

Abstract
Type III interferons (IFNλs) are secreted factors that are well-known for their antiviral activities. However, their regulation and functions during bacterial infections are unclear. Herein, we report that the regulation of IFNλ genes did not track with mechanisms that control type I IFN expression in response to Toll-like Receptors (TLRs). While type I IFNs were only expressed from TLRs present on endosomes, type III IFNs could be induced by TLRs that reside at the plasma membrane and that detect various bacterial products. The mechanisms that regulate type III IFN gene expression tracked with those that promote inflammatory cytokine and chemokine expression. Importantly, recombinant IFNλs enhanced epithelial barriers in vitro, preventing transcellular bacteria dissemination. We therefore propose that in addition to their functions in cell-intrinsic antiviral immunity, type III IFNs protect epithelial barrier integrity, an activity that would benefit the host during any infectious encounter.
Introduction
Type III interferons (IFNs) are factors that are secreted by immune cells during microbial infections (1, 2). In contrast to their more extensively studied counterparts, the type I IFNs, we know little of how these genes are regulated or the functions of their products in host defense. Much of the study of type III IFNs (also known as IFNλs) has focused on their roles in viral infections. Early studies demonstrated that these genes are co-regulated with type I IFNs during many infectious encounters (3, 4). However our recent work established that during viral infections, type I and III IFNs can be induced via distinct pathways (5). Functionally, type III IFNs are most distinct from type I IFNs in that they are particularly important at barrier surfaces. These locations are rich in epithelial cells that express high levels of the IFNλ receptor (6), and produce various factors that protect the host from microbial encounters. Type III IFNs also have roles in protecting endothelial cells at the blood-brain barrier from viral infection (7). Thus, this family of proteins has increasingly important functions in host defense.
Recent work has suggested a role for the type III IFN family in bacteria-host interactions. For example, infection with a number of bacterial pathogens and stimulation with lipopolysaccharides (LPS) from the outer membrane of gram-negative bacteria induce the expression of type III IFN genes (8–10). In addition, Listeria monocytogenes encodes virulence determinants that modulate the expression of type III IFN–responsive genes (11). These findings suggest an important role of type III IFNs in anti-bacterial defense, but the spectrum of bacteria that induce expression of these genes, and the function of their products, have yet to be revealed.
Toll-like Receptors (TLRs) of the innate immune system are common sensors of bacterial infection. These receptors are expressed on various cells, in particular phagocytes and barrier epithelial cells. Detection of cognate microbial ligands, collectively known as pathogen-associated molecular patterns (PAMPs), by TLRs leads to inflammatory and adaptive immune responses that restrict bacterial spread and provide long-term immunity (12). The spectrum of activities induced by TLRs is an active area of research. While all TLRs induce the expression of inflammatory chemokines and cytokines, a limited set of receptors induce type I IFN gene expression (13).
TLRs are one of several families of pattern recognition receptors (PRRs) that sense and respond to PAMPs. PRRs can be found in various subcellular locations, including the plasma membrane, endosomes, cytosol, endoplasmic reticulum and the nucleus (14). Several of these families induce type I and III IFN expression, including the cytosolic DNA sensor cGAS and cytosolic RNA sensors of the RIG-I like Receptor (RLR) family (1, 2, 15). Our cell biological analysis of the RLR pathway revealed that these receptors can induce type III IFN or type I IFN expression from distinct subcellular locations (5). The mechanisms underlying these differential sites of signaling, and the generality of these observations as they relate to other receptor families, remain to be established. Like the RLRs, TLRs signal from diverse organelles. These receptors can signal from the plasma membrane or endosomes, and some receptors signal from both locations (16, 17). Mechanistically, TLR signaling pathways depend on the ability of these receptors to assembly large supramolecular organizing centers (SMOCs) in the cytosol after microbial detection (18). These organizing centers serve as the principal sites of signals that promote inflammatory and immunoregulatory gene expression. The most commonly used SMOC in the TLR pathway is the myddosome. The myddosome consists minimally of the adaptors TIRAP and MyD88, and several members of the IRAK family of serine/threonine kinases (19–22). The myddosome is best-understood to promote NF-κB and AP-1 dependent upregulation of genes encoding inflammatory cytokines and chemokines (23). A subset of TLRs also activate a myddosome-independent response that relies on the adaptor TRIF (24). TRIF signaling leads to the expression of type I IFNs. This TRIF-dependent pathway is thought to be only activated by TLR4 and TLR3, although recent studies have expanded the possible roles for TRIF to other TLRs (25).
Several TLRs induce the expression of type III IFN genes, most notably those that detect viral nucleic acids (4, 8, 15). However, the full spectrum of TLRs that induce type III IFN gene expression, and the signaling pathways that promote these putative responses, are unknown. In this study, we sought to identify potential functions of type III IFNs during host-bacteria encounters, and define the means by which the genes encoding these factors are induced by bacteria. Analogous to their ability to reinforce the endothelial blood-brain barrier to prevent viral dissemination (26), we found that recombinant type III IFNs increase the barrier functions of a polarized human intestinal epithelial cell line. This barrier reinforcement prevents the transcellular passage of infectious gram-negative bacteria. Further, we found that a variety of bacterial PAMPs activate individual TLRs to promote type III IFN expression, and that myddosome components are required for TLR-dependent type III IFN gene expression. Finally, we identify type III IFNs as the first example of IFN genes whose expression can be induced by plasma membrane localized receptors. Overall, these results highlight type III IFNs as factors that are commonly upregulated by bacteria-sensing TLRs, and suggest their general importance in barrier immunity.
Materials and Methods
Cell Culture
WT, MyD88, TRIF or TLR5 knockout (KO) mice were purchased from JAX. BMDCs were differentiated from mouse bone marrow in IMDM (GIBCO), 10% GM-CSF, and 10% FBS. 293T cells were cultured in complemented DMEM supplemented with 10% FBS. WT THP1, along with IRAK2 or IRAK4 CRISPR KO were a kind gift from Iain Fraser, NIH. MyD88 KO THP1 cells were constructed in WT/Dual THP1 background (Invivogen). THP1 cells were cultured in complete RPMI supplemented with 10% FBS and were differentiated into macrophages using 50 ng/ml phorbol 12-myristate 13-acetate (PMA) for 48h. T84 epithelial cells were obtained from the Harvard Digestive Disease Center. T84 cells were cultured and polarized as previously described (27). Briefly, cells were plated on collagen-coated 3.0 μm polyester filters and grown for 7 days until Transepithelial electrical resistance (TEER) reached 1650 to 2970 Ω/cm2. TEER was measured with an epithelial Voltohmmeter (WPI). Human peripheral blood mononuclear cells (PBMCs) were isolated from blood of healthy individuals using Lympholyte and SepMate columns, according to the manufacturer’s instructions. Cells were then plated on tissue culture plates in RPMI (Gibco) + 10% FBS and allowed to adhere for 24h followed by washing, to enrich the monocyte fraction.
ELISA and antibodies
A human IFNλ1 ELISA kit was purchased from ebioscience and manufacturer’s recommendations were followed. Anti phosphoSTAT1 (pSTAT1), Viperin and actin antibodies were obtained from BD Legend, pJAK2 and STAT1 antibodies from BD Transduction respectively.
