

Abstract
Transposons can be used to easily generate and label the location of mutations throughout bacterial and other genomes. Transposon insertion mutants may be screened for a phenotype as individual isolates, or by selection applied to a pool of thousands of mutants. Identifying the location of a transposon insertion is critical for connecting phenotype to the genetic lesion. In this unit, we present an easy and detailed approach for mapping transposon insertion sites using arbitrarily-primed PCR (AP-PCR). Two rounds of PCR are used to i) amplify DNA spanning the transposon insertion junction, and ii) increase the specific yield of transposon insertion junction fragments for sequence analysis. The resulting sequence is mapped to a bacterial genome to identify the site of transposon insertion. In this protocol, AP-PCR as it is routinely used to map sites of transposon insertion within Staphylococcus aureus, is used to illustrate the principle. Guidelines are provided for adapting this protocol for mapping insertions in other bacterial genomes. Mapping transposon insertions using this method is typically achieved in 2–3 days if starting from a culture of the transposon insertion mutant.
Keywords: Transposon, Arbitrarily Primed PCR, Anchored PCR, Gene-walking PCR
INTRODUCTION
Transposons are mobile genetic elements (MGE) that generally insert into new locations in the genome by a replicative process, where the original element is replicated, and an associated transposase mediates insertion of the copy into a new site. In addition to one or more genes for transposition, transposons often encode additional functions such as antibiotic resistance. Transposons are widespread throughout the branches of life, from prokaryotes (Kleckner, 1981), to archaea (Kapitonov et al., 2016) and eukaryotes (Burns and Boeke, 2012; Daboussi and Capy, 2003; McClintock). Techniques have evolved for the facile use of transposons as mutagenic agents in prokaryotic and eukaryotic organisms, allowing the ready generation of pools or libraries of strains with various individual transposon insertion mutations.
The power of transposon mutagenesis lies in the ability to insert a selectable and identifiable marker, such as an antibiotic resistance gene, at many locations in a bacterial chromosome. The marker allows for positive selection of insertion events, and also provides a known sequence tag to map the site of insertion. These attributes have led to the widespread use of transposons for high-throughput identification of genes involved, for example, in global transcriptional regulation (Hoskisson et al., 2006; Lauro et al., 2008; Luong et al., 2003; O’Toole et al., 2000; Peterson et al., 2006; Tsou et al., 2011; Worley et al., 2000), colonization of a host (Brooks et al., 2014; Chiang and Mekalanos, 1998; Dziva et al., 2004; Fuller et al., 2000; Goodman et al., 2004; Hava and Camilli, 2002; Kavermann et al., 2003; Morgan et al., 2004), biofilm formation (Ballering et al., 2009; Loo et al., 2000; O’Toole et al., 2000; Watnick and Kolter, 1999), and motility (Brennan et al., 2013; Graf et al., 1994; Hendrixson et al., 2001). Useful strategies that exploit the power of transposon-mediated mutagenesis have involved arrayed transposon mutant libraries (Cameron et al., 2008; Fernandez-Martinez et al., 2011; Hutchison et al., 1999; Liberati et al., 2006; Lin et al., 2012; Salama et al., 2004), signature-tagged mutagenesis (STEM) (Mazurkiewicz et al., 2006), transposon-insertion sequencing (Goodman et al., 2009; Goodman et al., 2011; van Opijnen et al., 2009), and recombination-based in vivo expression technology (RIVET) (Lee et al., 2001).
Because of the large diversity of potential insertion sites and the depth of understanding of its transposition mechanism, derivatives of the Escherichia coli Tn5 (Reznikoff, 1993) or the Drosophila melanogaster mariner (Hartl, 2001) transposons have been most widely applied to mutagenize prokaryotic genomes. Both transposons integrate mainly as single copies into the target chromosome, usually resulting in loss of function of an interrupted gene, sometimes with polar effects on co-transcribed or adjacent genes due to the introduction of transcription termination sites and other changes to the local genetic environment. Depending on the nature of the element, transposon insertion may also introduce novel promoter elements. The range of sites for transposon integration is dictated by the transposition mechanism (Craig, 1997). For example, insertion of Tn5 shows a bias toward methylated DNA, which is often enriched around the origin during chromosomal replication. Similarly, mariner inserts into TA sites (Plasterk et al., 1999). Although the range of targets for Tn5 and mariner derivatives in most bacterial genomes is very large, there may be value to using multiple transposons to achieve maximum mutagenesis of target genomes.
Connecting a phenotype of interest to changes in the function of a gene first requires localization of the site of transposon insertion within the bacterial genome. In this unit, we describe a simple and highly adaptable method called Arbitrarily-Primed PCR (AP-PCR) to map the sites of transposon integration. The general concept is that a short region of chromosomal sequence may be amplified by pairing a transposon-specific primer with a random oligonucleotide that serves to prime flanking DNA for amplification. This typically involves two rounds of PCR (Figure 1). Round 1 is used to amplify and enrich the DNA in double-stranded products produced from the priming that occurs at low stringency with a subset of the random oligonucleotides at one end, and a transposon-specific primer at the other (Figure 1a). Because of the low stringency, the relative abundance of the transposon junction fragment of interest is usually too low to be readily detected. Therefore, Round 2 of amplification involves a second transposon-specific primer, further enriching the population of amplified molecules for those containing the transposon insertion junction (Figure 1b). To identify the site of transposon integration, one or more major products of the AP-PCR amplification are sequenced, and the junction of the transposon and the chromosome is mapped to the genome sequence of the target organism (Figure 1c). We present a general protocol for mapping transposon insertion sites using sequences derived from AP-PCR, and discuss approaches to optimize and troubleshoot this protocol for organisms of interest. We also provide a supporting protocol for isolating genomic DNA. Using this protocol, sequences are routinely obtained and mapped in 2–3 days if starting from a culture of the strains of interest.
