

Abstract
Deformed epidermal autoregulatory factor-1 (DEAF1), a transcription factor essential for central nervous system and early embryonic development, has recently been implicated in a series of intellectual disability related neurodevelopmental anomalies termed, in this study, DEAF1-associated neurodevelopmental disorder (DAND). We identified six potentially deleterious DEAF1 variants in a cohort of individuals with DAND via clinical exome sequencing (CES) and in-silico analysis, including two novel de novo variants: missense variant c.634G>A p.Gly212Ser in the SAND domain and deletion variant c.913_915del p.Lys305del in the NLS domain, as well as c.676C>T p.Arg226Trp, c.700T>A p.Trp234Arg, c.737G>C p.Arg246Thr, and c.791A>C p.Gln264Pro. Luciferase reporter, immunofluorescence staining and electrophoretic mobility shift assays revealed that these variants had decreased transcriptional repression activity at the DEAF1 promoter and reduced affinity to consensus DEAF1 DNA binding sequences. In addition, c.913_915del p.K305del localized primarily to the cytoplasm and interacted with wild-type DEAF1. Our results demonstrate that variants located within the SAND or NLS domains significantly reduce DEAF1 transcriptional regulatory activities and are thus, likely to contribute to the underlying clinical concerns in DAND patients. These findings illustrate the importance of experimental characterization of variants with uncertain significance identified by CES to assess their potential clinical significance and possible use in diagnosis.
Keywords: deformed epidermal autoregulatory factor-1 (DEAF1) associated neurodevelopmental disorder (DAND), intellectual disability (ID), clinical exome sequencing (CES), SAND, nuclear localization signal (NLS)
Introduction
Intellectual disability (ID) is a common neurodevelopmental disorder with high clinical and genetic heterogeneity that has a world-wide prevalence of ~ 1% [Maulik et al., 2011]. ID is characterized by various degrees of cognitive dysfunction and maladaptive behaviors [American Psychiatric Association 2013] and can occur either in isolation (nonsyndromic) or in conjunction with other clinical and physical features. Trio- and proband-based clinical exome sequencing (CES) has greatly increased the number of candidate genetic loci contributing to ID [Rump et al., 2016], and the functional significance of these genetic variants is under active investigation. Recent CES studies have identified a potential role for deformed epidermal autoregulatory factor-1 (DEAF1; MIM# 602635; NM_021008.3) in both autosomal dominant and autosomal recessive modes of inheritance in a neurodevelopmental disorder comprising intellectual disability, speech impairment, motor developmental delay, (MRD24; MIM# 615828; MIM# 617171) [Rauch et al., 2012; Vissers et al., 2010; Vulto-van Silfhout et al., 2014], and additional phenotypes such as seizures, brain malformations, and autism [Faqeih et al., 2014; Gund et al., 2016; Rajab et al., 2015; Waltl 2014; Wenger et al., 2017], as well as a Smith-Magenis syndrome (SMS; MIM# 182290) -like phenotype that includes ID, dysmorphic features, and sleep disturbances [Berger et al., 2017]. In this study, we use DEAF1-associated neurodevelopmental disorder (DAND) to describe a series of ID related neurodevelopmental anomalies that associated with pathogenic DEAF1 variants and share a common set of clinical features.
DEAF1 encodes an autoregulatory transcription factor that activates or represses the expression of various target genes [Czesak et al., 2012; Czesak et al., 2006; Jensik et al., 2014]. The protein is comprised of multiple structural domains including a SAND domain (Sp-100, AIRE, NucP41/75, and DEAF1), which is required for DNA binding (via a KDWK motif that binds consensus TTCG sequences in gene regulatory regions [Huggenvik et al., 1998]); a monopartite nuclear localization signal (NLS) and a leucine-rich nuclear export signal (NES), and a cysteine-rich MYND domain (myeloid translocation protein 8, Nervy, and DEAF1) in the C-terminus that mediates protein-protein interactions [Vulto-van Silfhout et al., 2014]. DEAF1 is essential for early embryonic and neuronal development. Disruption of Deaf1 function results in embryonic arrest and severe defects in early embryonic patterning in drosophila [Veraksa et al., 2002], and neural tube defects [Hahm et al., 2004], as well as impaired contextual memory and increased anxiety-related behavior in mice [Vulto-van Silfhout et al., 2014]. DEAF1 is highly expressed in brain [Gross and McGinnis 1996; Vulto-van Silfhout et al., 2014; Waltl 2014] and has been associated with neuropsychiatric disorders such as major depression and suicide [Czesak et al., 2012; Czesak et al., 2006; Lemonde et al., 2003].
Previous studies identified six de novo heterozygous missense variants within the DEAF1 SAND domain. Variants c.670C>T p.Arg224Trp, c.762A>C p.Arg254Ser [Vulto-van Silfhout et al., 2014] and c.683T>G p.Ile228Ser [Vissers et al., 2010; Vulto-van Silfhout et al., 2014] were reported in three unrelated MRD24 (MIM# 615828) patients, and c.791A>C p.Gln264Pro [Rauch et al., 2012; Vulto-van Silfhout et al., 2014] in a nonsyndromic ID patient. Each variant produced an altered DEAF1 protein with absent or significantly reduced DNA binding and the inability to repress its own promoter. Two additional de novo variants, c.700T>A p.Trp234Arg [Berger et al., 2017] and c.737G>C p.Arg246Thr [Wenger et al., 2017], were recently identified in a SMS-like patient and an ID patient with autism and sleep difficulties, respectively. An inherited homozygous SAND variant c.676C>T p.Arg226Trp was found in two related individuals from a consanguineous Saudi Arabian family displaying ID, microcephaly, hypotonia, white matter abnormalities and seizure [Faqeih et al., 2014], and in a third individual from an unrelated consanguineous Saudi Arabian family with similar features but no evidence of white matter disease [Gund et al., 2016]. Functional properties of this variant, however, have not yet been reported. Another inherited homozygous c.997+4A>C p.(Gly292Profs*) splice acceptor change was found in three related individuals from a consanguineous Omani family with ID, autism, hypotonia, motor and speech delay, dyskinesia of the limbs, seizures and abnormal brain MRI. Functional analysis of this variant revealed exon skipping and a 95% reduction in DEAF1 mRNA [Rajab et al., 2015].