Infections and stimulations in vitro
Where needed, cells were treated with 1 μg/ml Cytochalasin D (Sigma) or 5 μM 34-2 Dynole (Applied biochemicals). Dynole was applied in serum free medium. Where indicated, 50 μg/ml 4kDa Fluorescein isothiocyanate–dextran (Sigma) was applied on the apical side of polarized T84 cells for 90 min. Cells were then treated with 0.1 μg/ml recombinant human IFNλ1 or IFNβ (Peprotech) for 2 h prior to infection as described below. At the time points indicated, TEER and fluorescence in the basal chamber were measured, or bacteria in the basal compartment were quantified by colony forming unit (CFU) plating and enumeration.
Salmonella SL1344 and flic mutant strains were obtained from Denise Monack (Stanford), Shigella M90T strain were obtained from Neal Alto (UT Southwestern) while a Shigella sonnei strain was obtained from Abigail Clements (Imperial College). Shigella flexneri was used for the majority of the experiments including the functional assays in Fig. 1. Shigella sonnei was used in Fig. 2C, 2D, 2H and 2I, to respect regulations on work with Shigella flexneri in the United Kingdom. Salmonella was grown in Luria Bertani broth while Shigella was grown in Tryptic soy broth (TSB). Unless otherwise specified, in order to minimize expression of type III secretion systems, overnight cultures were used directly for infection. When virulent bacteria were required (Fig. 1), Salmonella were subcultured 1:33 for 3 h OD600 nm = 2 and Shigella were subcultured 1:60 in TSB until OD600 nm = 0.6. LPS was supplied by Enzo (ALX-581-012-L002). Phosphorothioate-linked CpG DNA (TCCATGACGTTCCTGACGTT) was synthesized by IDT. All other synthetic ligands were supplied by Invivogen : polyIC (tlrl-pic), Pam3CSK4 (tlrl-pms), Pam2CSK4 (tlrl-pm2s-1), Zymosan (tlrl-zyn) and vaccigrade flagellin (vac-fla). Where indicated, ligands were treated with polymyxinB (Sigma) for 30 min at room-temperature.
Figure 1. Type III IFNs protect epithelial barrier integrity from bacterial infection.
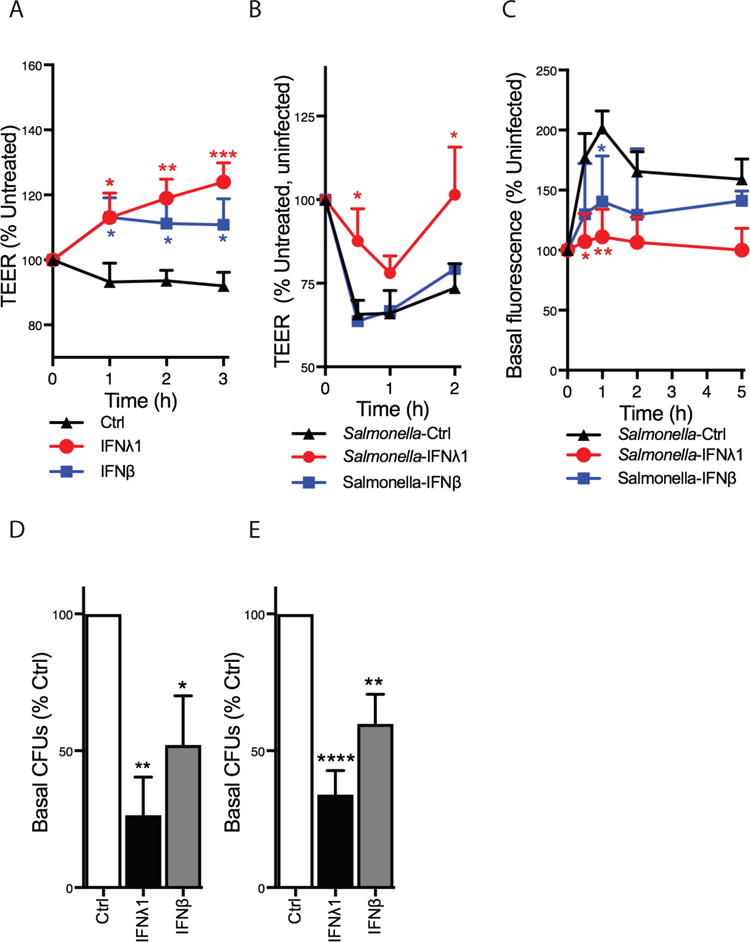
(A) Polarized T84 epithelial cells were treated with 0.1 μg/ml recombinant IFNλ1 or IFNβ, and transepithelial electrical resistance (TEER) was measured at the time points indicated. (B) Following IFN treatment for 2 h, cells were apically infected with wild-type Salmonella Typhimurium and TEER was measured at 30 min, 1 h and 2 h post-infection. (C) FITC-labeled 4kDa dextran was added to the apical side of cell monolayers. Cells were then incubated with IFNλ1 or IFNβ for 2 h, followed by infection with Salmonella. Fluorescence in the basal chamber was monitored over the next 5 h. (D) Similar to (B), T84 cells were incubated with recombinant IFNλ1 or IFNβ for 2 h, followed by a 2 h infection with Salmonella Typhimurium from the apical surface. Culture medium was collected from the basal chamber and colony/forming units (CFUs) were quantified to measure bacterial migration through the epithelial monolayer. (E), Similarly to (D) T84 cell were treated with recombinant IFNs and infected with wild-type Shigella flexneri. CFUs were collected and measured as described for Salmonella. Each panel except (C) represents averages ± SEM of at least 6 experiments. (C) shows one experiment representative of 6. Results are shown as % untreated or uninfected conditions, as indicated. Statistics were performed between Ctrl (not IFN treated) and IFN-treated samples. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (one or two-way ANOVA).
Figure 2. Type III IFNs are induced by bacterial infections in a MyD88-dependent manner.