Figure 1.
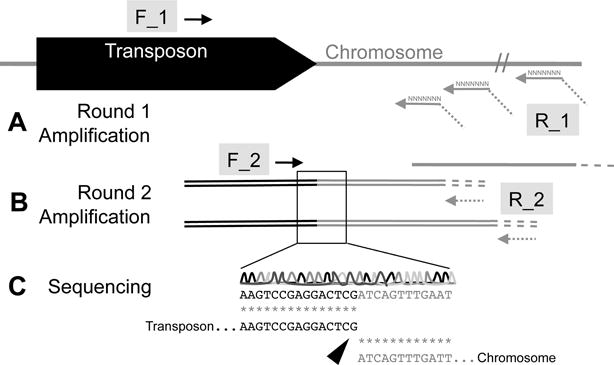
Mapping transposon-insertion sites with sequence generated by AP-PCR. A) During Round 1 of amplification, the transposon-specific F_1 primer is paired with an arbitrary primer R_1 to amplify double-stranded regions reading from the transposon into the site of chromosomal insertion. In addition to double-stranded DNA products, single-stranded DNA products may be primed from the pool of arbitrary R_1 sequences in regions of the chromosome distal from the transposon. At the end of the R_1 primers is a sequence of oligonucleotides that will prime R_2 in subsequent rounds of amplification. B) Following purification to remove single-stranded DNA and Round 1 primers, a Round 2 of amplification is performed. Primer F_1, which is nested within the region of the transposon amplified by F_1/R_1, pairs with primer R_2 to enrich for double-stranded DNA products containing transposon-chromosome sequence. C) These sequences are gel-purified, and the nucleotide sequence is obtained by the Sanger method. Alignment of the sequencing products with the transposon sequence is performed to identify the transposon. The remaining sequence is used to query the whole genome sequence of the mutagenized bacterial strain by BLAST. Alignment of the query to a unique, identical target defines the site of transposon insertion.
BASIC PROTOCOL: Mapping transposon insertion sites using AP-PCR
This protocol provides a general method that involves two rounds of PCR to identify the insertion site of a transposon mutation.
Materials
Q5 High-Fidelity DNA Polymerase (New England Biolabs, cat. no. M0491L)
dNTP Mix (New England Biolabs, cat. no. N0447S)
DNase-free Ultra Pure Water (VWR cat. no. 10128-466)
Oligonucleotide Primers (IDT)
- Qiaquick PCR Purification Kit (Qiagen, cat. no. 28106) containing:
- DNeasy mini spin columns in 2-mL collections tubes
- Buffer PB
- Buffer PE
- pH Indicator
- Buffer EB
ExoSAP-IT (NEB, cat. no. 95026-710)
Agarose (VWR, cat. no. VW1468-07)
100 bp DNA ladder (NEB, cat. co. N3231L)
- Components for TBE electrophoresis running buffer:
- Tris Base (VWR, cat. no. 71003-490)
- Boric Acid (VWR, cat. no. BDH9222-500G)
- EDTA pH 8.0 (VWR, cat. no. BDH9232-500G)
Ethidium Bromide (VWR, cat. no. 97064-602)
- QIAquick Gel Extraction Kit (Qiagen, cat. no. 28706) containing:
- DNeasy mini spin columns in 2 mL collections tubes
- Buffer QG
- Buffer PE
- Buffer EB
Isopropyl Alcohol (VWR, cat. no. BDH1174-4LP)
37°C Incubator
Tabletop Centrifuge
37°C, 56°C Water Baths
PCR 8-Well Strip Tubes (VWR, cat. no. 82006-606)
Low-Speed Centrifuge with PCR-Tube Adaptor
Thermal Cycler
Nanodrop Instrument
UV Trans-Illuminator
Electrophoresis Gel Tray
CAUTION Ethidium bromide is toxic and potentially mutagenic. Wearing gloves and working with this chemical in designated areas, and safe disposal of waste as specified by your institutional chemical safety guidelines are advised.
CAUTION UV, especially shorter wavelengths below 365 nm, can damage the eye, and exposure is carcinogenic for the skin. Wear proper protective equipment when visualizing ethidium-bromide stained agarose gels using UV illumination.
Round 1 PCR Amplification
-
1Select PCR primers (see Table 1) to amplify the junction of the transposon and flanking chromosomal sequence.
- Forward: primer specific to transposon
- Reverse: 35mer containing i) a primer anchor for Round 2 amplification (GGCCACGCGTCGACTAGTCA), a 10 bp random sequence, and an arbitrary pentameric sequence ending with a 3′ GC anchor
-
2
Prepare PCR reactions (total volume of 50 μl) in a PCR strip-tube as follows:
DNA 20 ng (or 5 μl of cell lysate [see annotation]) dNTP (10mM) 1 μl Forward Primer 1 (10uM) 1 μl Reverse Primer 1 (10uM) 1 μl 5X Q5 Reaction Buffer 10 μl Q5 HF DNA Polymerase 0.5 μl Nuclease-Free Water to 50 μl Mix reagents and briefly centrifuge with a PCR tube adaptor at low speed.
Optimal priming is achieved using purified genomic DNA at the indicated concentration. For many bacteria, a suitably prepared cell lysate may be used as template (see supporting protocol). Alternative DNA polymerases (such as Promega GoTaq, NEB Taq) may be used with appropriate optimized buffers for those enzymes.
-
3
For Round 1 of amplification, program a thermocycler as follows:
Initial denaturation: Denature: 5 min 95°C 6 cycles: Denature: 30 sec 95°C Anneal: 30 sec 30°C Extend: 60 sec 72°C 30 cycles: Denature: 30 sec 95°C Anneal: 30 sec 45°C Extend: 120 sec 72°C Final extension: Extend: 4 min 72°C Hold: ∞ 12°C Denaturation and extension temperatures may need to be varied for other enzymes.