In this study, we identified six DEAF1 variants within the SAND and NLS domains through CES of pediatric patients with developmental delay and other clinical features of unknown etiology. To ascertain whether these candidate variants are potentially pathogenic, we carried out molecular experiments to assess the effects of the amino acid substitutions or small deletions on human DEAF1 function. Our results demonstrate that these variants severely affect DEAF1 transcriptional activities, DNA binding and intracellular localization and thereby further support a role for DEAF1 as an underlying cause of clinical phenotypes in these individuals. These findings illustrate the importance of functional characterization of CES variants of uncertain significance (VUS) for revealing gene variant properties and improving diagnosis.
Materials and Methods
Clinical exome sequencing and patient information
Individuals with pediatric onset neurological abnormalities were referred for CES at Baylor Genetics (Houston, TX). Clinical data and peripheral blood samples from the patients and their parents were provided by the referring physician. Exome sequencing and variant calling were performed as previously described [Yang et al., 2013; Yang et al., 2014]. After data analysis, candidate DEAF1 variants were confirmed by Sanger Sequencing, at which point the parental samples were also analyzed (described in details in “Identification of candidate DEAF1 variants” of Results section). One DEAF1 variant c.737G>C p.Arg246Thr in one of the individuals (561320) was recently reported in an exome reanalysis cohort as a likely pathogenic variant, but without accompanying functional studies [Wenger et al., 2017].
Exome and clinical data for another individual (M2647) carrying a de novo DEAF1 variant c.700T>A p.Trp234Arg were provided for inclusion in this report by two co-authors (SB, ACMS) [Berger et al., 2017]. The patient was referred for Smith-Magenis syndrome (SMS; MIM# 182290)-like phenotype evaluated at the National Institutes of Health (NIH) National Human Genome Research Institute (NHGRI) under IRB-approved protocol 01-HG-0109. Exome sequencing was performed at the NIH Intramural Sequencing Center (NISC).
To exclude other possible causes for phenotypes observed in the six patients, additional genetic and biochemical tests were performed prior to CES, the results of which are summarized in Supp. Table S1. No abnormalities were found in metabolic investigations, studies for Angelman syndrome (AS), Prader-Willi syndrome (PWS) and fragile-X syndrome (FXS), fluorescence in situ hybridization (FISH) for SMS, DNA microarray and RAI1 coding region sequencing, etc. Additional gene variants of known/uncertain clinical significance (VUS) related/unrelated to clinical phenotypes were also detected in our ID cohort. Most of the variants, however, were not considered to be pathogenic since they were inherited or previously shown to have an autosomal recessive inheritance pattern, or not relevant to ID phenotype. A heterozygous de novo missense variant NM_001376.4:c.6272G>A p.Arg2091Gln) in the DYNC1H1 gene was detected in Subject 6 in addition to the DEAF1 variant p.K305del. Mutations in DYNC1H1 have previously been associated with spinal muscular atrophy, lower extremity, autosomal dominant (SMALED; MIM# 158600) [Harms et al., 2012], autosomal dominant mental retardation, type 13 (MRD13; MIM# 614563) [Willemsen et al., 2012], and Charot-Marie-Tooth disease type 2O (CMT2O; MIM# 614228) [Weedon et al., 2011]. Although the clinical phenotypes of Subject 6 were not fully represented by these disorders (e.g. ASD phenotype with family history was not reported in patients carrying DYNC1H1 variants), and most pathogenic DYNC1H1 variants reported are localized in tail domain (53 aa-1867 aa) and stalk domain (3189–3500) of this gene (ClinVar database, https://www.ncbi.nlm.nih.gov/clinvar) [Harms et al., 2012], further study is still required to elucidate whether DYNC1H1 plays a role in the patient’s phenotype.
Plasmid construction
Construction of pcDNA3-based expression plasmids encoding carboxyl-terminal FLAG or HA epitope-tagged human DEAF1 cDNA (RefSeq NM_021008.3) has previously been described [Jensik et al., 2004]. The DNA mutation numbering system used in this study is based on cDNA sequence. Nucleotide numbering designates the A of the ATG translation initiation codon in the reference sequence as +1 and the initiation codon as codon 1. Expression plasmids encoding mutated DEAF1 variants containing the amino acid substitutions NM_021008.3:c.634G>A p.Gly212Ser, NM_021008.3:c.676C>T p.Arg226Trp, NM_021008.3:c.700T>A p.Trp234Arg, NM_021008.3:c.737G>C p.Arg246Thr, or the deletion NM_021008.3:c.913_915del p.Lys305del were generated by PCR-mediated site-directed mutagenesis. PCR primers contained specific DEAF1 restriction endonuclease sites to facilitate subcloning (Supp. Table S2). Digested PCR fragments were used to replace the corresponding regions of wild-type (WT) DEAF1. All inserts were confirmed by standard Sanger sequencing.
Electrophoretic mobility shift assay (EMSA)
Epitope FLAG-tagged WT or variant DEAF1 recombinant proteins were purified from transfected human embryonic kidney (HEK293T) cells, as previously described [Jensik et al., 2012]. Proteins used in serial dilution EMSA were concentrated using Amicon Ultra 30K devices in storage buffer that included 0.1% deoxycholate and 1 mM DTT. EMSA analysis was performed using two verified probes: a IRDye800 labeled double-stranded (ds) DNA probe N52-69 (11 space probe) corresponding to the consensus human DEAF1 promoter binding sequence [Bottomley et al., 2001; Michelson et al., 1999] or a IRDye700 labeled probe S6con (6 space probe) corresponding to a high-affinity preferred DEAF1 binding motif [Jensik et al., 2004; Jensik et al., 2014; Vulto-van Silfhout et al., 2014]. Recombinant DEAF1 proteins (850 fmol) were incubated with 500 fmol of dsDNA probe and 1 μg of poly(dA-dT) (nonspecific competitor) in a 20 μl reaction containing 70 mM KCl, 35 mM Tris, pH 7.5, 0.7 mM DTT, 2.0% (v/v) glycerol for 20 min at room temperature. Protein-DNA complexes were separated in 4.0% non-denaturing polyacrylamide gels and probe migration was visualized on a Licor Odyssey CLx.