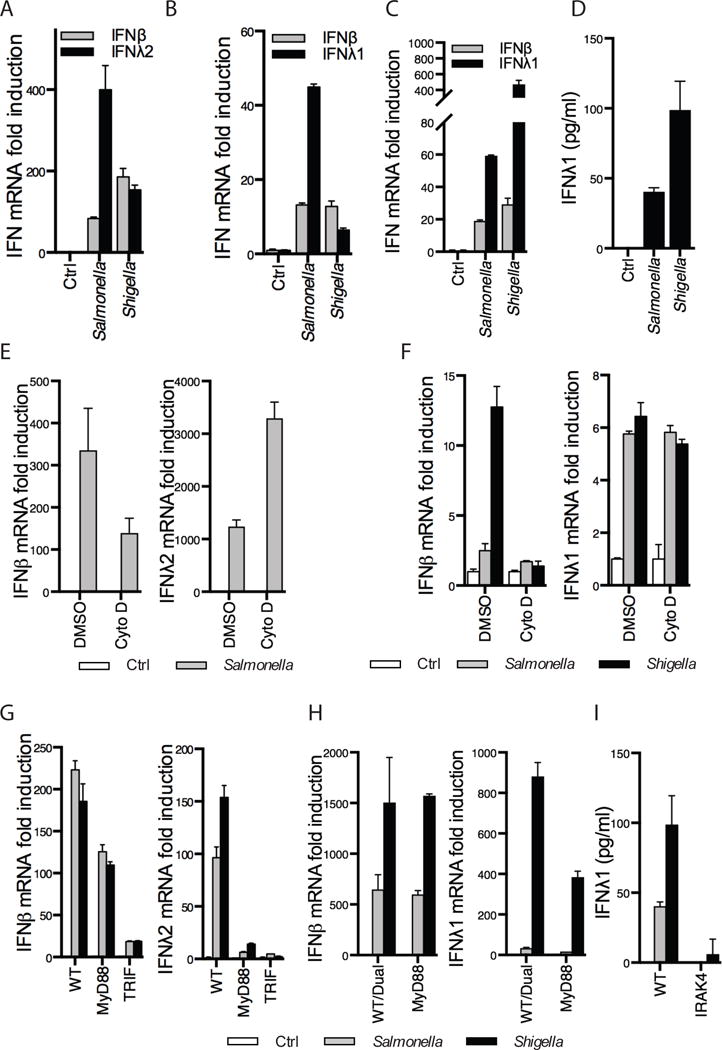
(A–C) Bone-marrow derived dendritic cells (BMDCs, A) polarized T84 epithelial cells (B) or PMA-differentiated THP1 cells (C) were infected for 2 h with wild-type (WT) Salmonella and Shigella from overnight cultures. IFNβ, IFNλ1 or IFNλ2 mRNA expression was measured by RT-qPCR. (D) Similar to (C) except IFNλ1 protein secretion was quantified by ELISA after a 5 h infection. (E) WT BMDCs were treated with 1 μg/ml of the bacterial invasion inhibitor Cytochalasin D for 30 min followed by 2 h infection with Salmonella. (F) Similar to (E), polarized T84 epithelial cells treated with Cytochalasin D were infected with bacterial strains for 2 h. (G) WT, MyD88 or TRIF KO BMDCs were infected with the indicated bacterial strains for 2 h. (H) WT/Dual or MyD88 KO THP1 cells were infected with Salmonella and Shigella for 1h. (I) WT or IRAK4 KO THP1 cells were infected with Salmonella and Shigella for 5h. IFNλ1 protein levels were quantified by ELISA. Where indicated, IFNβ, IFNλ1 or IFNλ2 mRNA expression was measured by RT-qPCR. Error bars represent mean ± SD of triplicate readings for one experiment representative of 2–4 or averages ± SEM of 3–6 independent experiments (D,H,I). Note that Shigella flexneri was used in A, B, F, G while Shigella sonnei was used in C, D, H, I. The two strains are similar for the purpose of these experiments, but later work had to be carried out with Shigella sonnei to keep in line with UK regulations on Shigella flexneri.
When indicated, cell culture media from BMDCs stimulated with TLR ligands was collected and incubated onto fresh 293T cells for 1 h. Cells were then lyzed and processed for western immunoblotting using standard techniques.
Quantitative real time PCR
Cells were lysed in RNA Lysis buffer (Ambion) supplemented with β-Mercaptoethanol and passed through Qiashredder columns (Qiagen). RNA was then isolated using Pure l Link RNA Mini Kit columns (Ambion) following the manufacturer’s recommendations. RNA samples were treated with DNAseI (ThermoScientific) for 30 min at 37°C followed by 10 min EDTA deactivation at 65°C. RT-qPCR was carried out with a Bio-Rad CFX384 real time cycler with Taqman probes as directed by the manufacturer. Data were normalized against the expression of a control gene (GAPDH) followed by the uninduced or uninfected (Control-Ctrl) sample (set to 1). When no counts were detected in a sample, an arbitrary number of 45 cycles was selected.
Results
Type III IFNs protect epithelial barrier integrity during bacterial infections
While much work has been focused on the role of type III IFNs in antiviral immunity at epithelial surfaces, the functions of these factors during bacterial infections are less clear. The IFNλ receptor protects the endothelial blood-brain barrier to prevent viral dissemination (26). In an analogous sequence of events, several enteric bacterial pathogens disrupt epithelial barriers to enable their spread to the underlying tissues of the body. We therefore determined if type III IFNs protect the intestinal barrier from damage inflicted by bacteria. As a control and complementary experiment, we studied the role of type I IFNs in the same assays.
For these studies, we examined polarized colonic human T84 cells, as these cells are routinely used as an in vitro model to study the interactions between enteric bacteria and intestinal epithelial cells (27, 28). Cells were treated with recombinant IFNλ and IFNβ, and transepithelial electrical resistance (TEER), a marker of epithelial barrier integrity, was monitored over time. We found that treatment with IFNλ led to a significant increase in TEER. IFNβ treatment also significantly increased TEER, but the effect was less sustained (Fig. 1A). As increases in TEER correspond to an increase in barrier integrity, we examined the role of type III IFNs in bacteria-induced epithelial disruptions. We first used Salmonella enterica serovar Typhimurium (Salmonella), a human pathogen that naturally disrupts intestinal epithelia to promote systemic dissemination (29, 30). Polarized T84 cells were treated with IFNβ or IFNλ for 2 h, followed by apical infection with Salmonella. These bacteria were grown to induce the SPI-1 type III secretion system (T3SS), which is required for bacterial invasion (31–33) and epithelial damage (30, 34). We found that Salmonella infection disrupted TEER (Fig. 1B, black line). In cells pretreated with IFNβ, TEER was disrupted to an extent similar to control conditions (Fig. 1B, blue line). By contrast, pretreatment with IFNλ protected T84 monolayers from Salmonella-induced TEER disruption, with the cells recovering by 2 h post-infection (Fig. 1B, red line). As an additional readout of epithelial barrier integrity, we measured the transcellular passage of fluorescent dextrans across the monolayer. Following apical IFN and dextran application, cells were infected apically, and fluorescence in the basal compartment was measured. Salmonella infection led to an increase in dextran passage into the basal compartment (Fig. 1C, black line), indicating a disruption of the epithelial barrier. Treatment with both IFNs reduced dextran passage across the monolayer, but the effect of IFNλ was more pronounced (Fig. 1C, red line). To determine if the ability of IFNλ to protect against dextran passage through the epithelial barrier correlated with bacterial passage, we quantified the number of bacteria in the basal chamber by plating and enumerating colony forming units (CFU). 2 h following apical infection, we found that 60% less bacteria were able to cross a monolayer treated with IFNλ, as compared to control conditions (Fig. 1D). Again, the effect of IFNβ treatment was less pronounced. To determine if the ability of IFNλ to protect epithelial barriers during Salmonella infections extended to other bacteria, we examined Shigella flexneri. Like Salmonella, Shigella is an enteric pathogen whose virulence relies on damage of the epithelial barrier (35). IFNλ efficiently prevented the transmigration of bacteria across the monolayer, as we measured 70% less CFUs in the basal chamber of monolayers treated with IFNλ, with IFNβ having a smaller effect (Fig. 1E). Taken together, these data establish that IFNλs protect epithelial barrier integrity from bacteria-induced damage, and efficiently block the ability of bacterial pathogens to cross the epithelium.