-
4
To verify amplification, separate 10 μl of the reaction product through 1% agarose in 0.5X TBE. Discrete bands of DNA should be visible (Figure 2a).
-
5
Purify amplified DNA using a QIAquick PCR Purification Kit, according to the manufacturer’s instructions. Determine concentration and purity of DNA using a spectrophotometer, such as a Nanodrop.
Separation of amplified DNA from unincorporated primers is important for successful Round 2 amplification. An alternative approach is to treat PCR products with a single-strand exonuclease, such as ExoSAP-It (Affymetrix), to remove Round 1 primers from the template.
Table 1.
Primer sequences
| Target | Primer | Name | Sequence (5′ to 3′)a | Tmb | Sourcec |
|---|---|---|---|---|---|
| Transposon | |||||
| Tn5 | 1_F | Tn5ext | GAACGTTACCATGTTAGGAGGTC | 55 | 1 |
| 2_F | Tn5int | CGGGAAAGGTTCCGTTCAGGACGC | 64 | ||
| Tn10d(Cm) | 1_F | CmExt | CAGGCTCTCCCCGTGGAGG | 63 | 2 |
| 2_F | CmInt | CTGCCTCCCAGAGCCTG | 58 | ||
| Tn10d(Kn) | 1_F | KnExt | CCGCGGTGGAGCTCC | 60 | 3 |
| 2_F | KnInt | ATGACAAGATGTGTATCCACC | 52 | ||
| Tn917(Em) | 1_F | MartnF | TTTATGGTACCATTCATTTTCCTGCTTTTTC | 57 | 4 |
| 2_F | MjmF | CCAAAATCCGTTCCTTTTTCATAGTTCCTATATAGTTATACGC | 61 | – | |
| Tn4351(Em) | 1_F | Tet-1 | TGTCGTAGTTGCTCTGTCGGGTAA | 60 | 5 |
| 2_F | 1030 | TAGCAAACTTTATCCATTCAG | 48 | ||
| Tn551(Em) | 1_F | Erm5.3 | TCTACATTACGCATTTGGAATAC | 51 | 6 |
| 2_F | Erm5.2 | AGATAATGCACTATCAACACACTC | 53 | ||
| Chromosome | |||||
| Any | 1_R | ARB1 | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT | – | 7 |
| 1_R | ARB6 | GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC | – | ||
| 2_R | ARB2 | GGCCACGCGTCGACTAGTAC | 60 |
Bold residues indicate the Round 2 reverse product. Underlined residues indicate the 3′ pentamer.
Tm, primer melting temperature in °C. Estimated in silico.
Sources as follows: 1, (Simon et al., 1989); 2, (Kleckner et al., 1991); 3, (Alexeyev and Shokolenko, 1995); 4,(Bae et al., 2008; Knobloch et al., 2003); 5, (Chen et al., 1999); 6, Li et al., 2009); 7, (O’Toole et al., 1999)
Figure 2.

Typical results of Round 1 and Round 2 AP-PCR amplifications. A) Following amplification of genomic DNA or lysate with Round 1 PCR primers, individual bands should be visible. Depending on the template and polymerase used, these bands may be faint. B) Following amplification of purified Round 1 PCR amplicons, a small number of amplicons should become brighter. Amplicons that are between the 200 bp and 500 bp standards (boxed) are suitable for PCR purification and sequencing. Amplicons that were chosen to sequence from this gel are indicated by >. L, ladder; 1–3 isolates; bp, base pair.
Round 2 Nested PCR Amplification
-
6
Select Round 2 primers, which include a new transposon-specific primer, and a second primer containing only the 20mer anchor sequence of the Round 1 arbitrary primer (Table 1).
-
7
Nested amplification of purified DNA from Round 1 PCR. Prepare PCR mix (total volume of 50 μl) as follows:
Template DNA 40 ng (or 1 μl of PCR product, treated with ExoSap according to the manufacturer’s instructions) dNTP (10mM) 1 μl Primer F_2 (10 μM) 1 μl Primer R_2 (10 μM) 1 μl 5X Q5 Reaction Buffer 10 μl Q5 HF DNA Polymerase 0.5 μl Nuclease-Free Water to 50 μl -
8
Run the following PCR Program:
Initial denaturation: Denature: 1 min 95°C 30 cycles: Denature: 30 sec 95°C Anneal: 30 sec X°Ca Extend: 120 sec 72°C Final extension: Extend: 4 min 72°C Hold: ∞ 12°C The denaturation and extension temperatures are specific to the polymerase chosen.
aThe annealing temperature is specific to the 2_F primer chosen (Table 1). -
9
Separate 10 μl of PCR product by size on a 1% Agarose Gel, with a 100 bp ladder (Figure 2b). If a single band is present, purify this PCR product as described in step 5. If multiple bands are present, excise the brightest two bands from each sample that are between 200 bp and 1 Kb in size, and purify the DNA using a QIAquick Gel Extraction Kit according to the manufacturer’s instructions. Determine concentration and purity of PCR products by Nanodrop spectrophotometry.
If multiple bands occur following Round 2 PCR amplification, it may be necessary to load more of the PCR product onto the gel to obtain sufficient quantities of excised DNA for sequencing. Sequencing multiple bands is often useful for revealing multiple sites of transposon insertion or for assuring that the plasmid vector (if applicable) is no longer present.
-
10
Prepare one or two Round 2 products for sequence determination. The forward primer from Round 2 amplification is used to prime the Sanger sequencing reaction (Table 1).
Sanger sequencing is readily available commercially and at DNA sequencing Core facilities. If performing sequencing in-house, follow sequencing facility protocols.