Luciferase reporter assay
Transcription assays using the pDEAF1 promoter-firefly luciferase construct have been previously described [Vulto-van Silfhout et al., 2014]. Briefly, HEK293T cells were plated in 1 ml DMEM in each well of 24-well plates (90,000 cells per well) and transfected in duplicate with 125 ng pcDNA3 (control) or DEAF1 (WT or DEAF1 variants) expression plasmids with 375 ng pDEAF1 promoter-firefly luciferase and 1.25 ng Renilla luciferase expression plasmid for normalization using the calcium phosphate technique. Luciferase assays were performed 24 h later using the Dual-Luciferase Reporter Assay System (Promega).
Immunofluorescence staining
HEK293T cells on 60 mm dishes were transfected with 1.0 μg of Flag epitope-tagged WT or DEAF1 variants expression constructs, or co-transfected with 500 ng of WT DEAF1-HA and 500 ng of p.K305del-Flag, by calcium phosphate precipitation. Twenty-four hours later, cells were fixed in 4% paraformaldehyde, permeabilized in methanol, and incubated with rabbit anti-DEAF1(1:1,000) followed by Cy3-conjugated goat anti-rabbit IgG (1:1,000) [Huggenvik et al., 1998], or rabbit anti-HA (1:1000) and mouse anti-FLAG (1:1000) followed by Cy3-conjugated goat anti-rabbit IgG and Alexafluor-488-conjugated donkey anti-mouse IgG (1:1,000) [Vulto-van Silfhout et al., 2014]. DNA was stained with Hoechst dye 33258 (1.0 μg/mL). Cells were visualized with an Olympus BW50 fluorescence microscope with a 60x water objective.
Results
Identification of candidate DEAF1 variants
To identify novel DEAF1 deleterious variants, we searched the Baylor Genetics internal CES database comprising 6067 individuals, and identified 248 individuals harboring a total of 83 distinct DEAF1 variants. To screen for candidate pathogenic DEAF1 variants, we focused on rare variants with frequencies less than 0.1% in the 1000 Genomes Project (www.1000genomes.org), dbSNP (www.ncbi.nlm.nih.gov/snp/), ExAC (exac.broadinstitute.org), and National Heart, Lung, and Blood Institute GO Exome Sequencing Project (ESP5400, http://evs.gs.washington.edu/EVS) databases. Synonymous variants likely to be benign or variants of uncertain significance (VUS) as assessed by PolyPhen 2 [Adzhubei et al., 2010] (http://genetics.bwh.harvard.edu/pph2/, HumVar or HumDiv scores < 0.95) and/or SIFT [Kumar et al., 2009] (http://sift.jcvi.org/) (Supp. Table S3) were removed. Variants listed in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/) were retained. Since both autosomal dominant and autosomal recessive modes of inheritance were reported for DEAF1-associated cases (Supp. Table S4), we focused on both de novo and biallelically inherited variants in probands. The retained variants were validated by Sanger sequencing, and tested for co-segregation by analyzing parental DNA samples.
CES data obtained at NHGRI NIH[Berger et al., 2017] were analyzed using ANNOVAR [Wang et al., 2010] (http://annovar.openbioinformatics.org/) (Supp. Table S3), focusing on rare variants with frequencies less than 0.1% in ExAC database and variants predicted to be deleterious based on a Combined Annotation Dependent Depletion (CADD) [Kircher et al., 2014] scores >20 for different Mendelian inheritance models in the trios.
Our final list of six candidate DEAF1 variants is shown in Figure 1a and Table 1. All variants reported in this study have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/: accession numbers: SCV000590953 - SCV000590958). Two of the six variants, c.634G>A p.Gly212Ser and c.913_915del p.Lys305del, are first reported here. Among the other four, c.700T>A p.Trp234Arg was recently identified in a suspected of Smith-Magenis syndrome patient (M2647) [Berger et al., 2017], and c.737G>C p.Arg246Thr in an exome reanalysis cohort [Wenger et al., 2017], respectively; c.676C>T (p.R226W) and c.791A>C (p.Q264P) were previously reported in association with brain malformations [Faqeih et al., 2014; Gund et al., 2016] and nonsyndromic ID [Vulto-van Silfhout et al., 2014], respectively (Supp. Table S3). Except for c.791A>C p.Gln264Pro [Vulto-van Silfhout et al., 2014], functional properties of these variants have not been reported.
Figure 1. Characteristics of DEAF1 variants identified in individuals with intellectual disability by clinical exome sequencing (CES).

A. Dark grey boxes within the diagram of human DEAF1 (NM_021008.3, NP_066288.2) show the locations of the SAND, NLS, NES and MYND domains. Arrows indicate the locations of the six potentially pathogenic DEAF1 variants identified in the CES dataset for individuals with ID. B. Alignments of DEAF1 SAND and NLS domains sequences for five vertebrates, Human (Homo sapiens: NP_066288.2), Chimpanzee (Pan troglodytes:NP_00100897), Rhesus monkey (Macaca mulatta: XP_014968734), Mouse (Mus musculus: NP_058570), and Rat (Rattus norvegicus: NP_113989), show that the six genetic variants affect conserved amino acid residues (indicated with asterisks).
Table 1.
Candidate pathogenic DEAF1 variants in SAND and NLS domains identified through clinical exome sequencing in this study
| Variant | Position | Variant type | Zygosity | Allele frequency Ø | Putative impact § | Functional evidence |
Assessment | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| DNA alteration | Protein alteration† |
location | Protein domain |
De novo status * |
Novelty | ID referrals to CES |
ExAC | 1000 genome |
ESP 5400 |
PolyPhen2 (HumDiv / HumVar) |
SIFT | ACMG† | |||
|
|
|
|
|
|
|||||||||||
| c.634G>A | p.Gly212Ser | Exon 4 | SAND | De novo | Novel | Het | 8.24E-05 | 0 | 0 | 0 | PD 1.000/PD 0.995 | D 0.01 | PS2 | This study | D |
| c.676C>T | p.Arg226Trp | Exon 5 | SAND | Inherited | [Faqeih, 2014; Gund, 2016] | Hom | 1.65E-04 | 8.28 E-06 | 0 | 0 | PD 1.000/PD 0.999 | D 0.00 | PM1 | This study | D |
| c.700T>A | p.Trp234Arg | Exon 5 | SAND | De novo | [Berger, 2017] | Het | 0 # | 0 | 0 | 0 | PD 1.000/PD 1.000 | D 0.00 | PS2 | This study | D |
| c.737G>C | p.Arg246Thr | Exon 5 | SAND | De novo | [Wenge, 2017] | Het | 8.24E-05 | 0 | 0 | 0 | PD 0.999/PD 0.997 | D 0.01 | PS2 | This study | D |
| c.791A>C | p.Gln264Pro | Exon 5 | SAND | De novo | [Rauch, 2012; Vulto, 2014] | Het | 8.24E-05 | 0 | 0 | 0 | PD 0.999/PD 0.972 | D 0.01 | PS2 | [Rauch, 2012; Vulto, 2014] | D |
| c.913_915del | p.Lys305del | Exon 7 | NLS | De novo | Novel | Het | 8.24E-05 | 0 | 0 | 0 | N.A. | N.A. | PS2 | This study | D |
The DNA variant numbering system used in this study is based on human DEAF1 cDNA sequence (RefSeq NM_021008.3). Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Verified by Sanger sequencing.