Extracellular bacteria induce type III IFN expression
We reasoned that if type III IFNs are important for protecting against bacteria-induced epithelial damage, then the genes encoding these factors may be expressed during bacterial encounters. We therefore analyzed the transcriptional response of cells infected with Salmonella and Shigella. Specifically, quantitative RT-PCR was used to examine the expression of the type III IFN IFNλ1 in human cells, as it is the most prominent type III IFN produced (36). However, as IFNλ1 (IL29) is a pseudogene in mice (37), we measured IFNλ2 (IL28A) in mouse cells. IFNβ was used to monitor type I IFN expression. Basal expression of each IFN gene was first established. We found that basal type I and III IFN expression differed from one another in different cell types, with mouse bone marrow-derived dendritic cells (BMDCs) and PMA-differentiated human THP1 monocytes containing higher levels of basal IFNβ transcripts, and polarized T84 cells containing higher levels of IFNλ1 (Supplemental Fig. 1A–C). Based on this differential basal expression, we monitored relative fold-induction over control, rather than transcript abundance, in all subsequent experiments. Relative fold-induction was calculated as the ratio of mRNA present in resting cells as compared to infected or stimulated cells. Infection of BMDCs (Fig. 2A), polarized T84 cells (Fig. 2B) and PMA-differentiated human THP1 cells (Fig. 2C) demonstrated that IFNλ fold-induction was at least as strong as IFNβ during Salmonella and Shigella infection. In addition, we observed secreted IFNλ1 protein by ELISA in Salmonella and Shigella-infected THP1 cells (Fig. 2D). To determine if phagocytosis was required for bacteria-induced IFNλ expression, we blocked bacterial entry using the actin polymerization inhibitor Cytochalasin D. Cytochalasin D treatment did not affect baseline levels of IFNβ or IFNλ1/2 in BMDCs (Supplemental Fig. 1D) or T84 cells (Supplemental Fig. 1E). Cytochalasin D-treatment of BMDCs (Fig. 2E) or T84 cells (Fig. 2F) prevented IFNβ expression, as compared with vehicle treatment. Interestingly Cytochalasin D did not inhibit the upregulation of the IFNλ in response to Salmonella in BMDCs (Fig. 2E), or Salmonella or Shigella in T84 cells (Fig. 2F). These results suggest that bacterial entry is not required for IFNλ expression.
As bacteria can activate multiple innate immune receptor families, we sought to determine the receptor(s) responsible for IFN expression during infection. TLR-dependent pathways can be identified by the examination of cells lacking the signaling adaptor proteins MyD88 and TRIF. We therefore infected BMDCs lacking MyD88 or TRIF (Fig. 2G) and THP1 cells that were rendered MyD88-deficient using CRISPR/Cas (Fig. 2H). Baseline levels of IFN transcripts in WT, MyD88 knockout (KO) and TRIF KO cells were comparable in BMDCs and THP1 cells (Supplemental Fig. 1F, G). IFNβ fold induction during infection was largely TRIF-dependent, whereas IFNλ expression was strongly dependent on MyD88 and TRIF in BMDCs (Fig. 2G). Similarly, in THP1 cells, IFNλ expression upon Salmonella and Shigella infection was MyD88 dependent (Fig. 2H). IFNβ expression in response to Salmonella and Shigella was not mediated by MyD88. Finally, we monitored IFNλ secretion by ELISA following infection of THP1 cells that are deficient for IRAK4, a component of the MyD88 signaling pathway (38). IRAK4 KO cells secreted minimal amounts of IFNλ after bacterial infection (Fig. 2I). These data indicate that bacteria-induced type I and III IFN expression is TLR-dependent, and further reveal that these IFN genes are subject to differential regulation. IFNλ expression activity depends on MyD88 signaling, and does not depend on phagocytosis.
Endosomal and cell surface resident TLRs activate type III IFN gene expression
To better understand the means by which bacteria induce IFNs, we took the reductionist approach of identifying individual PAMPs that promote type I and III IFN expression. PMA-differentiated THP1 monocytes (Fig. 3A), polarized T84 cells (Fig. 3B) and BMDCs (Fig. 3C) were treated with cognate ligands for all well-defined TLRs. Endosome-localized TLR3, TLR7/8 and TLR9 were respectively activated with polyIC (pIC), R848 and CpG DNA. These ligands induced IFNβ and IFNλ similarly (Fig. 3A–C). As expected, these ligands also upregulated the inflammatory cytokines IL6 and IL8 (Supplemental Fig. 2A). TLR4 activation by bacterial LPS also induced expression of both IFN species. TLR2/1 heterodimers were activated with the bacterial lipoprotein mimic Pam3Cys (P3C) and TLR2/6 heterodimers with Pam2Cys (P2C). Interestingly, TLR2 promoted a greater increase in the expression of IFNλ than IFNβ in these different cell types. We obtained comparable results with the yeast cell wall component zymosan, an additional TLR2 agonist (Fig 3A–C). TLR5 was activated by Salmonella flagellin (FliC). Much like TLR2, TLR5 induced IFNλ and IFNβ expression, although IFNλ fold induction was an order of magnitude higher than that of IFNβ, especially in THP1 cells and DCs (Fig. 3A, C). These findings with TLR2 and TLR5 were somewhat unexpected, as cell surface localized receptors are not typically considered to be capable of inducing IFN expression.
Figure 3. Type III IFNs are induced by all TLRs.
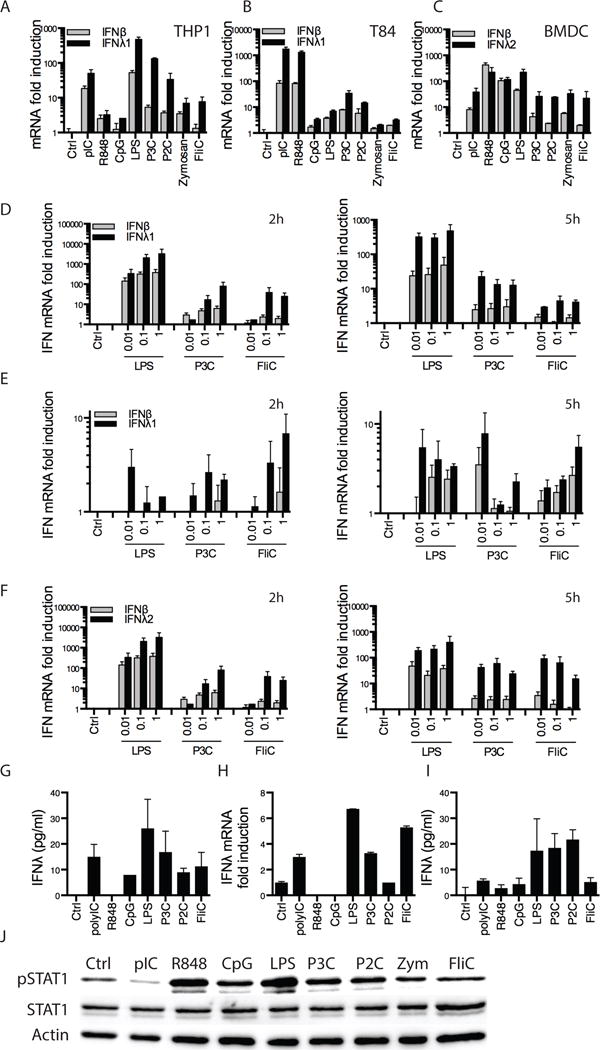
(A–C) PMA-differentiated THP1 macrophages (A), polarized T84 cells (B) and bone-marrow derived dendritic cells (BMDCs-C) were treated with the indicated ligands at the following concentrations: polyIC (pIC): 10 μg/ml, R848 0.1 μg/ml, CpG DNA 0.1 μg/ml, LPS 0.1 μg/ml, Pam3Cys (P3C): 0.01 μg/ml, Pam2Cys (P2C): 0.01 μg/ml, Zymosan 1 μg/ml, flagellin (FliC) 0.1 μg/ml for 5 h. As indicated, IFNβ, IFNλ1 or IFNλ2 mRNA expression was measured by RT-qPCR. (D–F) PMA-differentiated THP1 macrophages (D), polarized T84 cells (E) or BMDCs (F) were treated with the indicated concentrations of LPS, P3C or FliC. As indicated, IFNβ, IFNλ1 or IFNλ2 mRNA expression was measured by RT-qPCR. (G) Similar to (A) except IFNλ1 protein expression was measured by ELISA after 5h treatment. (H–I) Human blood-derived monocytes were treated with the indicated ligands at 0.1 μg/ml, except for pIC at 10 μg/ml, for 5 h. Cells were lyzed and IFNλ1 mRNA expression was measured by RT-qPCR (H) or cell culture supernatants were collected and IFNλ1 protein expression was measured by ELISA (I). (J) Cell culture supernatants from BMDCs challenged with ligands as in (C) were incubated onto human 293T cells. IFN activity was analyzed by western immunoblotting of phosphorylated STAT1 (pSTAT1). Total STAT1 and actin were used as loading controls. As mouse type I IFNs do not signal onto human cells but type III IFNs do (see Supplemental Fig. 2H), STAT1 phosphorylation was only due to IFNλ presence and activity. Data shown is from one experiment representative of 3 (J). Error bars represent mean ± SEM of triplicate readings for one experiment representative of 2 (A–C, H–I) or averages ± SEM of 3–6 independent experiments (D–G).