Mapping transposon insertion sites
-
11
Identify transposon-specific sequence by alignment with the nucleotide sequence of the transposon (Figure 1c). Alignment can be performed using a publicly available algorithm, such as the multiple sequence alignment tool MUSCLE (Edgar, 2004; Li et al., 2015; McWilliam et al., 2013).
-
12
Using a sequence editor, such as commercially available Geneious or open-source BioPython, remove the transposon-specific sequence so that only the sequence specific to the chromosome remains.
-
13
Trim the 3′ end of the sequence by removing any sequence containing >20% ambiguous bases. Usually, 50–300 nucleotides of sequence remain for use in probing databases for gene identity.
-
14
Where possible, create a BLAST directory of the annotated genome in which the transposon mutagenesis was performed. Also include the full sequence of any transposon-carrying plasmid or delivery vector used for mutagenesis.
The genome used for BLAST comparison is ideally isogenic to the strain background that has been mutagenized. Comparison to a conspecific genome may introduce ambiguity into insertion site determination.
-
15
Search this genome with the trimmed sequence query by performing nucleotide BLAST to locate the site of transposon insertion. Check the alignment score of the top hit, and ensure that the match is at least 90% identical (Figure 1c).
Depending on the polymerase used, the sequenced Round 2 products may contain a few polymorphisms. Therefore, correctly mapped hits are not always 100% identical to the target sequence.
-
16
Record the position where the first nucleotide of the query aligns to the target chromosome. This is the site of transposon insertion. In associating transposon location with a phenotype, it is important to consider the genetic environment, including potentially co-transcribed adjacent genes, because of the prospect of polar effects of the transposon insertion.
SUPPORT PROTOCOL: Isolation of bacterial DNA for AP-PCR
This supporting protocol describes how to isolate Staphylococcus aureus genomic DNA for use with the basic protocol for AP-PCR. Methods are described for extending the protocol for the isolation of DNA from other bacterial species.
Materials
Brain Heart Infusion (BHI) Broth (VWR, cat. no. 90003-032)
Technical Agar (VWR, cat. no. 90004-030)
Erythromycin (VWR, cat. no. TCE0751-25G)
Petri Dishes (VWR, cat. no. 25384-302)
Inoculating Loops (VWR, cat. no. 12000-810)
Sterile Culture Tubes (VWR, cat. no. 60818-689)
1.7 mL Centrifuge Tubes (VWR, cat. no. 87003-294)
- DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504) containing:
- DNeasy mini spin columns in 2-mL collections tubes
- 2-mL collection tubes
- Buffer ATL
- Buffer AL
- Buffer AW1
- Buffer AW2
- Buffer AE
- Proteinase K
Lysozyme (VWR, cat. no. AAJ60701-14)
Lysostaphin (Sigma-Aldrich, cat. no. L7386-5MG)
Dehydrated Alcohol, 200 Proof (VWR, cat. no. 89085-244)
Tris-EDTA pH 8.0 (VWR, cat. no. 97062-626)
CAUTION Toxin-expressing S. aureus is a select agent. Observe appropriate containment measures.
Isolation of genomic DNA
Growth of strains containing putative transposon insertions
-
1
Revive the strain of interest from frozen stocks by streaking a small amount of material onto BHI- or other nutrient agar, including antibiotic selection for the transposon, and incubate overnight at 37°C to obtain isolated colonies.
-
2
The next day, start a 2 – 5 mL culture of each strain of interest in BHI or nutrient broth with the corresponding antibiotic for transposon selection. Incubate cultures overnight at 37°C on a shaker set to 225 rpm to obtain approximately 109 CFU/ml.
These conditions can be modified to cultivate the organism of interest. It is important to start liquid cultures from single colonies, and to grow under appropriate antibiotic selection to insure the presence of the transposon.
Extraction of genomic DNA
Refer to the literature for optimized DNA isolation protocols for your bacterium, as methods for lysis vary according to the nature of the cell wall. For Gram-negative organisms, a small amount of overnight culture (~5 μl) or corresponding amount of a colony may simply be boiled at 95°C in 150 ul of water or TE to provide template of sufficient quality for AP-PCR. Otherwise, the protocol below may serve as a starting point.
-
3
The next day, transfer 1.4 mL of each overnight culture to a 1.5 mL tube, and centrifuge 2 min at 17000 × g at room temperature in a tabletop centrifuge.
-
4
To remove residual medium components, resuspend each pellet in 1 mL TE (Tris-EDTA pH 8.0) and centrifuge 2 min at 17000 × g at room temperature. Discard the supernatant.
-
5
For Staphylococcus aureus only, lyse the cells by resuspending the pellet in 196 μl of TE containing 200 μg/mL lysostaphin (4 μl of a 10 mg/mL stock solution). Incubate 30 min at 37°C.
For other bacteria, it is necessary replace lysostaphin with lysozyme or other cell wall lytic enzyme. A mix of 100 U/mL mutanolysin (Sigma Aldrich, cat. no. M9901-10KU) and 10 mg/ml of lysozyme is effective for lysing Enterococcus. For E. coli and many other Gram-negative organisms, step five may be skipped when buffer ATL is substituted for buffer AL in step 6 of the Qiagen DNeasy Blood and Tissue Kit protocol.
-
6
Proceed with manufacturer’s protocol from Step 2.
-
7
Measure DNA concentration on a Nanodrop device, noting the purity. Pure DNA has a 260:280 ratio of about 1.8 depending on base composition.
Purified genomic DNA may be stored at 4°C for up to a week, or longer at −20°C.
REAGENTS AND SOLUTIONS
Lysozyme Solution
Dissolve 500 mg into 10 mL of deionized water for a 50 mg/mL solution and store up to 6 months at −20°C in 1 mL aliquots until use.
Lysostaphin Solution
For a 10 mg/mL stock solution add 500 μl of deionized water to a 5 mg container of lysostaphin, dissolve thoroughly and store 20 μl aliquots up to 6 months at −20°C until use.
