid was placed toward the positive electrode in the middle of 5 mm gap of electrode paddles (CUY650-5, Nepa Gene). Five pulses (40 V, 50 ms in duration with a 950 ms interval) were delivered by a square wave electroporator (CUY21SC, Nepa Gene). After electroporation, organoids were transferred back to the SpinΩ bioreactor for further culturing.
Analysis of cell cycle progression by EdU pulse labeling
Analyses of cell cycle progression of mouse NPCs, hNPCs, and dissociated human forebrain organoids were performed as described previously (Terry and White, 2006; Wang et al., 2014b). In brief, mouse or human NPCs were pulsed by 10 μM EdU (ThermoFisher) for 30 min and washed thoroughly with NPC media. For human forebrain organoids, 10 μM EdU directly applied to culture media and organoids were incubated in the SpinΩ bioreactor for 1 hr to ensure complete penetrance, then washed thoroughly with culture media. After defined time points, cells were dissociated by Accutase, fixed with 4% paraformaldehyde in PBS for 20 min at 4°C, stained with Click-iT EdU Alexa 647 Flow Cytometry Kits (ThermoFisher) for Flow Cytometry following manufacturer’s protocol. Cells were stained with Vybrant DyeCycle Violet (ThermoFisher) or 7-AAD (ThermoFisher) for DNA content and applied to flow cytometry using BD LSR II Flow Cytometer (BD Bioscience). EdU+ or GFP+EdU+ cells were gated and DNA content of those cells was analyzed compared to that of whole cell population. Percentages of divided cells among EdU+ or GFP+EdU+ population (G1 or G0 phase determined by DNA content) during defined time intervals were quantified from four independent experiments.
Time-lapse live imaging of mouse NPCs
96-well glass bottom microplates (655892, Geiner bio-one) were coated with phenol red-free Matrigel (356237, Corning). After electroporation of mNPCs with 10 μg CDK2-sensor plasmid (pPGK-H2B-mCherry-DHB (aa994-1087)-GFP), cells were plated onto the microplates at a density of 3,000 cells per well and allowed to adhere overnight. Cells were imaged using a Nikon Eclipse Ti fluorescent microscope controlled by Metamorph microscopy automation software. Temperature (37°C), CO 2 (5%), and humidity were held constant throughout experiments. Five blank positions in a well containing Matrigel and media only were used to flat field mNPC images using custom software. ImageJ was used to merge the green and red channels. To quantify the total cell cycle length, time was measured from the first cell division to the next cell division of one or both daughters. To quantify the G1 phase length, time was measured from one cell division to the time point of significant reduction in the ratio of green/red intensity in the nucleus of the cell. S phase entry was quantitatively defined as the time when the cytoplasmic/nuclear ratio of green/red was approximately 1, as previously described (Spencer et al., 2013). A nuclear marker, H2B-mCherry, was used in the plasmid sensor to accurately segment the cytoplasm and the nucleus. The time point from S phase entry through the second cell division was then quantified as S-G2-M length.
RNA purification and quantitative RT-PCR analysis
For gene expression analysis, total RNA fraction was isolated from cultured NPC samples with RNeasy Mini Kit (Qiagen), treated with DNaseI and reverse-transcribed into the first-strand cDNA with SuperScript III (Invitrogen). cDNAs were used for SYBR-green based quantitative real-time PCR to measure the expression level of target genes with the T method (ABI). All the primers used for quantitative PCR were listed in Table S1.
Western blot analysis
Forebrains from E17.5 embryos were quickly dissected out and homogenized in RIPA buffer (50 mM Tris pH 7.5, 120 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS, 5 mM EDTA, Phosphatase Inhibitor Cocktail (Cell Signaling), protease inhibitor cocktail (Sigma). Lysates were incubated for 15 min on ice and centrifuged at 15,000g for 15 min at 4°C. Supernatant was collected and boiled for 5 min in Laemmli sample buffer (Biorad), resolved by SDS PAGE, transferred to PVDF membrane, and immunoblotted. Primary antibodies are listed in KEY RESOURCE TABLE. Quantification of bands was performed using ImageJ software.
m6A dot blot assay
mRNA was harvested from homogenized forebrains at embryonic stages E15.5 and E17.5 using Dynabeads mRNA Direct Purification Kit (61011, Ambion). Four biological replicates were pooled for each sample to ensure sufficient concentration of mRNA. Dots were applied to an Amersham Hybond-N+ membrane (GE Healthcare) in duplicate as 100 ng mRNA per 1 μl dot. After complete drying, the mRNA was crosslinked to the membrane using a UV Stratalinker 2400 by running the auto-crosslink program twice. The membrane was then washed in PBST three times and blocked with 5% skim milk in PBST for 2 hr. The PBST wash was repeated and the membrane was incubated with primary anti-m6A antibody (212B11, Synaptic Systems) at 1:1000 dilution for 2 hr at room temperature. After 3 washes in PBST, the membrane was incubated in HRP-conjugated anti-mouse IgG secondary antibody for 2 hr at room temperature, then washed again 3 times in PBST. Finally, the membrane was visualized using SuperSignal West Dura Extended Duration Substrate (34075, Thermo Scientific). To confirm equal mRNA loading, the membrane was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) and quantified m6A levels were normalized to amount of mRNA loaded. Four biological samples in technical duplicates for each time point were used.
m6A-sequencing
m6A profiling was performed as previously described (Dominissini et al., 2012). For m6A profiling of mouse developing brain, forebrains from WT E13.5 embryos were dissected. For m6A profiling of human organoids, 25 to 30 forebrain organoids at day 47 were used. For m6A profiling of PCW11 fetal human brain, cortex from 2 PCW11 fetuses were dissected. The total RNA was extracted using RNeasy Mini Kit (Qiagen). mRNA was isolated using the Dynabeads mRNA Purification Kit (Invitrogen) and mRNA was fragmented via sonication to 100–200 base pairs. m6A pull-down was performed using a rabbit polyclonal anti-m6A antibody (Synaptic systems), and immunoprecipitation with protein G dynabeads (ThermoFisher). m6A-tagged mRNAs were competitively eluted from beads with free N6-methyladenosine. cDNA libraries from pulled-down RNA and input RNA were prepared using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina®. The experiment was performed with three technical replicates. For m6A profiling of day 47 human forebrain organoids, the same procedure was followed, with the exception that the experiment was performed with two technical replicates because of the amount of samples required.
