

Abstract
We examined if baseline level of cognitive reserve (CR) and of Alzheimer’s disease (AD) biomarkers modify the rate of change in cognition among individuals with normal cognition at baseline (n=303, mean baseline age = 57 years, mean follow-up = 12 years); 66 participants subsequently developed Mild Cognitive Impairment (MCI) or dementia due to AD. CR was indexed by years of education, reading, and vocabulary measures. AD biomarkers were measured with a composite score composed of measures of amyloid, phosphorylated tau and neurodegeneration. Higher CR scores were associated with better cognitive performance, but did not modify the rate of change in cognition among those who remained cognitively normal, nor among those who progressed to MCI prior to symptom onset, independent of baseline biomarker levels. However, higher CR scores were associated with faster cognitive decline after symptom onset of MCI. These results suggest that the mechanism by which CR mediates the relationship between pathology and cognitive function is by delaying the onset of symptoms rather than reducing the rate of cognitive decline.
Keywords: Cognitive reserve, preclinical Alzheimer’s disease, biomarkers, cognitive change, longitudinal
1. Introduction
It is now recognized that Alzheimer’s disease (AD) pathology begins to accumulate in the brain many years prior to the onset of Mild Cognitive Impairment (MCI), when individuals are still cognitively normal (Sperling et al., 2011). It is therefore of considerable clinical importance to identify factors that may delay progression from normal cognition to MCI. One potential factor is cognitive reserve (CR). Despite a number of previous studies, however, it remains unclear if CR modifies cognitive trajectories during this ‘preclinical phase’ of AD.
CR is a theoretical concept that proposes that greater lifetime engagement in cognitively stimulating activities modifies the brain in such a way that the negative effects of brain pathology on cognition are reduced (Stern, 2009). Supporting the concept of CR, many studies have reported a reduced risk of dementia among individuals with higher educational or occupational attainment (e.g., Stern et al., 1994) and greater engagement in cognitive leisure activities (e.g., Scarmeas et al., 2001; Wilson et al., 2002); for a meta-analysis, see Sachdev & Valenzuela (2006a).
Despite the evidence for the beneficial effects of CR on onset of dementia, previous studies have generated inconsistent findings with respect to rates of cognitive change over time. Some studies have reported that a higher level of CR is associated with a reduced rate of cognitive decline (e.g., Pool et al., 2016; Yaffe et al., 2011; Zahodne et al., 2015), while others have reported only baseline differences in cognition by CR level, but no difference in the rate of cognitive change (e.g., Karlamangla et al., 2009; Piccinin et al., 2013; Wilson et al., 2009; Zahodne et al., 2011); still others have found greater decline among individuals with higher CR for some cognitive measures but not all (Glymour et al., 2012; Gottesman et al., 2014; Proust-Lima et al., 2008; Singh-Manoux et al., 2011).
There are several possible explanations for these discrepancies. For example, many long-term longitudinal studies on this subject have likely included a range of non-demented individuals, with little or no screening for cognitive impairment at baseline, potentially resulting in a mixture of cognitively normal and MCI participants (e.g., Amieva et al., 2014; Muniz-Terrera et al., 2009; Pool et al., 2016; Sperling et al., 2011; Yaffe et al., 2011; Zahodne et al., 2015). If the relationship between CR and longitudinal cognitive performance differs by degree of cognitive impairment, studies with different proportions of cognitively normal and MCI participants may produce different results. It is also possible that participants who are cognitively normal when first examined differ in levels of underlying brain pathology, and these differences are responsible for differing rates of cognitive decline over time. Only one study, to our knowledge, has addressed this issue, but the follow-up was relatively short (mean follow-up 2.7 years, Vemuri et al., 2015).
The goal of the current study, therefore, was to examine the association between baseline level of CR, baseline biomarker levels of Alzheimer disease, and the long-term cognitive trajectories of 303 middle-aged and older individuals who were cognitively normal at baseline and have been followed for up to 20 years. The long follow-up period of the current study allowed us to examine additional issues not previously addressed. First, we tested if the relationship between CR and cognitive trajectories differed for individuals who were initially cognitively normal but subsequently developed MCI, compared to individuals who remained cognitively normal. Second, we tested whether baseline level of CR differentially modifies the cognitive trajectories prior to and after the emergence of the initial symptoms of MCI. This issue addresses an important theoretical prediction made by Stern’s (2009) hypothetical model of CR, which predicts that individuals with high levels of CR perform better on cognitive tests than individuals with low CR prior to the onset of cognitive impairment, but show a faster rate of cognitive decline after the onset of cognitive impairment because they tend to harbor more pathology. Lastly, we assessed whether the relationship between CR and cognitive change differed when accounting for individual differences in baseline AD biomarkers using a composite score that combines measures previously shown to predict progression from normal cognition to onset of symptoms of MCI in the same cohort; this biomarker composite contains elements of each of the three major biomarker categories in use in the field: amyloid accumulation, phosphorylated tau and neuronal injury (Jack et al., 2016).