Mutanolysin Solution
For a 2500 U/ml stock solution, add 4 mL of deionized water to a 10,000 U container of mutanolysin, and store 100 μl aliquots up to 6 months at −20 °C until use.
COMMENTARY
Background Information
AP-PCR was first developed as a strategy to obtain DNA fingerprints (Welsh and McClelland, 1990). The central theory behind this is that some primers within a mixed pool will by chance possess sufficient complementarity to the DNA template to initiate DNA synthesis (Caetano-Anolles, 1993; Caetano-Anollés and Bassam, 1993) leading to amplification of a distinct set of sequences (Figure 1). When mixed primers are paired with a defined primer specific to a known sequence, in a technique sometimes called targeted gene walking PCR (Parker et al., 1991) or anchored PCR (Loh et al., 1989), a region of unknown sequence directly flanking a known sequence will likely be amplified. This version of AP-PCR has been successfully applied to map unknown sites of transposon insertion in organisms with a fully-sequenced genome (O’Toole et al., 1999).
In practice, an initial less-specific round of amplification, followed by a second more specific round of nested amplification usually results in amplicons that may be sequenced to map Tn-insertion sites, and this is the principle of AP-PCR (Figure 1). In Round 1 of AP-PCR, six cycles of low stringency amplification are performed, using a low annealing temperature of 30°C. This generates a pool of single-stranded products primed by oligonucleotide primers that may not be perfectly complementary to the template. With one primer perfectly complementary to the transposon, the subset of amplification products including the amplified portion of the transposon will yield double-stranded products, which are amplified more efficiently. In addition, other double-stranded products generated by two random primers will also be present. A pentameric DNA sequence ending with guanine or cytosine at the 3′ end of the R1 primer (Table 1) increases amplification efficiency because of stronger GC annealing (Das et al., 2005). The transposon junction amplicons are selectively amplified under stringent amplification conditions in Round 2 using a nested primer specific to the transposon, and a second primer specific for the anchor sequence incorporated into the flanking DNA in Round 1 of amplification.
Critical Parameters
PCR template DNA quality, and primer design are two critical parameters that influence the yield of discrete bands in AP-PCR reactions. AP-PCR involves three types of templates: genomic DNA, Round 1 amplification products, and Round 2 amplification products.
Ensuring sequencing template quality
AP-PCR amplification is a function of: i) the quality of the DNA used in Round 1 amplification, ii) removal of excess primers and single-stranded DNA from Round 1 products, iii) isolation of discrete amplification products from the Round 2 amplification, and iv) the presence of multiple transposon insertions, or residual transposon delivery vector leading to competing reactions.
Successful Round 1 amplification hinges on the ability of the random oligonucleotides to anneal to the DNA and prime DNA synthesis. Using highly purified genomic DNA as Round 1 AP-PCR template is optimal to ensure efficient priming, but may impose a limitation if many isolates are to be analyzed. The use of commercially available kits (such as the DNEasy 96 Blood and Tissue Kit) may facilitate processing up to 96 samples in parallel. Crude colony lysate has also been successfully used as template for AP-PCR (O’Toole et al., 1999), although cell types recalcitrant to lysis may yield poor template from this approach. If cell lysis yields sufficient template DNA, the lysed material may be treated with resins such as Chelex-100 (Bio Rad) or Ampure (Agencourt), to remove salts and cellular debris, and improve the efficiency of Round 1 PCR priming.
To derive meaningful data from Sanger sequencing, all nucleotide fragments present in a sample must share the same coding sequence from a primer. In theory, all products of AP-PCR should share the same 5′ sequence from the transposon-specific Round 2 primer. However, if the transposon is also carried by a residual delivery vector, or has integrated at a second site in the chromosome, sequencing all of these products together will yield mixed sequences that are difficult to interpret. Sequencing separate bands can identify the location of insertions at multiple chromosomal sites, and may provide useful information about the residual presence of the transposon delivery vector. Routinely, target bands are excised and purified using commercially available gel extraction DNA purification systems (such as the Qiagen Gel Extraction Kit). Separating the entire Round 2 AP-PCR reaction through a single lane of an agarose gel usually yields sufficient DNA for Sanger sequencing. Therefore, for Round 2 AP-PCR amplifications that yield multiple products, purifying at least two individual bands from a gel for sequencing template is most likely to yield interpretable results.
Optimization of primer sequence and concentration
The primer sequences listed in Table 1 have been used successfully for mapping Tn-integration sites in both Gram-positive and Gram-negative bacterial chromosomes (O’Toole et al., 1999). However, other primers may be optimized and applied for this purpose, depending on the bacterial species, transposon, or transposon delivery plasmid used. Alternative primers should be designed, keeping in mind several parameters. First, the reverse Round 1 primer includes a 3′ pentameric sequence. A guanine or cytosine at the 3′ end of this region has been found to increase the efficiency of priming (Das et al., 2005), and a sequence matching the GC content of the organism (O’Toole et al., 1999), or even a sequence specific to an abundant pentameric motif (Chen et al., 1999; Das et al., 2005) may improve binding efficiency. Ten random nucleotides preceding this sequence are sufficient to prime specific annealing to many sites within most target chromosomes during Round 1 amplification (Das et al., 2005). Biasing the composition of these nucleotides to the GC content of the organism may also increase the number of oligonucleotides in the random pool that may prime DNA replication (O’Toole et al., 1999). An anchor sequence is incorporated at the 5′ end to provide a specific target for the Round 2 reverse primer. If this sequence needs to be re-designed, the target melting temperature should be compatible with the forward Round 2 primer (Table 1). In addition to these specific design parameters, all sequences should be checked to minimize competing amplification of secondary sites present in the chromosome.