To further study IFN expression by cell surface TLRs, we stimulated different cell types with varying concentrations of LPS, P3C and flagellin for 2 or 5 hr. In THP1 cells (Fig. 3D), polarized T84 cells (Fig. 3E) and BMDCs (Fig. 3F), IFNλ expression in response to TLR2 or 5 stimulation was higher than IFNβ, especially at the later time point (Fig. 3D, F). IL6 or IL8 expression was also measured under the same conditions. As expected, these cytokines were induced by all ligands (Supplemental Fig. 2B–D). To rule out LPS contamination in our flagellin preparations, we compared IFN induction in TLR5 KO BMDCs. Baseline expression levels of all genes examined were comparable in WT and TLR5 KO cells (Supplemental Fig. 1H). flagellin-, but not LPS-, mediated induction of IFNs and cytokines was completely dependent on TLR5 (Supplemental Fig. 2E). In addition, treatment with the LPS inhibitor polymyxin B only affected LPS-mediated responses (Supplemental Fig. 2F). These results indicate that all TLRs are capable of inducing type III IFNs, and that TLR signaling from the plasma membrane favors the induction of type III IFN genes.
To determine if the amount of IFNλ expressed by cell surface TLRs was functional, we took three approaches. We first examined the release of IFNλ protein from THP1 cells stimulated with various TLR ligands (Fig. 3G). We found that all TLRs ligands, including cell-surface TLRs, caused the secretion of IFNλ, as detected by ELISA (Fig. 3G). In addition, we monitored IFNλ protein secretion from primary human peripheral blood-derived monocytes stimulated with the same TLR ligands (Fig. 3H, I). Again, treatment with ligands against all TLRs caused the production of IFNλ (Fig. 3H, I). In fact, type III IFN production was stronger upon stimulation of TLR4 and TLR2 than of all endosome-localized TLRs. Second, we analyzed the phosphorylation of STAT1 in T84 cells treated with various TLR ligands (Supplemental Fig. 2G). We found that STAT1 was phosphorylated following P3C or flagellin treatment to levels at least comparable to LPS or pIC (Supplemental Fig. 2G). We also found that the IFN-stimulated gene viperin was induced by treatment with all TLRs (Supplemental Fig. 2G). These data demonstrate that functional IFN proteins were produced upon TLR activation. To specifically investigate type III IFN signaling, we analyzed JAK2 phosphorylation, as we and others had previously demonstrated that JAK2 regulates the type III IFN signaling pathway (5, 39). Interestingly, JAK2 phosphorylation was induced by P3C and flagellin to levels comparable to LPS (Supplemental Fig. 2G). Third, we took advantage of our finding that mouse IFNλ2 (but not IFNβ, IFNα or IFNγ) can signal in human cells (5) and (Supplemental Fig. 2H). Thus, we treated human 293T cells with cell culture supernatants from mouse BMDCs that had been challenged with TLR ligands. Bioactivity of mouse IFNs was measured by western analysis of STAT1 phosphorylation (Fig. 3J). Densitometric analyses were performed using Image J (Supplemental Fig. 2I). Interestingly, we found that stimulation with bacterial PAMPs that activate cell-surface TLRs led to the release of bioactive type III IFNs that induced STAT1 phosphorylation to levels comparable to that obtained with endosomal TLRs (Fig. 3J). Collectively, these data establish that a range of plasma membrane and endosomal TLRs induce IFNλ gene expression and induce the release of bioactive IFNλ proteins.
TLR5 preferentially induces type III IFNs
Our results have thus far indicated that flagellin was able to upregulate IFNλ (Fig. 3). To analyze this observation further, we used flagellin mutant Salmonella (fliC KO). We infected polarized T84 cells (Fig. 4A) and BMDCs (Fig. 4B) with WT and fliC KO Salmonella. Interestingly, despite the fact that Salmonella encodes many PAMPs, IFNλ induction during infection with this bacterium was dependent on TLR5, as fliC KO Salmonella induced marginal amounts of type III IFNs (Fig. 4A, B). Similar data were obtained in THP1 cells, by monitoring IFNλ1 secretion (Fig. 4C). As a complementary experiment, we compared the ability of WT and TLR5 KO BMDCs to respond to WT Salmonella and Shigella. IL6 induction was independent of TLR5, while some IFNβ was induced at slightly lower levels in TLR5 KO cells (Fig. 4D). Notably, IFNλ induction was almost completely TLR5-dependent (Fig. 4D). These data further demonstrate the ability of TLR5 to induce type III IFNs, and suggest that TLR5 is an important contributor of IFNλ expression in infections with gram-negative pathogens.
Figure 4. Type III IFNs are preferentially induced by TLR5.
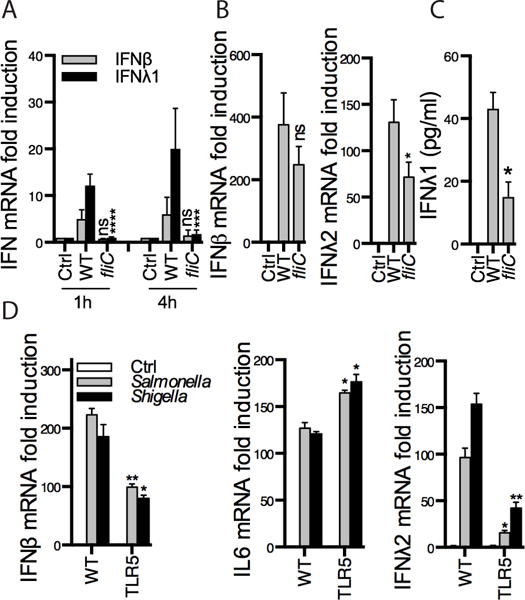
Polarized T84 cells (A) or BMDCs (B) were infected with WT or fliC mutant bacteria (lacking flagellin) for the time points indicated (A) or for 2 h (B). (C) PMA-activated THP1 cells were infected with the same bacterial strains for 5h, IFNλ1 protein expression was measured by ELISA. (D) WT or TLR5 KO BMDCs were infected with the indicated bacterial strains for 2 h. Where indicated, expression of the indicated genes was measured by RT-qPCR. Panels represent averages ± SEM of 2–4 independent experiments (A–C) or are representative of 2 independent experiments (D). Statistics were performed between WT and fliC mutant data (A–C) or WT and TLR5 KO (D). *P < 0.05; **P < 0.01; ***P < 0.001 ****P < 0.0001 (two-way ANOVA). ns, not significant.