2. Materials and Methods
2.1. Study Design
The present study reports on data from the BIOCARD study, which was initiated at the National Institutes of Health (NIH) in 1995. The overarching goal of the study was to identify variables among cognitively normal individuals that could predict the subsequent development of mild to moderate symptoms of AD. By design, approximately 75% of the participants had a first degree relative with a history of dementia of the Alzheimer type. The study was stopped in 2005 for administrative reasons and re-established at Johns Hopkins University (JHU) in 2009. While the study was at the NIH, participants were administered a comprehensive neuropsychological battery annually. MRI scans, CSF, and blood specimens were obtained approximately every two years. Since the study has been at JHU, participants have received annual clinical and cognitive assessments and provided blood specimens. In 2015, the biannual collection of MRI and CSF biomarkers was reinitiated, and amyloid imaging was begun. See Figure 1 for a timeline of the study. The JHU Institutional Review Board approved this study.
Figure 1. Timeline showing the design of the BIOCARD study.
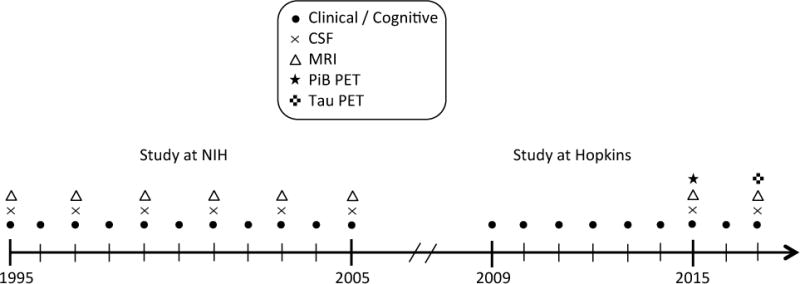
Types of data collected each year for the BIOCARD study between 1995 and 2016.
2.2. Selection of Participants
Participants were recruited by the staff of the geriatric psychiatry branch of the intramural program of the National Institute of Mental Health. A total of 349 participants were enrolled over time, beginning in 1995 and ending in 2005 and provided written informed consent. At baseline, all participants completed a comprehensive evaluation, consisting of a physical and neurological examination, an electrocardiogram, standard laboratory studies, and detailed neuropsychological testing. Individuals were excluded from participation if they were cognitively impaired, as determined by cognitive testing, or had significant medical problems such as severe cerebrovascular disease, epilepsy, or alcohol or drug abuse. See Supplementary Materials 1 for details regarding the selection of participants. MRI scans were obtained from 325 participants and CSF samples were from 307 participants via lumbar puncture.
Participants were excluded from analysis for the following reasons: 31 had not yet re-enrolled in the study or had withdrawn; 12 had an estimated age of onset of symptoms of MCI at or prior to their baseline visit; 3 had missing CR composite scores. Thus, analyses involving clinical and cognitive data are based on 303 individuals. Analyses involving the biomarker composite score are based on 171 individuals who provided both CSF and MRI data within 12 months of their baseline cognitive evaluation.
2.3. Clinical and Cognitive Assessments
Since the study was re-established at JHU, annual visits have included neuropsychological testing and a clinical evaluation consisting of a physical and neurological examination, record of medication use, behavioral and mood assessments, family history of dementia, history of symptom onset, and a Clinical Dementia Rating (CDR) with the participant and a collateral source (Hughes et al., 1982; Morris, 1993). The cognitive and clinical assessments at the NIH covered similar domains (for details, see Albert et al., 2014). Each participant included in our analyses received a consensus diagnosis by the staff of the JHU BIOCARD Clinical Core, prospectively starting in 2009 and retrospectively for the NIH visits. The diagnostic criteria followed the recommendations incorporated in the National Institute on Aging and the Alzheimer’s Association working group reports for the diagnosis of MCI (Albert et al., 2011) and dementia due to AD (McKhann et al., 2011). Details regarding the diagnostic process can be found in the Supplementary Materials 2. If a participant was impaired, the likely etiology of the impairment was identified and the age at which the clinical symptoms began was estimated, based primarily on the reports of the participant and collateral source derived from the CDR.