Listed minor allele frequencies are from the ExAC browser (exac.broadinstitute.org), 1000 Genomes Project (www.1000genomes.org), and ESP5400 data of the National Heart, Lung, and Blood Institute GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS)
Predicted by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT (http://sift.jcvi.org/), variant function interpreted according to the guidelines from the American College of Medical Genetics and Genomics (ACMG)31. According to the ACMG guidelines, PS2 indicates “De novo (both maternity and paternity confirmed) in a patient with the disease and no family history” and PM1 indicates “ Located in a mutational hot spot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation”.
Patient was found in a cohort of 6 patients with SMS-like phenotypes, 1 sibling, and 12 parents
Abbreviations: PD (Probably damaging), D (Damaging), N.A. (not available), Het (heterozygote), Hom (homozygote)
In silico analysis of DEAF1 variants
Among the variants mentioned above, five are missense variants located within the SAND domain, an essential region for transcriptional activation or repression, and the other one, p.K305del, is a 3-bp in-frame deletion within the NLS domain (Figure 1a). With the exception of p.R226W, which was homozygous and inherited from each parent, all of the remaining variants were heterozygous in the probands and not detected in their parents. All variants affected evolutionarily conserved amino acids (Figure 1b). Polyphen 2 and SIFT predictions and conservation scores suggest that these variants are potentially damaging to protein function. To assess changes in protein conformation, protein 3D-structures for three SAND missense variants, p.G212S, p.W234R and p.R246T, were obtained using the HOPE project analysis program (Have Our Protein Explained, http://www.cmbi.ru.nl/hope/) [Venselaar et al., 2010] (Supp. Table S3), based on the structure of the SAND domain of the homologous human SP100B (Protein Data Bank Entry 1H5P) (Supp. Figure S1). Together with previously published protein conformational changes in p.R226W [Faqeih et al., 2014] and p.Q264P [Vulto-van Silfhout et al., 2014], the variants identified in this study are likely to change DEAF1 protein conformation based on changes in amino acid charge, steric effect, hydrophobic or ionic interactions, and thus potentially damage protein function. The predicted pathogenicities of the variants were classified according to American College of Medical Genetics and Genomics (ACMG) guidelines [Richards et al., 2015] (Table 1).
Clinical features of individuals with DEAF1 variants
The key clinical features of six unrelated subjects harboring the potentially deleterious DEAF1 variants described above are summarized in Table 2. All of the subjects, four female and two males with an age range from 2–15 years, displayed developmental abnormalities including intellectual disability (n = 6), motor delay (n = 5), absent/delayed speech (n = 4), regression (n = 2), and/or prenatal growth retardation (n = 1). Additional neurological features included seizures (n = 5), hypotonia (n = 4), ataxia (n = 3), sleeping problems (n = 2) and hearing loss (n = 1). Previous reports have indicated that some individuals with pathogenic DEAF1 variants display a high threshold to pain, however, this feature was not reported for individuals in our cohort at the time of evaluation. Neurobehavioral abnormalities observed in our cohort included autism (n = 5), aggression (n = 2) and self-injury (n = 2), with one individual displaying pica. Previous studies have reported that a majority of individuals harboring DEAF1 mutations experience mood swings and excessive irritability, but these features were not indicated in our patients. Other dysmorphic features frequently observed in our cohort included brain abnormalities (n = 4), mild craniofacial features (n = 3), and various skeletal abnormalities. Several subjects were also noted to have frequent Infections, feeding difficulties and digestive abnormalities (Table 2).
Table 2.