m6A mRNA immunoprecipitation and Q-PCR
Total RNA from NPCs cultured from WT E13.5 mouse forebrain was extracted using RNeasy Mini Kit (Qiagen) and mRNA was isolated using the Dynabeads mRNA Purification Kit (Invitrogen). 1% of input mRNA was reserved for reverse transcription. Full length m6A tagged transcripts were pulled-down using a rabbit polyclonal anti-m6A antibody (Synaptic systems) and a mock pull-down was done with normal rabbit IgG (Cell Signaling Technologies). Immunoprecipitation was performed with protein G dynabeads (Thermo Fisher). m6A-tagged mRNAs were competitively eluted from beads with free N6-methyladenosine. Reverse transcription of input, m6A pull-down and mock pull-down mRNA was performed using the SuperScript® III First-Strand Synthesis System for RT-PCR (Thermo Fisher). cDNA was used for SYBR-green based quantitative real-time PCR. Enrichment of m6A tagged genes in m6A pull-down over input was calculated by comparing relative concentrations using Ct values (2−Ct) and dividing each concentration by the relative concentration of the input. The concentrations of the immunoprecipitated RNA were then divided by the concentration in the input RNA and multiplied by 100, to obtain the percentage of transcripts in the m6A immunoprecipitation relative to the input. This value was then normalized to enrichment in the mock (IgG) pull-down, which was also calculated using relative concentrations to determine a percentage of the input. Primers used are listed in Table S1.
Bioinformatic analyses of m6A-seq
cDNA libraries from input and m6A pull-down were sequenced on the Illumina Nextseq platform, using a 50-cycle single-end run. Pre-processing of reads was performed using the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), namely adapters were clipped, poor quality reads were filtered out, and identical reads were collapsed. Pre-processed reads from E13.5 mouse forebrains were aligned to the mouse genome (build GRCm38/mm10), and reads from the human organoids and fetal brain to the human genome (build GRCh37/hg19), using Tophat2 (Kim et al., 2013) with default settings. m6A-tagged regions were identified using the MACS2 peak calling algorithm (Zhang et al., 2008), with the input library as background. For identifying high confidence m6A regions, peaks were intersected in a pairwise fashion among all replicates using the BedTools package (Quinlan and Hall, 2010). Peaks that overlap in at least 50% of their length among 2 or more samples were designated as high confidence m6A regions.
For representative coverage plots of m6A and input libraries, RNA-seq read alignments in bam format were transformed to bedGraph format and normalized for library size using the genomecov function from the BedTools package (Quinlan and Hall, 2010). Analysis of m6A peak enrichment was performed based on 5 non-overlapping transcript segments defined as follows: Transcription start site (TSS) segment [TSS, TSS+200bp], 5′UTR [TSS+201bp, CDS start-1bp], coding region (CDS) [CDS start, CDS stop-101bp], stop codon segment [CDS stop−100bp, CDS stop+100bp], 3′ UTR [CDS stop+101bp, TTS]. Each high confidence peak was annotated to one of these regions using the BedTools package and fold enrichment was calculated from the ratio between observed peaks per region and expected number of peaks normalized by average region size. For analysis of correlation between gene expression levels and m6A peak fold change, we calculated RPKMs from input RNA seq libraries, using gene counts obtained with the htseq-count function from the HTSeq python package (Anders et al., 2015) that were normalized by library size and gene length defined as the length of its longest transcript. Fold changes for m6A peaks were obtained from MACS2 output.
Functional annotation and disease ontology
To assess enrichment of GO terms specific to a biological process, the ToppFunn module of the ToppGene Suite (Chen et al., 2009) was used. A hypergeometric probability mass function with Benjamini Hochberg FDR correction was used to identify significant enrichment for GO terms. Analysis of enrichment for Wikipathways terms was performed using ConsensusPathDB (Herwig, 2016), which calculates enrichment p-values using the Wilcoxon’s matched-pairs signed-rank test, and Benjamini Hochberg FDR correction.
Disease association analysis was performed using WebGestalt (Zhang et al., 2005), which uses a hypergeometric method and Benjamini Hochberg FDR correction. Protein interaction network figures were generated using Cytoscape 3.3.0 (Shannon et al., 2003), with the Reactome FI plugin.
RNA degradation assay
cDNA libraries were prepared from cultured NPCs from E13.5 WT and Mettl14 cKO cortex, at 0 and 5 hr post Actinomycin D treatment, using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina®. The experiment was performed with three replicates per condition. Sequencing was performed on the Illumina Nextseq platform, using a 100-cycle single-end run. Pre-processing of reads was performed using the FASTX toolkit. Gene expression levels were quantified using the RSEM package (Li and Dewey, 2011), which maps reads to the transcriptome using the aligner tool Bowtie2 (Langmead and Salzberg, 2012). Expected counts per gene per sample were combined into a count matrix, and this matrix was used as input for differential expression analysis using the EBSeq package (Leng et al., 2013), which uses empirical Bayesian methods to calculate the posterior probability of a gene being differentially expressed (PPDE). Posterior fold changes per gene between cKO and WT were obtained at time 0 and 5 hr after Actinomycin D treatment. Fold changes at 5 hr were normalized by fold changes at 0 hr (no Actinomycin D treatment) to specifically identify genes that degrade at a different slower rate in the cKO compared to WT, regardless of baseline changes in gene expression between two conditions. Genes with a normalized fold change higher than 2 in cKO over WT at 5 hr were considered as to be differentially degraded (Table S4).