The main outcome variable was an a priori—derived cognitive composite score based on 4 measures that were identified previously to be the best combination of cognitive predictors of the time to progress from normal cognition to clinical symptom onset of MCI (Albert et al., 2014) in this cohort. These measures were (1) Paired Associates immediate recall of the Wechsler Memory Scale–Revised (Wechsler, 1987), (2) Logical Memory delayed recall (Story A) of the Wechsler Memory Scale–Revised, (3) Boston Naming (Kaplan et al., 1983), and (4) Digit-Symbol Substitution from the Wechsler Adult Intelligence Scale–Revised (WAIS-R, Wechsler, 1981). These measures were administered at the NIH and are part of the annual battery at JHU. To calculate the cognitive composite score, the individual measures were transformed to z-scores and then averaged, with the requirement that at least two of the four scores were present at a given visit. If more than two scores were missing for a given visit, the cognitive composite score was set to missing for that visit.
2.4. Cognitive Reserve Composite Score
Baseline CR was operationalized by a composite score based on three measures thought to reflect lifetime cognitive experiences: 1) baseline scores from the National Adult Reading Test (NART; H. E. Nelson, 1982); 2) baseline scores on the vocabulary subtest of the WAIS-R (Wechsler, 1981); and 3) years of education. These measures were z-scored and then averaged. As reported by Soldan et al. (2013), the individual measures were strongly correlated and loaded on a single factor in factor analysis. Composite scores such as these may be more sensitive proxies of CR than years of education alone (Manly et al., 2005; Pettigrew et al., 2013).
2.5. APOE Genetic Analysis
APOE ε4 genetic status, the main genetic risk factor for AD (Corder et al., 1993), was established in all but one of the participants (n=348). APOE alleles were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics, Worcester, MA). We coded APOE ε4 carrier status as 1 if an individual had at least one ε4 allele and as 0 otherwise. Eight individuals with one APOE ε4 and one APOE ε2 allele were excluded from analyses involving APOE ε4 status, since the APOE ε4 allele increases dementia risk (Corder et al., 1993), whereas the APOE ε2 allele reduces dementia risk (Corder et al., 1994).
2.5. Biomarker Composite Score
A biomarker composite score was developed to estimate the severity of the underlying pathophysiological processes associated with the development of MCI due to AD. It was composed of five measures, each of which has been shown to be associated with the time to progress from normal cognition to onset of symptoms of MCI in this cohort and, as noted above, also reflect the major biomarker categories in use in the field: Aβ1–42 and p-tau as measured in CSF (Moghekar et al., 2013), the volume of the right hippocampus and thickness of the right entorhinal cortex (Soldan et al., 2015), and mean thickness of seven cortical regions vulnerable to AD-related atrophy (Pettigrew et al., 2016). A brief description of each of these measures is provided below, with further details in the Supplementary Materials section. To create the composite score, the measures were converted to z-scores and then averaged. Prior to averaging, all z-scores, except those for p-tau were multiplied by −1 so that more positive biomarker composite scores indicate more pathology.
2.6. Cerebrospinal Fluid Assessments
The CSF samples were analyzed with the same protocol used in the Alzheimer Disease Neuroimaging Initiative. This protocol used a kit (xMAP-based AlzBio3; Innogenetics) run on a suspension array system (Bio-Plex 200; Bio-Rad). Details about the CSF assay are described in Supplementary Materials 3; additional details have been published elsewhere (Moghekar et al. (2012). For participants included in the current analysis, the mean time between the baseline cognitive assessment and CSF draw was 8.2 days (SD = 35.9, range = 0 to 363).
2.7. MRI Assessments and Regional Brain Reconstructions
Baseline MRI scans were acquired at the NIH on a GE 1.5T scanner using a standard multi-modal protocol. The baseline MRI measures that were used as part of the biomarker composite score were reconstructed from coronal SPGR (Spoiled Gradient Echo) scans (TR = 24, TE = 2, FOV = 256 × 256, thickness/gap = 2.0/0.0 mm, flip angle = 20, 124 slices). For participants included in the current analysis, the mean time between the baseline MRI scan and the baseline cognitive assessment was 8.6 days (SD = 40.4, range = 0 to 362).