Clinical features of the six individuals with intellectual disability harboring pathogenic DEAF1 variants
| Subject | #1 (597158) | #2 (615391) | #3 (M2647) [Berger et al., 2017] |
#4 (561320) [Wenge et al., 2017] |
#5 (563832) | #6 (538820) | Subjects reported in the literature (n = 10) [Vissers et al., 2010; Rauch et al., 2012; Vulto et al., 2014; Faqeih et al., 2014; Rajab et al., 2015; Gund et al., 2016] |
|
|---|---|---|---|---|---|---|---|---|
| Profiles | DEAF1 variants* | p.Gly212Ser | p.Arg226Trp | p.Trp234Arg | p.Arg246Thr | p.Gln264Pro | p.Lys305del | p.Arg224Trp, p.Arg226Trp, p.Ile228Ser, p.Arg254Ser, p.Gln264Pro and p.(Gly292Profs*) |
| Age at evaluation | 15y | 2y | 4y | 8y | 12y | 3y | 2–20y | |
| Gender | F | M | M | F | F | F | 8 M and 2 F | |
| Family history | N.A. | + | N.A. | N.A. | N.A. | +, Brother mild autism | 6/10 | |
| Development abnormalities | ID | + | + | + | + | + | + | Moderate to severe ID (10/10) |
| Motor delay | + | + | + | + | N.A. | + | Mild-to-moderate motor problem and developmental delay (10/10) | |
| Speech problems | Absent | N.A. | Expressive speech delay (non-verbal) | Speech delay | N.A. | Speech delay | Severe speech delay and severely affected expressive speech (10/10) | |
| Regression | + | N.A. | 1y skill regression | N.A. | N.A. | N.A. | 2/10 | |
| Prenatal growth retardation | N.A. | N.A. | N.A. | Slight prenatal growth retardation | N.A. | N.A. | N.A. | |
| Neurological abnormalities | Seizures | + | N.A. | +, normal EEG | Partial seizures | + | + | 7/10 |
| Hypotonia | N.A. | + | + | N.A. | + | Central hypotonia | 7/10 | |
| Ataxia | N.A | N.A | Slight tremor | N.A | + | + | Abnormal walking pattern (4/10), involuntary movements and drooling (3/10), dyskinesia (3/10), brisk reflexes without spasticity (1/10) | |
| Sleeping problems | N.A. | N.A. | + | Episodic | N.A. | N.A. | Sleep problems (5/10) | |
| Hearing loss | N.A. | N.A. | N.A. | Mild low frequency hearing loss | N.A. | N.A. | 2/10 | |
| Other | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | High pain threshold (4/10) | |
| Behavioral abnormalities | Autistic phenotype | + | N.A. | +, Repetitive behavior | + | + | + | 6/10, Poor eye contact (4/10), repetitive behavior (4/10) |
| Aggressive behavior | N.A. | N.A. | + | + | N.A. | N.A. | 6/10 | |
| Self-injurious behavior | N.A. | N.A. | +, Head banging, hits/bites self | + | N.A. | N.A. | 1/10 | |
| Others | N.A. | N.A. | Pica | N.A. | N.A. | N.A. | Mood swing and excessive irritability (7/10), happy predisposition (4/10), self-stimulatory behavior (4/10), hyperactivity (3/10), compulsive behavior (3/10), fascinations (2/10), | |
| Dysmorphologies | Brain abnormality | Borderline macrocephaly | Microcephaly | OTC WNL, normal MRI | Brain MRI showed slightly low cerebellar tonsils and EEG showed left-sided central parietal interictal spikes | N.A. | N.A. | Microcephaly (3/10), bilateral and symmetrical white matter abnormalities (3/10), symmetric T2 lesions in the basal ganglia (2/10), abnormal brain MRI (1/10), volume loss of the corpus callosum and brainstem in MRI (1/10) |
| Craniofacial abnormality | Mild dysmorphology | N.A. | Mild dysmorphology | Left-sided ear tag | + | N.A. | Straight eyebrows (4/10), full nasal tip (4/10), prominent chin (3/10), cupid’s bow in upper lip (3/10), full lower lip (3/10), high arched palate (2/10), triangular face (1/10), widow’s peak (1/10), flat face (1/10), frontal bossing (1/10), upslant (1/10), epicanthic folds (1/10) | |
| Skeletal abnormality | Mild dysmorphology, skeletal large hands | N.A. | Pes planus, mild 2–3 syndactyly, 4th toe overlaps, 3rd joint laxity | History of congenital left hip dislocation | N.A. | N.A. | Skin syndactyly in toes 2 and 3 (4/10), brachydactyly (2/10), fetal finger pads (2/10), clinodactyly (2/10), hyperlaxity (2/10), sacral dimple (2/10), flat feet (1/10), sandal gap (1/10) | |
| Others | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | Thin fair hair (3/10), low-set hairline (1/10), left nipple inverted (1/10), dry skin (1/10), scrotal raphe (1/10) | |
| Additional phenotypes | Infections | N.A. | N.A. | Frequent otitis media | N.A. | N.A. | Recurrent infections (4/10) | |
| Feeding difficulties | N.A. | N.A. | Early feeding issues | N.A. | N.A. | N.A. | Feeding difficulties (3/10) | |
| Digestive abnormality | Gastroesophageal reflux | N.A. | Gastroesophageal reflux | Constipation | N.A. | N.A. | N.A. |
Abbreviations: Y (years), F (female), M (male), fs (frame shift), ID (intellectual disability), OTC (orbitofrontal cortex) WNL (within normal limits), EEG (electroencephalogram), MRI (magnetic resonance imaging), N.A. (not available)
The DNA variant numbering system used in this study is based on human DEAF1 cDNA sequence (RefSeq NM_021008.3). Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Functional characterization of DEAF1 variants
We performed functional assays to assess the effects of the amino acid substitutions or deletion on human DEAF1 protein activity. Previous studies have shown that the WT DEAF1 protein represses its own promoter activity through binding to a site in its promoter region [Vulto-van Silfhout et al., 2014]. Using a luciferase reporter assay, we showed that DEAF1 transcriptional repression activity is significantly impaired in p.G212S, p.W234R, p.R246T and p.K305del variants relative to WT DEAF1 (*p<0.01, one-way ANOVA with Dunnett’s multiple comparison; Figure 2). By contrast, the p.R226W variant showed no significant change in transcriptional repression activity. A DEAF1 mutation in the KDWK DNA binding motif of the SAND domain, ADWA (NM_021008.3:c.[748A>G;749A>C;757A>G;758A>C] p.[Lys250Ala;Lys253Ala]), that abolishes DEAF1 DNA binding [Bottomley et al., 2001; Vulto-van Silfhout et al., 2014], was used as a positive control for impaired DEAF1 transcriptional repression activity (Figure 2). To identify potentially subtle differences in DEAF1 transcriptional repression activity due to the p.R226W variant, luciferase assays were performed using cells transfected with decreasing quantities of WT DEAF1 or p.R226W expression constructs. A dose-dependent decrease in repression activity was observed for both WT and p.R226W, however no significant alterations in repression activity were observed when comparing WT and p.R226W activities at similar expression plasmid concentrations (Supp. Figure S2).
Figure 2. DEAF1 variants have impaired transcriptional repression activity.