The annealing temperature of the first six cycles of Round 1 AP-PCR amplification largely determines the number of major products enriched by subsequent cycles of amplification (Welsh and McClelland, 1990). It is theoretically possible to titrate the number of bands that might be amplified by AP-PCR by performing a gradient PCR during the first 6 cycles of Round 1 amplification. An initial annealing temperature range between 30°C and 50°C should be chosen as a starting point.
The ratio of primer to template is another parameter to consider in optimizing AP-PCR. It may be necessary to experimentally determine the optimum concentration of primers used in Rounds 1 and 2 AP-PCR reactions (Table 1), to accommodate differences in the efficiency of amplification from the genomic DNA of an organism of interest. In particular, increasing the ratio of arbitrary primers to transposon-specific primers in the Round 1 amplification is one strategy to increase the yield of products produced in the Round 2 amplification (Das et al., 2005). As a starting point, 20 ng of purified chromosomal DNA should be used as a template for Round 1amplification, and 40 ng of purified Round 1 AP-PCR product should be used as a template for Round 2 amplification. Optimizing primer concentrations around these amounts of template should result in the amplification of products sufficient quantity for Sanger sequencing.
Troubleshooting
In addition to the detailed protocol and discussion of critical parameters, additional advice for troubleshooting common problems is presented (Table 2). As an alternative to AP-PCR, inverse PCR is also routinely used to map transposon insertion sites (Ochman et al., 1988). In inverse PCR, a restriction enzyme that cuts the chromosome, but not the transposon, is used to digest chromosomal DNA, which is then ligated back onto itself. Outward-reading transposon-specific primers are used to amplify across the ligated junction, and identify the regions flanking the transposon. If the restriction site chosen yields annealed segments of chromosomal DNA too large for PCR amplification, then the approach cannot be applied. Thus, the utility of inverse PCR is limited by the availability of suitable restriction enzyme sites in the target chromosome.
Table 2.
Troubleshooting guide
| Step | Problem | Possible cause | Solution |
|---|---|---|---|
|
| |||
| 4 | Round 1 PCR products are not discreet bands | Annealing temperature too low for Round 1 of amplification | Increase annealing temperature to 35 °C If colony template is used, switch to genomic DNA |
| 9 | Round 2 PCR products are faint | Major products not abundant in Round 1 of amplification | Increase amount of Round 1 PCR product used as template from 1 μl to 10 μl |
| 11 | Sequencing read contains only ambiguous bases | A single PCR product was not sequenced from the Round 2 amplification | Perform Round 2 amplification and agarose gel electrophoresis again, running the gel longer, and increasing the % agarose from 1% to 1.5% |
| 15 | Chromosome-specific sequence contains only ambiguous bases | Product selected to sequence may be too small to contain good-quality sequence reading into the chromosome Strain may contain multiple transposon insertion sites. Isolate may contain multiple strains Transposon may have been lost |
Select a larger product to gel purify and sequence Sequence multiple bands to identify alternate insertion sites Re-derive single colonies, and perform AP-PCR and mapping on these Grow isolates without selection, attempt to amplify transposon-specific sequence |
|
Support 2 |
Strains do not grow | Concentration of antibiotic may be too high | Check primary literature to confirm the correct concentration of antibiotic is used to maintain integrants |
| 7 | DNA yield low | Too few cells used for isolation | Check the optical density of cultures used, and the specifications for the amount of cells to use for the DNA isolation method selected |
| DNA purity low | Too many cells used for isolation | See above | |
Anticipated Results
If PCR conditions are optimized, sequencing the gel-extracted bands will yield a single product. The resulting sequence will contain transposon-specific sequence, as well as 30–500 bp of chromosomal sequence corresponding to sequence downstream of the site of transposon integration. Depending on the fidelity of the enzyme used, the sequence at the transposon junction may contain a few errors. However, even short sequences may accurately identify unique sites in a chromosome (e.g. a sequence as short as 11 nucleotides will only occur by chance one time in > 4 million bases), meaning that AP-PCR may be used to map the site of transposon insertion within a bacterial genome with high confidence.
Time Considerations
Once Round 1 PCR template has been obtained, AP-PCR, Sanger sequencing, and insertion site mapping may be accomplished in as little as a day or two, depending on how quickly sequencing results are obtained. AP-PCR amplification requires approximately 8 h: two rounds of PCR (4 h each round: 0.5 h to set up, 2 h reaction time, 1 h to separate and visualize products on a gel, 1–0.5 h to purify and quantify products). If the process has been optimized and time is limiting, visualization of the Round 1 products on an agarose gel may be omitted. Gel purification of samples for Sanger sequencing requires approximately 2 h. Mapping the sequences to the chromosome to identify transposon insertion sites requires approximately 1 h of set-up time to create a custom BLAST directory, and about 5 minutes per sample to trim and map sequences. The protocol is routinely completed in three days if both amplifications are performed on the first day (steps 1 to 8), the Round 2 products are prepared and submitted for sequencing on the second day (steps 9 and 10), and sequences are analyzed on the third day (steps 11 through 16). If the scale of the project requires more than 50 insertion sites to be mapped, consider breaking the work into batches, as processing more than this number of samples may introduce other inefficiencies, and possibly affect the quality of results. The AP-PCR protocol can be scaled up to map thousands of transposon insertions and to create a transposon insertion database (Liberati et al., 2006; Urbach et al., 2009).
Acknowledgments
We would like to acknowledge Elaine Menezes, and Jenna Wurster for technical assistance; D. Van Tyne, B. Fiore, F. Lebreton, A. Gaca, and the members of the Gilmore laboratory for critical evaluation of this protocol; and G. Roberts for historical insight. This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, AI083214.