Type III IFN induction by TLRs depends on the myddosome components MyD88 and IRAK4
We next sought to determine the signaling pathways that differentially drive type I and III IFN production in response to TLR activation. We first focused on TLR4, as we found it induces IFNs of both families (Fig. 3). In the TLR4 pathway, genes encoding inflammatory cytokines are induced by myddosome component MyD88 (40). However, TLR4 also activates a myddosome-independent pathway that is required for TLR4-induced type I IFN expression (41). We sought to determine the role of the myddosome in TLR4-dependent IFNλ expression. We therefore examined murine BMDCs that were genetically deficient for MyD88 (Fig. 5A). We found that LPS-induced IFNβ expression was MyD88-independent while IL6 expression was MyD88-dependent. These findings are consistent with the work of Akira and colleagues, who generated MyD88-deficient mice (42). Interestingly, we observed a requirement of MyD88 for LPS-induced IFNλ expression (Fig. 5A). To corroborate these findings, similar stimulations were performed in THP1 monocytes lacking MyD88. MyD88-deficient THP1 cells retained the ability to express IFNβ. In contrast, IFNλ expression in response to LPS was reduced in the absence of MyD88 (Fig. 5B). In addition, the myddosome components IRAK2 and IRAK4 were not required for IFNβ expression in THP1 cells, while expression of the inflammatory chemokine IL8 was dependent on these kinases (Fig. 5C). Importantly, IFNλ expression in these cells was IRAK2- and IRAK4-dependent (Fig. 5C). Baseline levels of IFN and IL8 transcripts are shown in Supplemental Fig. 1I. The small differences we observed in IFNλ baseline levels did not affect the effects we observed upon cell stimulation. Finally, IRAK4 was required for LPS-induced IFNλ secretion, as assessed by ELISA in THP1 cells (Fig. 5D). Collectively, these studies in human and mouse cells reveal a fundamental difference between the signaling pathways used by TLR4 to promote type I and III IFN expression, with IFNλ (but not IFNβ) expression depending on the myddosome.
Figure 5. TLR induction of type III IFNs is MyD88-dependent.
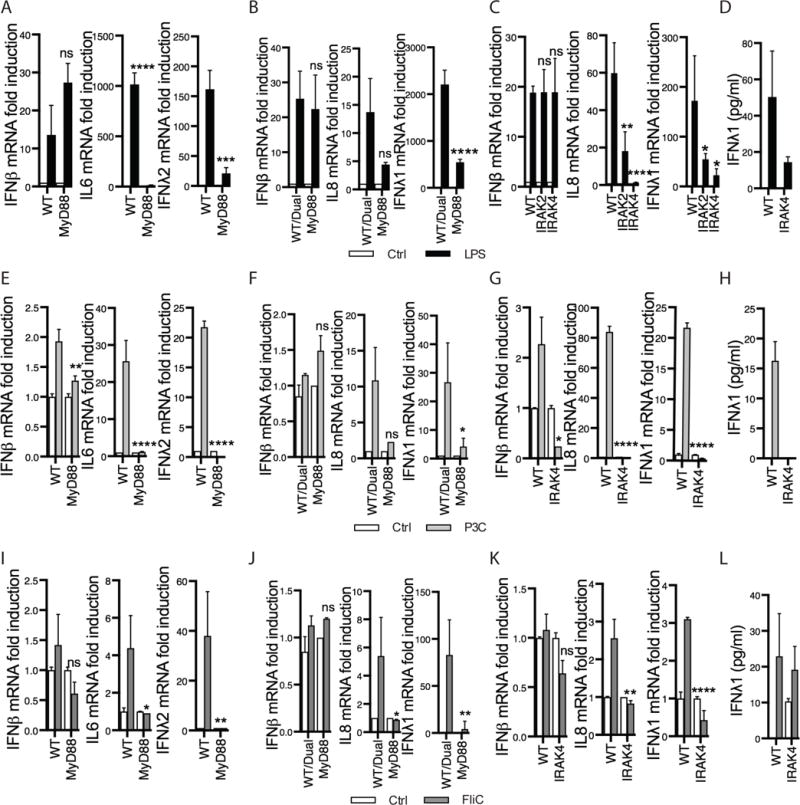
(A, E, I) WT and MyD88 KO bone-marrow derived dendritic cells (BMDCs) were challenged with 0.01 μg/ml LPS (A), Pam3Cys (P3C, E) or flagellin (FliC, I) for 5 h. (B,F,J) WT/Dual or MyD88 KO PMA-differentiated THP1 macrophages were treated with 0.1 μg/ml LPS (B), P3C (F) or FliC (J) for 5 h. (C,G,K) WT, IRAK2 or IRAK 4 KO PMA-differentiated THP1 macrophages were treated with ligands as in B,F,J. Expression of the indicated genes was measured by RT-qPCR. (D,H,L) IFNλ1 protein expression was measured by ELISA following 5 h 0.1 μg/ml LPS, P3C or FliC treatment of WT or IRAK4 KO PMA-differentiated THP1 cells. Each panel represents averages ± SEM of 3–6 independent experiments. Statistics were performed between WT and KO cell lines for each condition. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (two-way ANOVA). ns, not significant.
To determine if other TLRs also require the myddosome components to promote type III IFN expression, we examined the requirement of MyD88 on the ability of TLR2 and 5 to induce IFNλ expression. We found that MyD88 was required for the induction of IFNλ in response to P3C and flagellin in BMDCs and THP1 cells (Fig. 5E–F, I–J). As expected, IL6/IL8 induction in response to both ligands was MyD88-dependent. Interestingly, the low induction of IFNβ observed in response to these ligands also required MyD88. Similar findings were obtained when we examined IRAK2 and IRAK4 deficient THP1 monocytes (Fig. 5G, K), and IFNλ secretion upon P3C or flagellin treatment was also IRAK dependent (Fig. 5H, L). Overall, these findings likely explain how a broad range of TLRs can induce type III IFN expression, as IFNλ is induced by the signaling hub most commonly used by TLRs—the myddosome.
TLR4 induces type III IFNs from the cell surface
Although TLR4 detects LPS at the plasma membrane, and induces myddosome-signaling from this location, type I IFN induction does not occur until TLR4 reaches endosomes (43). In fact, no receptor is known to induce the expression of any IFN gene from the plasma membrane. Our finding that TLR4-induced IFNλ expression depends on the myddosome therefore raised the possibility that the signals inducing this gene emanate from the cell surface. This possibility was examined by preventing endocytosis with Dynole, a chemical inhibitor of dynamin family GTPases (44). Baseline levels of all genes in BMDCs and T84 epithelial cells were not affected by Dynole treatment (Supplemental Fig. 1J, K). As expected, Dynole treatment blocked the LPS-induced expression of IFNβ in BMDCs (Fig. 6A) and polarized T84 cells (Fig. 6B). Also as expected, the LPS-induced expression of the myddosome-dependent genes IL6 and IL8 was insensitive to endocytosis inhibition (Fig. 6A, B). Interestingly, IFNλ expression was not blocked by endocytosis inhibition (Fig. 6A, B). These data suggest that pharmacological inhibition of TLR4 endocytosis differentially affects type I and III IFN expression in response to LPS. To complement this analysis, a genetic approach was used by taking advantage of the fact that TLR4 endocytosis is dependent on CD14 (45). We therefore examined the requirement of CD14 for LPS-induced gene expression. CD14 KO cells had baseline levels of all genes comparable to WT cells (Supplemental Fig. 1L). CD14 was required for IFNβ, but not IL6, expression (Fig. 6C). Most notably, IFNλ expression was independent of CD14 (Fig. 6C). These collective data establish a unique aspect of type III IFNs, in that they can be induced by TLRs present at the plasma membrane (Fig. 6D).