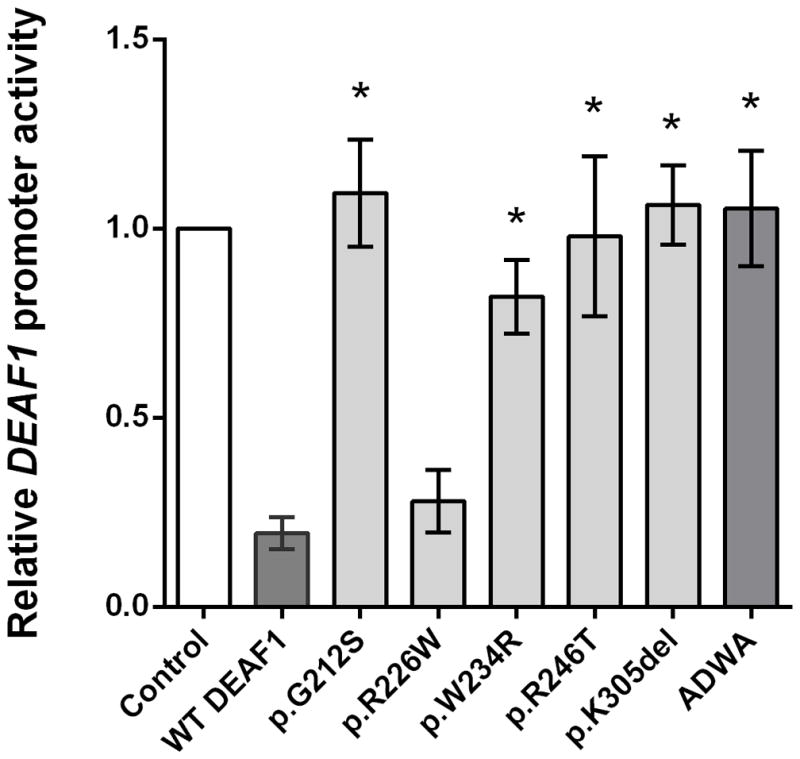
Expression of a luciferase reporter gene linked to the DEAF1 promoter was repressed in HEK293T cells by co-expressed WT DEAF1, but not by co-expressed DEAF1 variants p.G212S, p.W234R, p.R246T or p.K305del. A DEAF1 SAND domain variant, ADWA, located within the KDWK DNA binding motif and previously shown to be defective in transcriptional repression activity [Bottomley et al., 2001; Vulto-van Silfhout et al., 2014], was used as a control. Each bar represents the mean +/− SEM of the normalized luciferase activities from at least three independent experiments with the activity of the DEAF1 promoter co-transfected with the control plasmid pcDNA3 set to 1. Data were analyzed by one-way ANOVA followed by Dunnett’s multiple comparison post hoc test. *p < 0.01 for expression in the presence of co-expressed DEAF1 variants vs. WT DEAF1. Co-expression of the previously identified DEAF1 variant p.R226W did not show a significant difference in repression activity compared to WT DEAF1.
Proteins were tested for DNA binding affinity to the DEAF1 consensus sequences in electrophoretic mobility shift assays (EMSA) using two verified probes, N52-69 [Bottomley et al., 2001; Michelson et al., 1999] and S6con [Jensik et al., 2014; Vulto-van Silfhout et al., 2014]. Compared to WT DEAF1, the variants p.G212S, p.W234R, p.R246T showed significantly decreased binding to both dsDNA probes. By contrast, the p.K305del variant displayed binding similar to that of WT-DEAF1. The control ADWA variant showed severely diminished binding to both dsDNA probes (Figure 3). EMSA was also performed using a series of decreasing amounts of WT, p.R226W, or p.K305del proteins to detect changes in relative binding affinity to the N52-69 probe. A dose-dependent decrease in DNA binding was observed for each protein, however no observable differences in relative DNA binding affinity were observed comparing WT DEAF1 and p.K305del or p.R226W proteins (Supp. Figure S3 and S4).
Figure 3. DEAF1 variants have altered DNA binding.

Electrophoretic mobility shift assays (EMSA) showed decreased DNA binding affinity of the four novel DEAF1 variants to the 11-space and 6-space double stranded DNA DEAF1 binding sequences [Vulto-van Silfhout et al., 2014], compared to WT DEAF1. The DEAF1 ADWA variant, which has previously been shown to be defective in binding DNA [Bottomley et al., 2001], was used as a negative control.
Immunofluorescence staining in transfected cells was used to assess whether the DEAF1 variants had altered subcellular localizations. WT DEAF1, p.G212S, p.W234R, p.R246T and pR226W localized exclusively to the nucleus, while the p.K305del variant was predominantly localized to the cytoplasm (Figure 4a). Possible protein interactions between WT DEAF1 and the p.K305del variant were examined using an immunofluorescence interaction assay in cells co-expressing WT DEAF1 and p.K305del. Most of the p.K305del protein re-localized to the nucleus in cells co-expressing WT DEAF1, with a fraction of the WT DEAF1 localized to the cytoplasm (Figure 4b). These observations suggest that, in the heterozygous condition, interactions between WT and mutant DEAF1 can alter the localization of both proteins.
Figure 4. Subcellular localization of DEAF1 variants and changes in localization of WT DEAF1 and p.K305del in cells co-expressing both proteins.

A. Immunofluorescence analysis of WT DEAF1 and DEAF1 variants in transfected HEK293T cells revealed nuclear localization for variants p.G212S, p.R226W, p.W234R and p.R246T, and cytoplasmic localization for p.K305del. DEAF1 protein is shown in red in the upper panels, nuclei in blue in the middle panels, and merged signals in the bottom set of panels. B. Protein interactions between WT DEAF1 and p.K305del were monitored by immunofluorescence analysis. Co-expression of these two proteins resulted in nuclear and cytoplasmic localization of both proteins (columns 3 and 4). HA epitope-tagged DEAF1 protein is shown in red in the upper panel, FLAG epitope-tagged p.K305del in green in the middle, and nuclei in blue in the bottom.
Discussion
Trio-based CES is increasingly becoming a first-tier clinical test to identify genetic variants associated with pediatric neurological abnormalities, which are frequently the result of de novo variants in probands [Rauch et al., 2012; Vissers et al., 2010]. To determine whether de novo variants within protein coding regions are pathogenic, it is essential to perform functional assays to assess the effect of the amino acid substitution on properties of the encoded protein. In this study, we identified two previously unreported de novo DEAF1 variants within the SAND and NLS domains, p.G212S and p.K305del, in a cohort of individuals with neurological abnormalities of an unknown etiology through CES. We provide molecular evidence suggesting that these variants and two reported DEAF1 variants (p.W234R [Berger et al., 2017], p.R246T [Wenger et al., 2017]) impair DEAF1 transcriptional activity, impair DNA-binding, and alter subcellular localization, and as a consequence, likely contribute to the associated phenotypes in individuals with DAND.