Half-life measurement of m6A-tagged transcripts
Mouse NPCs were cultured in standard 6 well culture plates to approximately 80% confluence. Actinomycin D (Sigma) was added at a concentration of 5 μM. Cells were collected at three time points after addition (0, 3 hr, 5 hr) by washing once with PBS, then lysing the cells in Buffer RLT from the RNeasy Kit (Qiagen) with 1% β-Mercaptoethanol. A cell scraper was used to remove all cells from the well plate. Each sample was normalized for cell number by quantifying DNA content using a Quant-IT PicoGreen dsDNA Assay Kit (ThermoFisher) according to manufacturer instructions. Equal amounts of cellular contents, as measured by DNA quantity, were taken from each sample and 1 pg of luciferase control RNA (Promega) was added to each sample before RNA purification. Total RNA was then purified using an RNeasy Kit and reverse transcribed using the SuperScript II First-Strand Synthesis System (Thermo Fisher). Real time PCR was performed on a Step One Plus cycler from Applied Biosystems with Fast SYBR® Green Master Mix. Standard curves were generated by plotting CT values against the known initial concentration of luciferase control RNA, and then used to derive mRNA concentration of each target gene at each time point. The lnmRNA concentrations at time point 0, 3 and 5 hr were then used to perform a linear regression as a function of time, and identify the slope of said line as the decay rate (k) (Figure S4F). Half life was calculated with the following formula: t1/2=ln2/kdecay (Chen et al., 2008).
Metabolic labeling and purification of nascent RNA
4sU labeling of nascent RNA was performed as previously described (Duffy et al., 2015). Mouse NPCs from E13.5 WT and Mettl14 cKO forebrain were cultured in standard 6 well culture plates to approximately 80% confluence, treated with 500 μM of 4sU (Carbosynth) for 1 hr, washed with PBS, and harvested with TRIzol reagent (ThermoFisher). Samples were extracted by chloroform twice and precipitated with isopropanol. Biotinylation of 4sU-RNA were carried out in a total volume of 250 μl, containing 70 μg total RNA, 10 mM HEPES (pH 7.5), 1 mM EDTA, and 5 μg MTSEA biotin- XX (Biotium) freshly dissolved in DMF (final concentration of DMF = 20%). Reactions were incubated at RT for 30 min in the dark, and excess biotin reagents were removed by chloroform extraction twice. Purified RNA was dissolved in 50 μl RNase-free water and denatured at 65°C for 10 min, followed by rapid cooling on ice for 5 min. Biotinylated RNA was separated from non-labeled RNA by incubating with 100 μl Streptavidin Magnetic Beads (ThermoFisher) for 20 min at RT. Beads were washed twice with high-salt wash buffer (500 μl each, 100 mM Tris- HCl pH 7.4, 10 mM EDTA, 1 M NaCl, and 0.1% Tween-20). 4sU-RNA was eluted with 100 μl freshly prepared 100 mM DTT followed by a second elution with an additional 100 μl 5 min later. RNA was recovered using the MinElute Spin columns (Qiagen) according to the instructions of the manufacturer, and applied for Q-PCR analysis.
Comparison between human and mouse m6A-seq datasets
For comparison of m6A sequencing data from day 47 human forebrain organoids, PCW11 fetal human cortex, and mouse E13.5 forebrains, we restricted our analysis to expressed genes with a one-to-one ortholog between species. For determining expressed genes, we calculated RPKMs (as stated above) from input libraries, and used a threshold of RPKM > 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data in figure panels reflect several independent experiments performed on different days. An estimate of variation within each group of data is indicated using standard error of the mean (SEM). We performed unpaired Student’s t-test for assessing the significance of differences between two treatments (See each figure for details).
Analyses of mouse cortical neurogenesis in vivo
For quantitative analysis of electroporated neocortices, only GFP+ cells localized within the dorso-lateral cortex were examined. 3 × 3 tiled images were obtained to cover the electroporated region of each coronal section with a 20x or 40x objective by scanning microscope (Zeiss LSM 800) and compared with equivalent sections in littermate counterparts. Quantifications were performed using Imaris software (Bitplane). Specifically, for quantification of cell fate after in utero electroporation, GFP+ cells were marked, and GFP+Pax6+ cells were defined and counted based on the intensity of Pax6 immunofluorescence in GFP+ cells measured with the same criteria among different groups using Imaris software. For the distribution of GFP+ cells in each layer, the borders between different layers were defined by Pax6 immunofluorescence (VZ/SVZ) and DAPI staining (SVZ/IZ and IZ/CP). For quantification of cell fate in WT and Mettl14 cKO mice at E17.5, P0 and P5, the regions of the primary somatosensory cortex were identified and the numbers of Pax6+, Tbr2+, S100β+, Ctip2+, Satb2+ or Tbr1+ cells were counted in each vertical column with 100 μm width. For distribution plots, the distances between soma of EdU+Pax6+, Pax6+, Tbr2+ or Neurod1+ cells and the ventricular surface were calculated. Only EdU+Pax6+ cells within 200 μm distance from the ventricular surface were measured, and the histogram of cell location in every 20 μm interval from the ventricular surface was plotted as a percentage. For distribution plots of Pax6+Tbr2+ or Pax6+Neurod1+ cells, the distances between soma of cells and the ventricular surface were calculated and the numbers of cells per 100 μm2 area in every 20 μm interval from the ventricular surface were plotted as density distribution. All quantifications were performed with 4–10 brain sections from at least 4 animals. Data are presented as the mean ± SEM and statistical significance was assessed using unpaired Student’s t-test.
DATA AND SOFTWARE AVAILABILITY
The access number for the data for m6A-seq reported in this study is NCBI GEO: GSE99017.