Figure 6. Type III IFN expression by TLR4 does not require receptor endocytosis.
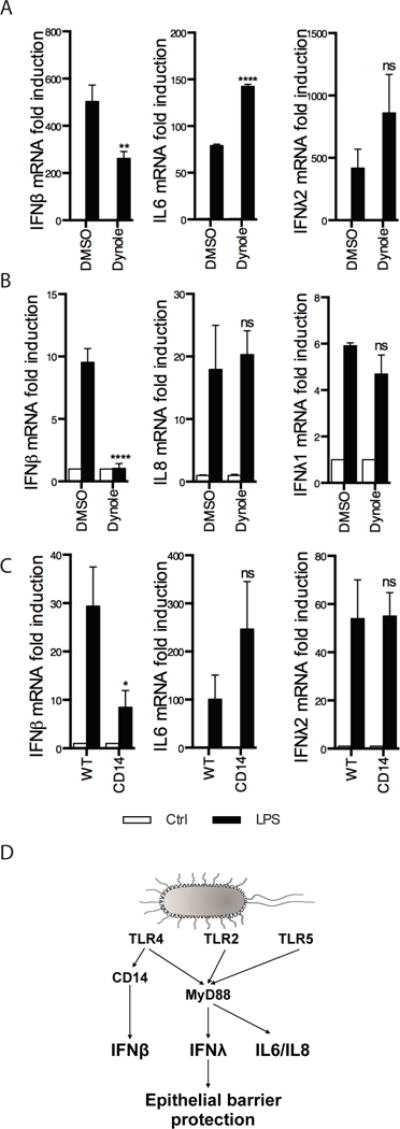
(A) Bone-marrow derived dendritic cells (BMDCs) were treated with the endocytosis inhibitor Dynole for 30 min followed by challenge with 1 μg/ml LPS for 5 h. (B) Similar to (A) except polarized T84 cells were used. (C) WT or CD14 KO BMDCs were treated with 1 μg/ml LPS for 5 h. Expression of the indicated genes was measured by RT-qPCR. Each panel represents averages ± SEM of 3 independent experiments. Statistics were performed between DMSO and Dynole (A, B) or WT and CD14 (C) LPS-treated samples. *P < 0.05; **P < 0.01;****P < 0.0001 (unpaired t-test). ns, not significant. (D) Model depicting differential regulation of IFNλ and IFNβ in response to TLR activation. TLR4, TLR2 and TLR5 activation from the cell surface induces IFNλs via the myddosome, while IFNβ is produced by TLR4 in a CD14-dependent manner.
Discussion
In this study, we describe an unexpected aspect of the TLR signaling pathways, in that these pathways are general inducers of type III IFNs during bacterial encounters. Much of the work on type III IFNs has focused on the regulation of their expression and functions during viral infections. However, several recent reports suggest a role for this IFN family in bacteria-host interactions as well. These reports include observations that some bacterial infections or LPS challenges induce type III IFN expression (8–10, 46). In addition, the bacterium Listeria monocytogenes encodes virulence factors that fine tunes the expression of IFNλ-responsive genes (11). The generality of these observations, and the means by which type III IFN gene expression is regulated by TLRs, has been unclear.
TLRs have been examined extensively for their abilities to induce cytokine, chemokine and type I IFN expression. The collective studies from several laboratories have led to a model whereby all TLRs are potent inducers of cytokine/chemokine expression, and that the signals that induce these genes can emanate from the plasma membrane or endosomes (14, 47). In contrast, a subset of TLRs are strong inducers of type I IFNs, and the expression of these genes can only be initiated by TLRs that are present on endosomes. This statement applies to TLRs that detect viral nucleic acids, which sense their ligands within endosomes, but also applies to TLR4. Indeed, TLR4 can detect bacterial LPS at the plasma membrane, but cannot induce the expression of type I IFN genes until after CD14-dependent endocytosis (43, 45, 48). Studies in recent years have extended this principle to TLR2, which induces the weak (but detectable) expression of type I IFN genes (25, 49–51). Like TLR4, TLR2 must be internalized into endosomes before type I IFN can be induced (25, 49–51). In considering this paradigm, our finding that type III IFN genes can be induced by TLR4 at the plasma membrane is notable, as it provides an intriguing exception to the apparent cell biological rule that IFNs cannot be induced from the plasma membrane.
Several lines of evidence support this conclusion. First, LPS-induced IFNλ expression is dependent on signaling proteins that TLR4 engages at the plasma membrane, such as the myddosome components MyD88, IRAK2 and IRAK4 (Fig. 6D). In contrast, LPS-induced IFNβ expression is mainly myddosome-independent. Second, LPS-induced IFNλ expression is not sensitive to the endocytosis inhibitor Dynole, whereas IFNβ expression is. Third, IFNλ expression induced by LPS proceeds normally in the absence of CD14, which is an essential regulator of TLR4 endocytosis. IFNβ expression, in contrast, is lost in the absence of CD14. Thus, at least in the case of TLR4, this receptor induces type I and III IFNs from different organelles. Our recent studies on virus-induced type III IFN expression led to a similar conclusion, but with different receptors and organelles (5). During viral infections, RLRs induce the expression of type I and III IFN genes. These receptors can signal from peroxisomes and mitochondria (52, 53). Signaling from mitochondria induces both sets of IFNs, whereas signaling from peroxisomes induces the expression of type III IFNs specifically. Thus, distinct receptor families in the innate immune system can separate the subcellular sites of type I and III IFN expression.
The aspects of IFNλ gene regulation described herein are reminiscent of those governing the expression of gene encoding inflammatory cytokines and chemokines, such as IL8 and IL6 (Fig. 6D). These aspects include the ability to be transcriptionally upregulated by all TLRs, the dependence on myddosome components and the ability to be induced from the plasma membrane. Unlike type I IFNs, which are most important for antiviral defense, inflammatory cytokines and chemokines are important to defend against any microbial infection. It therefore stands to reason that the common regulatory mechanisms governing the expression of cytokines, chemokines and type III IFNs reflect a function of IFNλs that extends beyond antiviral defense.
What could be the function of type III IFNs be in general antimicrobial defense? While this answer is likely to be context-dependent, our observation that recombinant IFNλ can reinforce the barrier functions of polarized T84 epithelial cells provides some clues. IFNλ treatment was sufficient to increase the intrinsic TEER of T84 cells, and prevent the barrier damage induced by pathogenic Salmonella and Shigella. This protection from epithelial damage resulted in a diminished number of bacteria that can pass into the basal chamber of our in vitro culture system. These findings complement recent findings that the IFNλ receptor is required to prevent virus-induced damage to the blood brain barrier (26) and that type III IFN signaling appears to have a protective role in colitis models (54). Thus, using in vitro and in vivo systems with viruses and bacteria, type III IFNs appear to play an important role of reinforcing epithelial and endothelial barriers. Based on the diverse functions of other immunomodulatory cytokines, it is likely that additional activities of type III IFNs exist. This study should provide a mandate to explore these possibilities in various infectious settings.
Supplementary Material
Acknowledgments
We thank members of the Kagan and Zanoni laboratories for helpful discussions, and in particular Dr Charles Rosadini for his instrumental help purifying mouse dendritic cells.