Previous studies have shown that the SAND domain mediates DNA-binding, multimerization, and protein–protein interactions required for proper DEAF1 function [Bottomley et al., 2001; Jensik et al., 2004; Jensik et al., 2012]. In particular, the DEAF1 SAND domain is required to repress its own promoter activity [Bottomley et al., 2001] and stimulate or repress the expression of various target genes, e.g. eukaryotic translation initiation factor four gamma three (Eif4g3) [Jensik et al., 2014] and 5-hydroxytryptamine receptor 1A (5-HT1A) [Czesak et al., 2012; Czesak et al., 2006]. The SAND domain also mediates DEAF1 multimerization, which is required for DNA binding [Jensik et al., 2004] and interactions with X-ray repair cross-complementing protein 6 (XRCC6 or Ku70) [Jensik et al., 2012]. Functional assays in this study show that the de novo SAND variants p.G212S, p.W234R and p.R246T variants, eliminate both DEAF1 transcriptional repression activity at the DEAF1 promoter and DEAF1-DNA interactions with DEAF1 consensus sequences. The alterations in DEAF1 function due to the p.G212S, p.W234R, and p.R246T variants are similar to the previously described SAND domain variants p.R224W, p.I228S. p.R254S, and p.Q264P [Vulto-van Silfhout et al., 2014].
Homozygosity of the p.R226W variant was previously identified in three individuals with ID, microcephaly, and hypotonia from two consanguineous families. Two of these patients presented with white matter disease and cortical gyri abnormalities, and one experienced fatal complications, but no functional consequences of the p.R226W variant have been reported [Faqeih et al., 2014; Gund et al., 2016]. Homozygosity of the same variant was also identified in our cohort. In silico analysis revealed that the tryptophan substitution may sterically hinder normal protein folding [Faqeih et al., 2014]. In this study, we showed that p.R226W localizes to the nucleus (Figure 4) and observed no significant alterations in DEAF1 transcriptional activity (Figure 2). Although deleterious effects of the p.R226W variant on DEAF1 function have not yet been detected, it is plausible that this amino acid substitution produces pathogenic effects by disrupting transcriptional regulation of specific downstream DEAF1 target genes or modulating DEAF1-protein interactions. Further study will be needed to identify alterations in DEAF1 function/activity due to the p.R226W substitution.
The monopartite NLS in DEAF1 is necessary for proper subcellular localization and protein-protein interactions [Jensik et al., 2004]. Here, we identified a deletion within the DEAF1 NLS domain, p.K305del, that causes the mutant protein to localize primarily to the cytoplasm (Figure 4). Although p.K305del does not appear to affect the binding of DEAF1 to DNA in vitro, the observed inability of exogenously expressed p.K305del DEAF1 to repress transcriptional activity at the DEAF1 promoter (Figure 2) is likely the consequence of its cytoplasmic localization and limited access to the DEAF1 promoter in the nucleus. The lack of access to DNA in the nucleus may result in similar transcriptional outcomes to those observed with the de novo DEAF1 SAND variants. When we exogenously co-expressed WT DEAF1 and p.K305del in HEK293T cells to mimic the heterozygous condition in Subject 6, nuclear and cytoplasmic localizations were observed for both proteins (Figure 4b). These data likely indicate that DEAF1-DEAF1 protein interactions can re-localize p.K305del DEAF1 to the nucleus and partially localize WT protein to the cytoplasm. These observations suggest that individuals heterozygous for p.K305del may have constitutively insufficient quantities of nuclear WT DEAF1, possibly resulting in impaired transcriptional repression activity. The cytoplasmic DEAF1 may also re-localize some as-yet-to-be-determined cofactor that is needed for proper DEAF1 function in the nucleus. Further functional studies are needed to elucidate the mechanisms underlying possible pathogenic effects of this variant.
In summary, this study identified novel de novo DEAF1 variants in patients with intellectual disability, developmental delay and autism spectrum disorder, expanding the phenotype associated with this neurodevelopmental condition and provides molecular evidence that not only variants in the SAND domain but also in the NLS domain may contribute to DAND. Together, these findings illustrate the importance of elucidating the functional consequences of CES genetic variants of uncertain significance (VUS) to provide correct variant annotation and facilitate clinical diagnoses.
Supplementary Material
Acknowledgments
Grant Sponsors: Smith-Magenis Syndrome Research Foundation, National Science Foundation-China grant (31200937), Shanghai Health and Family Planning Commission grant (20144Y0106), China Scholarship Council (201306050007), National Institutes of Health Grant NINDS (5R21NS091724) and Intramural Research Program of the National Human Genome Research Institute, NIH.
We thank the patients and families for their participation in this study, and Jill Mokry, M.S.in CGC BCM for the clinical whole exome sequencing and data analysis. This work was supported by the Smith-Magenis Syndrome Research Foundation (SHE), National Science Foundation-China grant 31200937 (LC), Shanghai Health and Family Planning Commission grant 20144Y0106 (LC), China Scholarship Council 201306050007 (LC), and National Institutes of Health Grant NINDS 5R21NS091724 (PJ) Support of the contributions of ACMS and SB was provided by the Intramural Research Program of the National Human Genome Research Institute, NIH.