Supplementary Material
(A) Expression of molecular mediators of m6A signaling based on a published single-cell RNA-seq dataset of embryonic mouse cortical neurogenesis (Telley et al., 2016). Shown are the expression profiles of selected genes as violin plots, generated using the Seurat package of R (http://genebrowser.unige.ch/science2016/) (Macosko et al., 2015). AP: Apical progenitors/RGCs; BP: daughter basal progenitors/IPCs; EN: early neurons; LN: late neurons.
(B) Depletion of Mettl14 protein in the forebrain of Nestin-Cre;Mettl14f/f cKO mice. Shown are the genetic deletion strategy (left) and sample Western blot images from WT or cKO E17.5 forebrain lysates (right). Because Mettl14 was only deleted in the nervous system, the minor non-neural cells contributed to the residual Mettl14 proteins (faint bands).
(C) Appearance of WT and cKO pups at P5 and P14. Note the impairment in the P14 cKO pup to maintain body balance. Scale bars: 1 cm.
(D) Survival curve of WT (n = 45), Het (n = 23) and cKO (n = 22) pups.
(E–F) Deficits in the production of upper-layer neurons in cKO cortices at P5. Shown in (E) are sample confocal images of staining for Satb2 (layer 2/3), Ctip2 (layer 5) and DAPI, or Ctip2 (layer 5), Tbr1 (layer 6) and DAPI. Scale bars: 100 μm. Quantification is shown in (E). Values represent mean ± SEM (n = 6; **: P < 0.01; unpaired Student’s t-test).
(G–H) Deficits in the production of lower-layer neurons in cKO cortices at E17.5. Shown in (E) are sample confocal images of staining for Ctip2 and DAPI. Scale bar: 100 μm. Quantification is shown in (H). Values represent mean ± SEM (n = 6; **: P < 0.01; unpaired Student’s t-test).
Table S1. List of primers used in the current study, related to Figure S3, 4, S4, 5, S5, and S6. (See Excel file)
Table S2. Dataset from m6A-seq of E13.5 mouse forebrain, day 47 human forebrain organoids, and PCW11 fetal human cortex, related to Figure 4, 7, and S7. (See Excel file)
Table S3. GO analysis of m6A-tagged genes in E13.5 mouse forebrain, related to Figure 4 (See Excel file)
Table S4. Dataset from RNA decay assay of WT and Mettl14 cKO NPCs, related to Figure 4 (See Excel file)
Table S5. Gene and disease ontology analysis of m6A-tagged genes in mouse and human, related to Figure 7 and S7 (See Excel file)
(A) Schematic diagrams of the dual reporter system used to track cell cycle status by time-lapse imaging. Nuclear localized H2B-mCherry and a GFP-tagged Cdk2 substrate DHB are co-expressed in the individual cell. Cdk2 becomes active during the G1-S transition and phosphorylates DHB-GFP, which is then translocated from the nucleus to the cytoplasm. The presence of GFP in the mCherry+ nucleus indicates cells in the G1 phase, whereas translocation to the cytoplasm indicates the initiation of the S phase, and continual buildup of cytoplasmic GFP occurs until mitosis.
(B–D) Flow cytometry analysis of cell cycle progression of WT and Mettl14 cKO NPCs. NPCs were pulse-labeled with EdU (10 μM) for 30 min, cultured for 0 or 5 hr, followed by EdU and DNA content (7AAD) staining and flow cytometry analysis. Shown in (B) are sample dot plots at 0 and 5 hr after EdU pulsing. Cells in a specific cell cycle phase were marked in a box. Note that EdU+ cells (S phase at 0 hr) were segregated into divided (G1*) and non-divided (S/G2*/M*) populations. Shown in (C) are sample histograms of DNA content from EdU+ cells and the total cell population (as a reference). Quantification is shown in (D). Values represent mean ± SEM (n = 4; ***: P < 0.01; unpaired Student’s t-test).
(A) Efficacy of the shRNA against mouse Mettl3. Mouse B16F10 cells were transfected with shRNA-control and shRNA-Mettl3. The amount of Mettl3 mRNA was assessed by Q-PCR 3 days later. Values represent mean ± SEM (n = 3; ***: P < 0.001; unpaired Student’s t-test).
(B–C) Depletion of m6A mRNA methylation by Mettl3 KD. Shown are sample images of m6A dot blot and methylene blue staining (as loading controls; B) and quantification (C). Data were normalized to the averaged levels of WT samples. Values represent mean ± SEM (n = 3; ***: P < 0.01; unpaired Student’s t-test).
(D) Flow cytometry analysis of cell cycle status of mouse NPCs. Mouse NPCs were electroporated to co-express GFP and shRNA-control, or shRNA-Mettl3. After 4 days, NPCs were pulse-labeled with EdU (10 μM) for 30 min, cultured for 9 hr, followed by EdU and DNA content (DyeCycle Violet) staining and flow cytometry analysis. GFP+ and GFP− cells were gated separately and shown as dot plots. Note that GFP+ cells with Mettl3 KD showed accumulation of non-divided (S/G2*/M*) population.
(A) Venn diagram showing intersection among m6A peaks identified in 3 independent m6A-seq experiments. 4,055 high confidence peaks shared by 2 out of 3 replicates, corresponding to 2,059 genes, were used for downstream analysis.
(B) Enrichment of m6A peaks in 5 non-overlapping transcript segments. Pie chart shows percentage of peaks annotated to each segment. Bar plot shows fold enrichment of peaks for each segment, normalized for the segment length.
(C) m6A peaks do not correlate with transcript expression levels. Scatter plot shows gene expression levels (lnRPKM) of m6A-tagged genes plotted against m6A peak lnfold change. Histogram shows distribution of gene expression levels (lnRPKM) for all transcripts detected in 3 RNA-seq input libraries.