Footnotes
INTERNET RESOURCES
Documentation and source code for Python programming language
Biopython documentation and source code Note that a useful instruction manual called the Biopython cookbook is available in the documentation.
http://www.ebi.ac.uk/Tools/msa/muscle/
Web host for the MUSCLE algorithm
http://www.ncbi.nlm.nih.gov/books/NBK279688/
Instructions for making a local database using command line programming.
LITERATURE CITED
- Alexeyev MF, Shokolenko IN. Mini-Tn10 transposon derivatives for insertion mutagenesis and gene delivery into the chromosome of Gram-negative bacteria. Gene. 1995;160:59–62. doi: 10.1016/0378-1119(95)00141-R. [DOI] [PubMed] [Google Scholar]
- Bae T, Glass EM, Schneewind O, Missiakas D. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. In: Osterman Andrei L, Gerdes Svetlana Y., editors. Microbial Gene Essentiality: Protocols and Bioinformatics. Humana Press; Totowa, NJ, USA: 2008. pp. 103–116. [DOI] [PubMed] [Google Scholar]
- Ballering KS, Kristich CJ, Grindle SM, Oromendia A, Beattie DT, Dunny GM. Functional genomics of Enterococcus faecalis: multiple novel genetic determinants for biofilm formation in the core genome. J Bacteriol. 2009;191:2806–2814. doi: 10.1128/JB.01688-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CA, Mandel MJ, Gyllborg MC, Thomasgard KA, Ruby EG. Genetic determinants of swimming motility in the squid light-organ symbiont Vibrio fischeri. Microbiologyopen. 2013;2:576–594. doi: 10.1002/mbo3.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JF, Gyllborg MC, Cronin DC, Quillin SJ, Mallama CA, Foxall R, Whistler C, Goodman AL, Mandel MJ. Global discovery of colonization determinants in the squid symbiont Vibrio fischeri. Proc Natl Acad Sci USA. 2014;111:17284–17289. doi: 10.1073/pnas.1415957111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KH, Boeke JD. Human transposon tectonics. Cell. 2012;149:740–752. doi: 10.1016/j.cell.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anolles G. Amplifying DNA with arbitrary oligonucleotide primers. Genome Res. 1993;3:85–94. doi: 10.1101/gr.3.2.85. [DOI] [PubMed] [Google Scholar]
- Caetano-Anollés G, Bassam BJ. DNA amplification fingerprinting using arbitrary oligonucleotide primers. Appl Biochem Biotechnol. 1993;42:189–200. doi: 10.1007/BF02788052. [DOI] [PubMed] [Google Scholar]
- Cameron DE, Urbach JM, Mekalanos JJ. A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proc Natl Acad Sci USA. 2008;105:8736–8741. doi: 10.1073/pnas.0803281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Yong R, Dong H, Duncan MJ. A general method for direct sequencing of transposon mutants by randomly primed PCR. Tech Tips Online. 1999;4:58–61. doi: 10.1016/S1366-2120(08)70140-5. [DOI] [Google Scholar]
- Chiang SL, Mekalanos JJ. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol. 1998;27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- Craig NL. Target site selection in transposition. Annu Rev Biochem. 1997;66:437–474. doi: 10.1146/annurev.biochem.66.1.437. [DOI] [PubMed] [Google Scholar]
- Daboussi MJ, Capy P. Transposable elements in filamentous fungi. Annu Rev Microbiol. 2003;57:275–299. doi: 10.1146/annurev.micro.57.030502.091029. [DOI] [PubMed] [Google Scholar]
- Das S, Noe JC, Paik S, Kitten T. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Methods. 2005;63:89–94. doi: 10.1016/j.mimet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Dziva F, van Diemen PM, Stevens MP, Smith AJ, Wallis TS. Identification of Escherichia coli O157: H7 genes influencing colonization of the bovine gastrointestinal tract using signature-tagged mutagenesis. Microbiology. 2004;150:3631–3645. doi: 10.1099/mic.0.27448-0. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez L, Del Sol R, Evans M, Fielding S, Herron P, Chandra G, Dyson P. A transposon insertion single-gene knockout library and new ordered cosmid library for the model organism Streptomyces coelicolor A3 (2) Antonie Van Leeuwenhoek. 2011;99:515–522. doi: 10.1007/s10482-010-9518-1. [DOI] [PubMed] [Google Scholar]
- Fuller TE, Kennedy MJ, Lowery DE. Identification of Pasteurella multocida virulence genes in a septicemic mouse model using signature-tagged mutagenesis. Microb Pathog. 2000;29:25–38. doi: 10.1006/mpat.2000.0365. [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Wu M, Gordon JI. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc. 2011;6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf J, Dunlap PV, Ruby EG. Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J Bacteriol. 1994;176:6986–6991. doi: 10.1128/jb.176.22.6986-6991.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL. Discovery of the transposable element mariner. Genetics. 2001;157:471–476. doi: 10.1093/genetics/157.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hava DL, Camilli A. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol. 2002;45:1389–1406. doi: 10.1046/j.1365-2958.2002.03106.x/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrixson DR, Akerley BJ, DiRita VJ. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol. 2001;40:214–224. doi: 10.1046/j.1365-2958.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- Hoskisson PA, Rigali S, Fowler K, Findlay KC, Buttner MJ. DevA, a GntR-like transcriptional regulator required for development in Streptomyces coelicolor. J Bacteriol. 2006;188:5014–5023. doi: 10.1128/JB.00307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286:2165–2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Makarova KS, Koonin EV. ISC, a novel group of bacterial and archaeal DNA transposons that encode Cas9 homologs. J Bacteriol. 2016;198:797–807. doi: 10.1128/JB.00783-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavermann H, Burns BP, Angermüller K, Odenbreit S, Fischer W, Melchers K, Haas R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813–822. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner N. Transposable elements in prokaryotes. Annu Rev Genet. 1981;15:341–404. doi: 10.1146/annurev.ge.15.120181.002013. [DOI] [PubMed] [Google Scholar]