Footnotes
Disclosure statement: The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript. The authors declare no competing interests.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental health disorders: DSM-5. 5. American Psychiatric Publishing; 2013. [Google Scholar]
- Berger SI, Ciccone C, Simon KL, Malicdan MC, Vilboux T, Billington C, Fischer R, Introne WJ, Gropman A, Blancato JK, Mullikin JC, Gahl WA, Huizing M, Smith AC NISC Comparative Sequencing Program. Exome analysis of Smith-Magenis-like syndrome cohort identifies de novo likely pathogenic variants. Hum Genet. 2017;136(4):409–420. [Google Scholar]
- Bottomley MJ, Collard MW, Huggenvik JI, Liu Z, Gibson TJ, Sattler M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat Struct Biol. 2001;8(7):626–633. doi: 10.1038/89675. [DOI] [PubMed] [Google Scholar]
- Czesak M, Le Francois B, Millar AM, Deria M, Daigle M, Visvader JE, Anisman H, Albert PR. Increased serotonin-1A (5-HT1A) autoreceptor expression and reduced raphe serotonin levels in deformed epidermal autoregulatory factor-1 (Deaf-1) gene knock-out mice. J Biol Chem. 2012;287(9):6615–6627. doi: 10.1074/jbc.M111.293027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czesak M, Lemonde S, Peterson EA, Rogaeva A, Albert PR. Cell-specific repressor or enhancer activities of Deaf-1 at a serotonin 1A receptor gene polymorphism. J Neurosci. 2006;26(6):1864–1871. doi: 10.1523/JNEUROSCI.2643-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faqeih EA, Al-Owain M, Colak D, Kenana R, Al-Yafee Y, Al-Dosary M, Al-Saman A, Albalawi F, Al-Sarar D, Domiaty D, Daghestani M, Kaya N. Novel homozygous DEAF1 variant suspected in causing white matter disease, intellectual disability, and microcephaly. Am J Med Genet A. 2014;164A(6):1565–1570. doi: 10.1002/ajmg.a.36482. [DOI] [PubMed] [Google Scholar]
- Gross CT, McGinnis W. DEAF-1, a novel protein that binds an essential region in a Deformed response element. EMBO J. 1996;15(8):1961–1970. [PMC free article] [PubMed] [Google Scholar]
- Gund C, Powis Z, Alcaraz W, Desai S, Baranano K. Identification of a syndrome comprising microcephaly and intellectual disability but not white matter disease associated with a homozygous c.676C>T p.R226W DEAF1 mutation. Am J Med Genet A. 2016;170(5):1330–1332. doi: 10.1002/ajmg.a.37580. [DOI] [PubMed] [Google Scholar]
- Hahm K, Sum EY, Fujiwara Y, Lindeman GJ, Visvader JE, Orkin SH. Defective neural tube closure and anteroposterior patterning in mice lacking the LIM protein LMO4 or its interacting partner Deaf-1. Mol Cell Biol. 2004;24(5):2074–2082. doi: 10.1128/MCB.24.5.2074-2082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM, Miller LJ, Jani-Acsadi A, Pestronk A, Shy ME, Muntoni F, Vallee RB, Baloh RH. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78(22):6. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenvik JI, Michelson RJ, Collard MW, Ziemba AJ, Gurley P, Mowen KA. Characterization of a nuclear deformed epidermal autoregulatory factor-1 (DEAF-1)-related (NUDR) transcriptional regulator protein. Mol Endocrinol. 1998;12(10):1619–1639. doi: 10.1210/mend.12.10.0181. [DOI] [PubMed] [Google Scholar]
- Jensik PJ, Huggenvik JI, Collard MW. Identification of a nuclear export signal and protein interaction domains in deformed epidermal autoregulatory factor-1 (DEAF-1) J Biol Chem. 2004;279(31):32692–32699. doi: 10.1074/jbc.M400946200. [DOI] [PubMed] [Google Scholar]
- Jensik PJ, Huggenvik JI, Collard MW. Deformed epidermal autoregulatory factor-1 (DEAF1) interacts with the Ku70 subunit of the DNA-dependent protein kinase complex. PLoS One. 2012;7(3):e33404. doi: 10.1371/journal.pone.0033404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensik PJ, Vargas JD, Reardon SN, Rajamanickam S, Huggenvik JI, Collard MW. DEAF1 binds unmethylated and variably spaced CpG dinucleotide motifs. PLoS One. 2014;9(12):e115908. doi: 10.1371/journal.pone.0115908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, JS A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lemonde S, Turecki G, Bakish D, Du L, Hrdina PD, Bown CD, Sequeira A, Kushwaha N, Morris SJ, Basak A, Ou XM, Albert PR. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci. 2003;23(25):8788–8799. doi: 10.1523/JNEUROSCI.23-25-08788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32(2):419–426. doi: 10.1016/j.ridd.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Michelson RJ, Collard MW, Ziemba AJ, Persinger J, Bartholomew B, Huggenvik JI. Nuclear DEAF-1-related (NUDR) protein contains a novel DNA binding domain and represses transcription of the heterogeneous nuclear ribonucleoprotein A2/B1 promoter. J Biol Chem. 1999;274(43):30510–30519. doi: 10.1074/jbc.274.43.30510. [DOI] [PubMed] [Google Scholar]
- Rajab A, Schuelke M, Gill E, Zwirner A, Seifert F, Morales Gonzalez S, Knierim E. Recessive DEAF1 mutation associates with autism, intellectual disability, basal ganglia dysfunction and epilepsy. J Med Genet. 2015;52(9):607–611. doi: 10.1136/jmedgenet-2015-103083. [DOI] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Ropke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schrock E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380(9854):1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rump P, Jazayeri O, van Dijk-Bos KK, Johansson LF, van Essen AJ, Verheij JB, Veenstra-Knol HE, Redeker EJ, Mannens MM, Swertz MA, Alizadeh BZ, van Ravenswaaij-Arts CM, Sinke RJ, Sikkema-Raddatz B. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med Genomics. 2016;9:7. doi: 10.1186/s12920-016-0167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veraksa A, Kennison J, McGinnis W. DEAF-1 function is essential for the early embryonic development of Drosophila. Genesis. 2002;33(2):67–76. doi: 10.1002/gene.10090. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA. A de novo paradigm for mental retardation. Nat Genet. 2010;42(12):1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, Raghavan R, Reardon SN, Jarrett K, McIntyre T, Bulinski J, Ownby SL, Huggenvik JI, McKnight GS, Rose GM, Cai X, Willaert A, Zweier C, Endele S, de Ligt J, van Bon BW, Lugtenberg D, de Vries PF, Veltman JA, van Bokhoven H, Brunner HG, Rauch A, de Brouwer AP, Carvill GL, Hoischen A, Mefford HC, Eichler EE, Vissers LE, Menten B, Collard MW, de Vries BB. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. Am J Hum Genet. 2014;94(5):649–661. doi: 10.1016/j.ajhg.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltl S. Intellectual disability: novel mutations in DEAF1 cause speech impairment and behavioral problems. Clin Genet. 2014;86(6):507–508. doi: 10.1111/cge.12475. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, Williams M, King C, Greenhalgh L, Newbury-Ecob R, Ellard S. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89(2):308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19(2):209–214. doi: 10.1038/gim.2016.88. [DOI] [PubMed] [Google Scholar]
- Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, de Vries BB, Schoots J, Lugtenberg D, Hamel BC, van Bokhoven H, Brunner HG, Veltman JA, Kleefstra T. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. 2012;49(3):179–183. doi: 10.1136/jmedgenet-2011-100542. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.