(D) Validation of m6A-tagging in specific transcripts in cortical NPCs. The enrichment of m6A-tagged transcripts by IP with anti-m6A antibodies over lgG was quantified by Q-PCR. Values represent mean ± SEM (n = 3; ***: P < 0.01; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test)
(E) Representative coverage plots from the RNA-seq analysis at 0 or 5 hr after treatment with ActD showing increased stability of m6A-tagged genes (Sox1 and Emx2), but not a non m6A-tagged gene (Rad17) in Mettl14 cKO compared to WT NPCs.
(F) Representative plot for calculating half-life of transcripts in WT and cKO NPCs. Data for Emx2 is plotted as an example. The ln of the transcript concentration at each time point, (0, 3, and 5 hr after Actinomycin D treatment) was plotted, and a linear regression was used to determine the slope of the resultant line. The half-life was then calculated as the ln2 divided by the absolute value of the slope of the line.
(A) Q-PCR analysis of pre-mRNA from WT and Mettl14 cKO NPCs using pre-mRNA specific primers. All Ct values were first normalized to the Actin control (not m6A-tagged), which were similar in both WT and cKO NPCs. The ratio (cKO over WT) was calculated for each experiment and values represent mean ± SEM (n = 4 cultures; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(B) Single-cell transcriptome analysis (Telley et al., 2016) reveals the expression of neuronal lineage genes in mouse embryonic cortical RGCs in vivo. Shown are the expression profiles of neural lineage genes as violin plots, similarly as in Figure S1A.
(C–D) Increased expressions of neuronal lineage genes in FlashTag+ (FT+) RGCs 3 hr after pulse labeling. Shown are sample confocal images (C; scale bars: 20 μm) and quantifications of the percentage of FT+Tbr2+Pax6+ cells, or FT+Neurod1+Pax6+ cells (D), among total FT+ cells. Values represent mean ± SEM (n = 5 sections from 2 animals; ***: P < 0.01; unpaired Student’s t-test).
(E–F) Precocious expression of Tbr2 and Neurod1 proteins in RGCs upon knockdown of mRNA deadenylase components in vivo. Shown are sample confocal images (E; scale bars: 20 μm) and quantifications of the percentage of GFP+Tbr2+Pax6+ cells, or GFP+Neurod1+Pax6+ cells, among total GFP+Pax6+ cells and the density distribution of GFP+Tbr2+Pax6+, or GFP+Neurod1+Pax6+ cells from the ventricular surface (F). Values represent mean ± SEM (n = 5 sections from 3 animals; ***: P < 0.001; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(A) Validation of hNPC differentiation from human iPSCs. Shown is a sample confocal image. Scale bar: 50 μm.
(B) Efficacy of the shRNA against METLL14. Human NPCs were electroporated to co-express GFP and shRNA-control, or shRNA-METLL14, and dissociated 3 days later. Amount of METLL14 mRNA in FACS-purified GFP+ cells was assessed by Q-PCR. Values represent mean ± SEM (n = 3; ***: P < 0.01; unpaired Student’s t-test).
(C) Flow cytometry analysis of cell cycle progression of hNPCs with METTL14 KD. Similar to Figure S3D.
(D) Comparison of neuronal differentiation among day 47 human forebrain organoids and embryonic mouse cortical development at E13.5, E15.5 and E17.5. Shown are confocal images of immunostaining for CTIP2 and SATB2 and DAPI. Scale bar: 50 μm. Note that day 47 human forebrain organoids exhibit a differentiation pattern most similar to E13.5 mouse cortex.
(E) Electroporation of human forebrain organoid with shRNA-expressing plasmid. Day 45 forebrain organoids were electroporated to co-express GFP and shRNA-control, or shRNA-METTL14, by microinjection into the lumen of organoids. After 7 days, organoids were pulse-labeled with EdU (10 μM) for 30 min, and cultured further for 14 hr. Shown are sample confocal images at day 52. Scale bar: 100 μm.
(F) Flow cytometry analysis of cell cycle progression with METTL14 KD in human forebrain organoids. Shown are sample dot plots 14 hr after EdU pulse. Cells in a specific cell cycle phase were marked within a box.
(A) Venn diagram showing intersection between m6A peaks identified in 2 independent m6A-seq of day 47 human forebrain organoids. 11,994 high confidence peaks corresponding to 4,702 genes were used for downstream analysis.
(B) Venn diagram showing intersection between m6A peaks identified in 3 independent m6A-seq of PCW11 human fetal brain. 10,980 high confidence peaks corresponding to 5,049 genes were used for downstream analysis.
(C–D) Enrichment of m6A peaks in 5 non-overlapping transcript segments for day 47 human forebrain organoids (C) and PCW11 fetal human brain (D). Same as in Figure S4B.
(E) Pie charts showing the percentage of m6A-tagged genes among all expressed genes in each samples.
(F) GO analysis for m6A-tagged genes shared between human forebrain organoids and fetal brain, but not in mouse E13.5 forebrain (left panel), and GO analysis of m6A-tagged genes shared among all three samples (right panel).
Movie S1. Time-lapse imaging of WT NPCs using a dual fluorescence reporter system, related to Figure 2.
Movie S2. Time-lapse imaging of Mettl14 cKO NPCs using a dual fluorescence reporter system, related to Figure 2.
Highlights.
m6A depletion leads to prolonged cell cycle progression of cortical neural progenitors
m6A promotes decay of tagged neurogenesis-related transcripts
Transcriptional pre-patterning for normal cortical neurogenesis
Acknowledgments
We thank K.M. Christian and J. Schnoll for comments, members of Ming and Song laboratories for discussion, L. Liu, Y. Cai, and D.G. Johnson for technical assistance, and T. M. Hyde and D. R. Weinberger for dissected fetal human brain samples. K-J.Y. was partially supported by a Young Investigator Award from Brain & Behavior Research Foundation and a postdoctoral fellowship from Maryland Stem Cell Research Found (MSCRF); C.V. was partially supported by an NSF pre-doctoral fellowship and NIH T32GM007445. The research was supported by grants from NIH (R37NS047344 to H.S., U19MH106434 to H.S and G-l.M., P01NS097206 to H.S., G-l.M., P.J and C.H., R01MH105128 and R35NS097370 to G-l.M., U19AI131130 to G-l.M. and P.J., R01NS051630 and R01MH102690 to P.J, RM1HG008935 to C.H., H.S and P.J), Simons Foundation (to H.S. and G-l.M.), and The Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G-l.M.). C.H. is a Howard Hughes Medical Institute Investigator.