J.C.K. is supported by US National Institutes of Health grants AI093589, AI072955 and P30 DK34854. J.C.K. received an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. C.O. is supported by a King’s College London Prize Fellowship and a Sir Henry Dale Fellowship from the Royal Society and the Wellcome Trust (Grant number 206200/Z/17/Z).
References
- 1.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 2.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2002;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 3.Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, Fujita T. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 4.Mäkelä SM, Osterlund P, Julkunen I. TLR ligands induce synergistic interferon-β and interferon-λ1 gene expression in human monocyte-derived dendritic cells. Molecular immunology. 2011 doi: 10.1016/j.molimm.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, Boulant S, Gehrke L, Cossart P, Kagan JC. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15:717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathogens. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazear HM, Nice TJ, Diamond MS. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, Cella M, Lande R, Uzé G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 9.Thomson SJP, Goh FG, Banks H, Krausgruber T, Kotenko SV, Foxwell BMJ, Udalova IA. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci USA. 2009;106:11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bierne H, Travier L, Mahlakõiv T, Tailleux L, Subtil A, Lebreton A, Paliwal A, Gicquel B, Staeheli P, Lecuit M, Cossart P. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS ONE. 2012;7:e39080. doi: 10.1371/journal.pone.0039080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, Matteï P-J, Regnault B, Nahori M-A, Cabanes D, Gautreau A, Ait-Si-Ali S, Dessen A, Cossart P, Bierne H. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science. 2011;331:1319–1321. doi: 10.1126/science.1200120. [DOI] [PubMed] [Google Scholar]
- 12.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 13.Noppert SJ, Fitzgerald KA, Hertzog PJ. The role of type I interferons in TLR responses. Immunol Cell Biol. 2007;85:446–457. doi: 10.1038/sj.icb.7100099. [DOI] [PubMed] [Google Scholar]
- 14.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ank N, Iversen M, Bartholdy C. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. The Journal of. 2008 doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 16.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014;14:821–826. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, Robinson CV, Latz E, Gay NJ. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. Journal of Biological Chemistry. 2009;284:25404–25411. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-likereceptor signalling complexes. Nat Rev Immunol. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 21.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell. 2014;156:705–716. doi: 10.1016/j.cell.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NJ Gay, Gangloff M, O’Neill LAJ. What the Myddosome structure tells us about the initiation of innate immunity. Trends in Immunology. 2011;32:104–109. doi: 10.1016/j.it.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003 doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 25.Nilsen NJ, Vladimer GI, Stenvik J, Orning MPA, Zeid-Kilani MV, Bugge M, Bergstroem B, Conlon J, Husebye H, Hise AG, Fitzgerald KA, Espevik T, Lien E. A Role for the Adaptor Proteins TRAM and TRIF in Toll-Like Receptor 2 Signaling. Journal of Biological Chemistry. 2014;290:3209–3222. doi: 10.1074/jbc.M114.593426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M, Klein RS, Diamond MS. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med. 2015;7:284ra59–284ra59. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lencer WI, Delp C, Neutra MR, Madara JL. Mechanism of cholera toxin action on a polarized human intestinal epithelial cell line: role of vesicular traffic. J Cell Biol. 1992;117:1197–1209. doi: 10.1083/jcb.117.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 29.Köhler H, Sakaguchi T, Hurley BP, Kase BJ, Reinecker HC, McCormick BA. Salmonella enterica serovar Typhimurium regulates intercellular junction proteins and facilitates transepithelial neutrophil and bacterial passage. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2007;293:G178–G187. doi: 10.1152/ajpgi.00535.2006. [DOI] [PubMed] [Google Scholar]
- 30.Hallstrom K, McCormick BA. Salmonella Interaction with and Passage through the Intestinal Mucosa: Through the Lens of the Organism. Front Microbiol. 2011;2:1–10. doi: 10.3389/fmicb.2011.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Betts J, Finlay BB. Identification of Salmonella typhimurium invasiveness loci. Can J Microbiol. 1992;38:852–857. doi: 10.1139/m92-138. [DOI] [PubMed] [Google Scholar]
- 32.Mills DM, Bajaj V, Lee CA. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Molecular Microbiology. 1995;15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 33.Galan JE, Curtiss R. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerlach RG, Cláudio N, Rohde M, Jäckel D, Wagner C, Hensel M. Cooperation of Salmonellapathogenicity islands 1 and 4 is required to breach epithelial barriers. Cell Microbiol. 2008;10:2364–2376. doi: 10.1111/j.1462-5822.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- 35.Sakaguchi T, Köhler H, Gu X, McCormick BA, Reinecker HC. Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol. 2002;4:367–381. doi: 10.1046/j.1462-5822.2002.00197.x. [DOI] [PubMed] [Google Scholar]
- 36.Kotenko SV. IFN-λs. Current Opinion in Immunology. 2011 doi: 10.1016/j.coi.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lasfar A, Lewis-Antes A, Smirnov SV, Anantha S, Abushahba W, Tian B, Reuhl K, Dickensheets H, Sheikh F, Donnelly RP, Raveche E, Kotenko SV. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 2006;66:4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Li N, Oh KS, Dutta B, Vayttaden SJ, Lin B, Ebert TS, De Nardo D, Davis J, Bagirzadeh R, Lounsbury NW, Pasare C, Latz E, Hornung V, Fraser IDC. Comprehensive RNAi-based screening of human and mouse TLR pathways identifies species-specific preferences in signaling protein use. Sci Signal. 2016;9:ra3–ra3. doi: 10.1126/scisignal.aab2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SJ, Kim WJ, Moon SK. Role of the p38 MAPK signaling pathway in mediating interleukin-28A-induced migration of UMUC-3 cells. Int J Mol Med. 2012;30:945–952. doi: 10.3892/ijmm.2012.1064. [DOI] [PubMed] [Google Scholar]
- 40.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 42.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-Deficient Mice to Endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 43.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson MJ, Deane FM, Robinson PJ, McCluskey A. Synthesis of Dynole 34-2, Dynole 2-24 and Dyngo 4a for investigating dynamin GTPase. Nat Protoc. 2014;9:851–870. doi: 10.1038/nprot.2014.046. [DOI] [PubMed] [Google Scholar]
- 45.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krupna-Gaylord MA, Liveris D, Love AC, Wormser GP, Schwartz I, Petzke MM. Induction of Type I and Type III Interferons by Borrelia burgdorferi Correlates with Pathogenesis and Requires Linear Plasmid 36. PLoS ONE. 2014;9:e100174. doi: 10.1371/journal.pone.0100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 48.Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS. 2008;368:94–99. doi: 10.1016/j.bbrc.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 49.Stack J, Doyle SL, Connolly DJ, Reinert LS, O’Keeffe KM, McLoughlin RM, Paludan SR, Bowie AG. TRAM Is Required for TLR2 Endosomal Signaling to Type I IFN Induction. The Journal of Immunology. 2014;193:6090–6102. doi: 10.4049/jimmunol.1401605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS ONE. 2010;5:e10250. doi: 10.1371/journal.pone.0010250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands : Abstract : Nature Immunology. Nat Immunol. 2009 doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 53.Dixit E, Boulant S, Zhang Y, Lee ASY, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rauch I, Rosebrock F, Hainzl E, Heider S, Majoros A, Wienerroither S, Strobl B, Stockinger S, Kenner L, Müller M, Decker T. Noncanonical Effects of IRF9 in Intestinal Inflammation: More than Type I and Type III Interferons. Mol Cell Biol. 2015;35:2332–2343. doi: 10.1128/MCB.01498-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.