- Kleckner N, Bender J, Gottesman S. [7] Uses of transposons with emphasis on Tn10. Methods Enzymol. 1991;204:139–180. doi: 10.1016/0076-6879(91)04009-D. [DOI] [PubMed] [Google Scholar]
- Knobloch JKM, Nedelmann M, Kiel K, Bartscht K, Horstkotte MA, Dobinsky S, Rohde H, Mack D. Establishment of an arbitrary PCR for rapid identification of Tn917 insertion sites in Staphylococcus epidermidis: characterization of biofilm-negative and nonmucoid mutants. Appl Environ Microbiol. 2003;69:5812–5818. doi: 10.1128/AEM.69.10.5812-5818.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro FM, Tran K, Vezzi A, Vitulo N, Valle G, Bartlett DH. Large-scale transposon mutagenesis of Photobacterium profundum SS9 reveals new genetic loci important for growth at low temperature and high pressure. J Bacteriol. 2008;190:1699–1709. doi: 10.1128/JB.01176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Butler SM, Camilli A. Selection for in vivo regulators of bacterial virulence. Procs Natl Acad Sci U S A. 2001;98:6889–6894. doi: 10.1073/pnas.111581598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Rigby K, Lai Y, Nair V, Peschel A, Schittek B, Otto M. Staphylococcus aureus mutant screen reveals interaction of the human antimicrobial peptide dermcidin with membrane phospholipids. Antimicrob Agents Chemother. 2009;53:4200–4210. doi: 10.1128/AAC.00428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucl Acids Res. 2015;43:W580–W584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Procs Natl Acad Sci U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, Chaconas G, Philipp MT, Norris SJ. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS ONE. 2012;7:e47532. doi: 10.1371/journal.pone.0047532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh EY, Elliott JF, Cwirla S, Lanier LL, Davis MM. Polymerase chain reaction with single-sided specificity: analysis of T cell receptor delta chain. Science. 1989;243:217–220. doi: 10.1126/science.2463672. [DOI] [PubMed] [Google Scholar]
- Loo C, Corliss D, Ganeshkumar N. Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol. 2000;182:1374–1382. doi: 10.1128/JB.182.5.1374-1382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong TT, Newell SW, Lee CY. Mgr, a novel global regulator in Staphylococcus aureus. J Bacteriol. 2003;185:3703–3710. doi: 10.1128/JB.185.13.3703-3710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurkiewicz P, Tang CM, Boone C, Holden DW. Signature-tagged mutagenesis: barcoding mutants for genome-wide screens. Nat Rev Genet. 2006;7:929–939. doi: 10.1038/nrg1984. [DOI] [PubMed] [Google Scholar]
- McClintock B. Mutable loci in maize. Year B Carnegie Inst Wash. 1948;47:155–169. [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucl Acids Res. 2013;41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan E, Campbell JD, Rowe SC, Bispham J, Stevens MP, Bowen AJ, Barrow PA, Maskell DJ, Wallis TS. Identification of host-specific colonization factors of Salmonella enterica serovar Typhimurium. Mol Microbiol. 2004;54:994–1010. doi: 10.1111/j.1365-2958.2004.04323.x. [DOI] [PubMed] [Google Scholar]
- O’Toole GA, Gibbs KA, Hager PW, Phibbs PV, Kolter R. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J Bacteriology. 2000;182:425–431. doi: 10.1128/JB.182.2.425-431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole GA, Pratt LA, Watnick PI, Newman DK, Weaver VB, Kolter R. [6] Genetic approaches to study of biofilms. Methods Enzymol. 1999;310:91–109. doi: 10.1016/S0076-6879(99)10008-9. [DOI] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JD, Rabinovitch PS, Burmer GC. Targeted gene walking polymerase chain reaction. Nucl Acids Res. 1991;19:3055–3060. doi: 10.1093/nar/19.11.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CN, Carabetta VJ, Chowdhury T, Silhavy TJ. LrhA regulates rpoS translation in response to the Rcs phosphorelay system in Escherichia coli. J Bacteriol. 2006;188:3175–3181. doi: 10.1128/JB.188.9.3175-3181.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH, Izsvák Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15:326–332. doi: 10.1016/S0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- Reznikoff WS. The Tn5 transposon. Annu Rev Microbiol. 1993;47:945–964. doi: 10.1146/annurev.mi.47.100193.004501. [DOI] [PubMed] [Google Scholar]
- Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Quandt J, Klipp W. New derivatives of transposon Tn5 suitable for mobilization of replicons, generation of operon fusions and induction of genes in Gram-negative bacteria. Gene. 1989;80:161–169. doi: 10.1016/0378-1119(89)90262-X. [DOI] [PubMed] [Google Scholar]
- Tsou AM, Liu Z, Cai T, Zhu J. The VarS/VarA two-component system modulates the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiology. 2011;157:1620–1628. doi: 10.1099/mic.0.046235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach JM, Wei T, Liberati N, Grenfell-Lee D, Villanueva J, Wu G, Ausubel FM. Using PATIMDB to Create Bacterial Transposon Insertion Mutant Libraries. Curr Protoc Mol Biol. 2009;86:19.7.1–19.7.34. doi: 10.1002/0471142727.mb1907s86. 19.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick PI, Kolter R. Steps in the development of a Vibrio cholerae El Tor biofilm. Mol Microbiol. 1999;34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J, McClelland M. Fingerprinting genomes using PCR with arbitrary primers. Nucl Acids Res. 1990;18:7213–7218. doi: 10.1093/nar/18.24.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley MJ, Ching KH, Heffron F. Salmonella SsrB activates a global regulon of horizontally acquired genes. Mol Microbiol. 2000;36:749–761. doi: 10.1046/j.1365-2958.2000.01902.x. [DOI] [PubMed] [Google Scholar]