Footnotes
AUTHOR CONTRIBUTIONS
K-J. Y. led the project and contributed to all aspects of the study. F.R.R. performed m6A-seq, RNA-seq, and bioinformatics analysis. C.V. contributed to most aspects of the study. F.J. contributed to human NPC and organoid analysis. M.P. and S.R. contributed to time-lapse imaging analysis of cell cycle. D.J-C., Y.S., N-S.K., Y.Z., L.Z., S.K., X.W. and P.J. contributed to additional data collection and analyses. L.C.D., X.Z. and C.H. contributed Mett1f/f mice. S.C. contributed to bioinformatics analyses. G-l.M., H.S. and K-J.Y. conceived the project. K-J. Y., F.R.R., C.V., G-l. M., and H.S. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS, Meyre D, Golzio C, Molinari F, Kadhom N, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–111. doi: 10.1016/j.ajhg.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ezzeddine N, Shyu AB. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008;448:335–357. doi: 10.1016/S0076-6879(08)02617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud H, Zhang D, McMurray F, Yu A, Luco SM, Vanstone J, Jarinova O, Carson N, Wickens J, Shishodia S, et al. Identification of a pathogenic FTO mutation by next-generation sequencing in a newborn with growth retardation and developmental delay. J Med Genet. 2016;53:200–207. doi: 10.1136/jmedgenet-2015-103399. [DOI] [PubMed] [Google Scholar]
- Desrosiers RC, Friderici KH, Rottman FM. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry. 1975;14:4367–4374. doi: 10.1021/bi00691a004. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nature communications. 2016;7:12626. doi: 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Rao S, Wu L, Ye N, Liu Z, Hu H, Xiu J, Shen Y, Xu Q. An association study of the m6A genes with major depressive disorder in Chinese Han population. J Affect Disord. 2015;183:279–286. doi: 10.1016/j.jad.2015.05.025. [DOI] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 2015;59:858–866. doi: 10.1016/j.molcel.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer ND, Chen B, Chou SJ, Hippenmeyer S, Nguyen L, Ghashghaei HT. Neural Stem Cells to Cerebral Cortex: Emerging Mechanisms Regulating Progenitor Behavior and Productivity. J Neurosci. 2016;36:11394–11401. doi: 10.1523/JNEUROSCI.2359-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, Soller M. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Jamuar SS, Walsh CA. Genomic variants and variations in malformations of cortical development. Pediatr Clin North Am. 2015;62:571–585. doi: 10.1016/j.pcl.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohwi M, Doe CQ. Temporal fate specification and neural progenitor competence during development. Nat Rev Neurosci. 2013;14:823–838. doi: 10.1038/nrn3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M, et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–247. doi: 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods. 2016;14:23–31. doi: 10.1038/nmeth.4110. [DOI] [PubMed] [Google Scholar]
- Ma DK, Chiang CH, Ponnusamy K, Ming GL, Song H. G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem cells. 2008;26:2131–2141. doi: 10.1634/stemcells.2008-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Drechsel D, Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nature reviews Molecular cell biology. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Pattabiraman K, Visel A, Rubenstein JL. Genomic perspectives of transcriptional regulation in forebrain development. Neuron. 2015;85:27–47. doi: 10.1016/j.neuron.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi K, Shimada T, Nitta Y, Kihara H, Okubo H, Uehara T, Kawasaki Y. Specific gene expression patterns of 108 schizophrenia-associated loci in cortex. Schizophr Res. 2016;174:35–38. doi: 10.1016/j.schres.2016.03.032. [DOI] [PubMed] [Google Scholar]
- Okano H, Temple S. Cell types to order: temporal specification of CNS stem cells. Curr Opin Neurobiol. 2009;19:112–119. doi: 10.1016/j.conb.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, Ruan Y, Smith J, van Heyningen V, Rubenstein JL, Livesey FJ. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009;5:e1000511. doi: 10.1371/journal.pgen.1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Gotz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502. doi: 10.1146/annurev-cellbio-101011-155801. [DOI] [PubMed] [Google Scholar]
- Tebbenkamp AT, Willsey AJ, State MW, Sestan N. The developmental transcriptome of the human brain: implications for neurodevelopmental disorders. Curr Opin Neurol. 2014;27:149–156. doi: 10.1097/WCO.0000000000000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Govindan S, Prados J, Stevant I, Nef S, Dermitzakis E, Dayer A, Jabaudon D. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science. 2016;351:1443–1446. doi: 10.1126/science.aad8361. [DOI] [PubMed] [Google Scholar]
- Terry NH, White RA. Flow cytometry after bromodeoxyuridine labeling to measure S and G2+M phase durations plus doubling times in vitro and in vivo. Nature Protocols. 2006;1:859–869. doi: 10.1038/nprot.2006.113. [DOI] [PubMed] [Google Scholar]
- Wang X, Huang J, Zou T, Yin P. Human m6A writers: Two subunits, two roles. RNA Biol. 2017;14:300–304. doi: 10.1080/15476286.2017.1282025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature cell biology. 2014a;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014b;29:217–232. doi: 10.1016/j.devcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CR, Cole PA, Meyers DJ, Kormish J, Dent S, Zaret KS. Chromatin “prepattern” and histone modifiers in a fate choice for liver and pancreas. Science. 2011;332:963–966. doi: 10.1126/science.1202845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, Tcw J, Kouznetsova J, Ogden SC, Hammack C, et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming GL, Song H. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci. 2016;17:537–549. doi: 10.1038/nrn.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, Makri G, Nauen D, Shin JH, Park Y, et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell. 2014;15:79–91. doi: 10.1016/j.stem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Song G, Qian X, Pan J, Xu D, Rho HS, Kim NS, Habela C, Zheng L, Jacob F, et al. Zika-Virus-Encoded NS2A Disrupts Mammalian Cortical Neurogenesis by Degrading Adherens Junction Proteins. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nature reviews Molecular cell biology. 2017a;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017e;542:475–478. doi: 10.1038/nature21355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Expression of molecular mediators of m6A signaling based on a published single-cell RNA-seq dataset of embryonic mouse cortical neurogenesis (Telley et al., 2016). Shown are the expression profiles of selected genes as violin plots, generated using the Seurat package of R (http://genebrowser.unige.ch/science2016/) (Macosko et al., 2015). AP: Apical progenitors/RGCs; BP: daughter basal progenitors/IPCs; EN: early neurons; LN: late neurons.
(B) Depletion of Mettl14 protein in the forebrain of Nestin-Cre;Mettl14f/f cKO mice. Shown are the genetic deletion strategy (left) and sample Western blot images from WT or cKO E17.5 forebrain lysates (right). Because Mettl14 was only deleted in the nervous system, the minor non-neural cells contributed to the residual Mettl14 proteins (faint bands).
(C) Appearance of WT and cKO pups at P5 and P14. Note the impairment in the P14 cKO pup to maintain body balance. Scale bars: 1 cm.
(D) Survival curve of WT (n = 45), Het (n = 23) and cKO (n = 22) pups.
(E–F) Deficits in the production of upper-layer neurons in cKO cortices at P5. Shown in (E) are sample confocal images of staining for Satb2 (layer 2/3), Ctip2 (layer 5) and DAPI, or Ctip2 (layer 5), Tbr1 (layer 6) and DAPI. Scale bars: 100 μm. Quantification is shown in (E). Values represent mean ± SEM (n = 6; **: P < 0.01; unpaired Student’s t-test).
(G–H) Deficits in the production of lower-layer neurons in cKO cortices at E17.5. Shown in (E) are sample confocal images of staining for Ctip2 and DAPI. Scale bar: 100 μm. Quantification is shown in (H). Values represent mean ± SEM (n = 6; **: P < 0.01; unpaired Student’s t-test).
Table S1. List of primers used in the current study, related to Figure S3, 4, S4, 5, S5, and S6. (See Excel file)
Table S2. Dataset from m6A-seq of E13.5 mouse forebrain, day 47 human forebrain organoids, and PCW11 fetal human cortex, related to Figure 4, 7, and S7. (See Excel file)
Table S3. GO analysis of m6A-tagged genes in E13.5 mouse forebrain, related to Figure 4 (See Excel file)
Table S4. Dataset from RNA decay assay of WT and Mettl14 cKO NPCs, related to Figure 4 (See Excel file)
Table S5. Gene and disease ontology analysis of m6A-tagged genes in mouse and human, related to Figure 7 and S7 (See Excel file)
(A) Schematic diagrams of the dual reporter system used to track cell cycle status by time-lapse imaging. Nuclear localized H2B-mCherry and a GFP-tagged Cdk2 substrate DHB are co-expressed in the individual cell. Cdk2 becomes active during the G1-S transition and phosphorylates DHB-GFP, which is then translocated from the nucleus to the cytoplasm. The presence of GFP in the mCherry+ nucleus indicates cells in the G1 phase, whereas translocation to the cytoplasm indicates the initiation of the S phase, and continual buildup of cytoplasmic GFP occurs until mitosis.
(B–D) Flow cytometry analysis of cell cycle progression of WT and Mettl14 cKO NPCs. NPCs were pulse-labeled with EdU (10 μM) for 30 min, cultured for 0 or 5 hr, followed by EdU and DNA content (7AAD) staining and flow cytometry analysis. Shown in (B) are sample dot plots at 0 and 5 hr after EdU pulsing. Cells in a specific cell cycle phase were marked in a box. Note that EdU+ cells (S phase at 0 hr) were segregated into divided (G1*) and non-divided (S/G2*/M*) populations. Shown in (C) are sample histograms of DNA content from EdU+ cells and the total cell population (as a reference). Quantification is shown in (D). Values represent mean ± SEM (n = 4; ***: P < 0.01; unpaired Student’s t-test).
(A) Efficacy of the shRNA against mouse Mettl3. Mouse B16F10 cells were transfected with shRNA-control and shRNA-Mettl3. The amount of Mettl3 mRNA was assessed by Q-PCR 3 days later. Values represent mean ± SEM (n = 3; ***: P < 0.001; unpaired Student’s t-test).
(B–C) Depletion of m6A mRNA methylation by Mettl3 KD. Shown are sample images of m6A dot blot and methylene blue staining (as loading controls; B) and quantification (C). Data were normalized to the averaged levels of WT samples. Values represent mean ± SEM (n = 3; ***: P < 0.01; unpaired Student’s t-test).
(D) Flow cytometry analysis of cell cycle status of mouse NPCs. Mouse NPCs were electroporated to co-express GFP and shRNA-control, or shRNA-Mettl3. After 4 days, NPCs were pulse-labeled with EdU (10 μM) for 30 min, cultured for 9 hr, followed by EdU and DNA content (DyeCycle Violet) staining and flow cytometry analysis. GFP+ and GFP− cells were gated separately and shown as dot plots. Note that GFP+ cells with Mettl3 KD showed accumulation of non-divided (S/G2*/M*) population.
(A) Venn diagram showing intersection among m6A peaks identified in 3 independent m6A-seq experiments. 4,055 high confidence peaks shared by 2 out of 3 replicates, corresponding to 2,059 genes, were used for downstream analysis.
(B) Enrichment of m6A peaks in 5 non-overlapping transcript segments. Pie chart shows percentage of peaks annotated to each segment. Bar plot shows fold enrichment of peaks for each segment, normalized for the segment length.
(C) m6A peaks do not correlate with transcript expression levels. Scatter plot shows gene expression levels (lnRPKM) of m6A-tagged genes plotted against m6A peak lnfold change. Histogram shows distribution of gene expression levels (lnRPKM) for all transcripts detected in 3 RNA-seq input libraries.
(D) Validation of m6A-tagging in specific transcripts in cortical NPCs. The enrichment of m6A-tagged transcripts by IP with anti-m6A antibodies over lgG was quantified by Q-PCR. Values represent mean ± SEM (n = 3; ***: P < 0.01; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test)
(E) Representative coverage plots from the RNA-seq analysis at 0 or 5 hr after treatment with ActD showing increased stability of m6A-tagged genes (Sox1 and Emx2), but not a non m6A-tagged gene (Rad17) in Mettl14 cKO compared to WT NPCs.
(F) Representative plot for calculating half-life of transcripts in WT and cKO NPCs. Data for Emx2 is plotted as an example. The ln of the transcript concentration at each time point, (0, 3, and 5 hr after Actinomycin D treatment) was plotted, and a linear regression was used to determine the slope of the resultant line. The half-life was then calculated as the ln2 divided by the absolute value of the slope of the line.
(A) Q-PCR analysis of pre-mRNA from WT and Mettl14 cKO NPCs using pre-mRNA specific primers. All Ct values were first normalized to the Actin control (not m6A-tagged), which were similar in both WT and cKO NPCs. The ratio (cKO over WT) was calculated for each experiment and values represent mean ± SEM (n = 4 cultures; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(B) Single-cell transcriptome analysis (Telley et al., 2016) reveals the expression of neuronal lineage genes in mouse embryonic cortical RGCs in vivo. Shown are the expression profiles of neural lineage genes as violin plots, similarly as in Figure S1A.
(C–D) Increased expressions of neuronal lineage genes in FlashTag+ (FT+) RGCs 3 hr after pulse labeling. Shown are sample confocal images (C; scale bars: 20 μm) and quantifications of the percentage of FT+Tbr2+Pax6+ cells, or FT+Neurod1+Pax6+ cells (D), among total FT+ cells. Values represent mean ± SEM (n = 5 sections from 2 animals; ***: P < 0.01; unpaired Student’s t-test).
(E–F) Precocious expression of Tbr2 and Neurod1 proteins in RGCs upon knockdown of mRNA deadenylase components in vivo. Shown are sample confocal images (E; scale bars: 20 μm) and quantifications of the percentage of GFP+Tbr2+Pax6+ cells, or GFP+Neurod1+Pax6+ cells, among total GFP+Pax6+ cells and the density distribution of GFP+Tbr2+Pax6+, or GFP+Neurod1+Pax6+ cells from the ventricular surface (F). Values represent mean ± SEM (n = 5 sections from 3 animals; ***: P < 0.001; **: P < 0.01; *: P < 0.05; unpaired Student’s t-test).
(A) Validation of hNPC differentiation from human iPSCs. Shown is a sample confocal image. Scale bar: 50 μm.
(B) Efficacy of the shRNA against METLL14. Human NPCs were electroporated to co-express GFP and shRNA-control, or shRNA-METLL14, and dissociated 3 days later. Amount of METLL14 mRNA in FACS-purified GFP+ cells was assessed by Q-PCR. Values represent mean ± SEM (n = 3; ***: P < 0.01; unpaired Student’s t-test).
(C) Flow cytometry analysis of cell cycle progression of hNPCs with METTL14 KD. Similar to Figure S3D.
(D) Comparison of neuronal differentiation among day 47 human forebrain organoids and embryonic mouse cortical development at E13.5, E15.5 and E17.5. Shown are confocal images of immunostaining for CTIP2 and SATB2 and DAPI. Scale bar: 50 μm. Note that day 47 human forebrain organoids exhibit a differentiation pattern most similar to E13.5 mouse cortex.
(E) Electroporation of human forebrain organoid with shRNA-expressing plasmid. Day 45 forebrain organoids were electroporated to co-express GFP and shRNA-control, or shRNA-METTL14, by microinjection into the lumen of organoids. After 7 days, organoids were pulse-labeled with EdU (10 μM) for 30 min, and cultured further for 14 hr. Shown are sample confocal images at day 52. Scale bar: 100 μm.
(F) Flow cytometry analysis of cell cycle progression with METTL14 KD in human forebrain organoids. Shown are sample dot plots 14 hr after EdU pulse. Cells in a specific cell cycle phase were marked within a box.
(A) Venn diagram showing intersection between m6A peaks identified in 2 independent m6A-seq of day 47 human forebrain organoids. 11,994 high confidence peaks corresponding to 4,702 genes were used for downstream analysis.
(B) Venn diagram showing intersection between m6A peaks identified in 3 independent m6A-seq of PCW11 human fetal brain. 10,980 high confidence peaks corresponding to 5,049 genes were used for downstream analysis.
(C–D) Enrichment of m6A peaks in 5 non-overlapping transcript segments for day 47 human forebrain organoids (C) and PCW11 fetal human brain (D). Same as in Figure S4B.
(E) Pie charts showing the percentage of m6A-tagged genes among all expressed genes in each samples.
(F) GO analysis for m6A-tagged genes shared between human forebrain organoids and fetal brain, but not in mouse E13.5 forebrain (left panel), and GO analysis of m6A-tagged genes shared among all three samples (right panel).
Movie S1. Time-lapse imaging of WT NPCs using a dual fluorescence reporter system, related to Figure 2.
Movie S2. Time-lapse imaging of Mettl14 cKO NPCs using a dual fluorescence reporter system, related to Figure 2.