The volume of the hippocampus and the thickness of the entorhinal cortex were obtained with a semi-automated method, based on large deformation diffeomorphic metric mapping (LDDMM) techniques (Miller et al., 2013). Hippocampal volume was normalized for head size by regressing it on total intracranial volume, and the standardized residual was used for further analyses. The mean cortical thickness measure of AD-vulnerable regions reflects the average of seven cortical regions, obtained using FreeSurfer 5.1 (http://surfer.nmr.mgh.harvard.edu/): inferior parietal cortex, superior parietal cortex, precuneus, inferior temporal cortex, middle temporal gyrus, temporal pole, and posterior cingulate cortex. Additional details concerning the MRI methods can be found in Supplementary Materials 4.
2.8. Statistical Analysis
Group differences in demographic variables at baseline were assessed by t-test or Wilcoxon rank sum test for continuous variables, as appropriate, or chi-square tests for dichotomous variables.
2.8.1. Cross-sectional analyses of baseline cognitive reserve, cognition, and biomarker composite score
Linear regressions were performed to test if the baseline biomarker composite score was associated with 1) the baseline cognitive composite score, 2) the baseline CR composite score, or 3) last follow-up diagnosis. Model 1 included the baseline cognitive composite score as the dependent variable, and the biomarker composite score, age, gender, diagnosis at last follow-up visit, and the diagnosis by biomarker interaction (i.e., cross-product) as independent variables. Follow-up diagnosis was coded dichotomously based on the diagnosis at the last follow-up visit (e.g., remained cognitively normal vs. progressed to MCI or dementia). Participants with a diagnosis of Impaired not MCI at last follow-up (n = 37) were included in the group of cognitively normal participants, but results were comparable when these participants were excluded from analysis. Model 2 used the baseline CR composite score as the dependent variable and included the same predictors as Model 1. Model 3 tested if individuals who progressed to MCI had higher baseline biomarker composite scores than the group who remained cognitively normal, covarying age, gender, and the CR composite score.
2.8.2. Cognitive reserve and longitudinal change in cognition: Relationship to follow-up diagnosis
To test if baseline level of CR influences the cognitive trajectories over time, we used general mixed regression models with linear effects of time and a random intercept and slope for each participant. In this type model, the intercept represents the estimated baseline cognitive score, the main effect of time represents the linear slope (i.e., rate of change) of the cognitive score over time, and the baseline cognitive score is not entered as a separate variable. The outcome variable was the cognitive composite score (including baseline and all follow-up scores); the predictors were baseline age, gender, CR composite score, last diagnosis, time, the interactions between each predictor and time (excluding the gender × time interaction, which was not significant in any model), and the interaction of CR composite score × last diagnosis × time. To determine if APOE ε4 status influences longitudinal cognitive performance, the model was re-run including the APOE ε4 indicator and its interaction with time as additional predictors.
Follow-up mixed effects models were run, separately by follow-up diagnostic status (normal vs. progressed). For participants who progressed to MCI, two additional models were run, including (1) only scores obtained prior to symptom onset of MCI and (2) only scores obtained after the onset of symptoms. Both models had the following predictors: baseline CR composite score, age, gender, time, and the age × time and CR × time interaction terms.
2.8.3. Cognitive reserve and longitudinal change in cognition: Relationship to baseline biomarker composite score
Linear mixed effects analysis was also used to examine whether baseline biomarker composite scores modified the association between baseline CR scores and cognitive change. This model was identical to the model including the diagnosis indicator variable (see above), except that the biomarker composite score was used instead of the diagnosis indicator.
3. Results
Table 1 shows baseline characteristics of the entire BIOCARD cohort as well as of individuals included in the analyses. Baseline characteristics for participants who remained cognitively normal and for those who progressed to MCI on follow-up are shown in Table 2. For the 66 who have progressed, the mean time from baseline to onset of symptoms of MCI was 7.1 years. Individuals who progressed to MCI were significantly older at baseline [t=5.0, p<0.0001], scored lower on all cognitive tests at baseline [all p<0.05], had lower CR composite scores [t=3.8, p=0.0003], and had higher biomarker composite scores [t=3.2, p=0.002].
Table 1.
Participant Characteristics at Baseline
| Variable | Cohort as a whole (N = 349) |
Participants in analyses of clinical data (N = 303) |
Participants in analysis of biomarker data (N = 170) |
|---|---|---|---|
| Age in years, mean (SD) | 57.3 (10.4) | 57.2 (10.2) | 56.5 (9.5) |
| Follow-up years, mean (SD) | 10.9 (4.6) | 12.1 (4.2) | 12.1 (3.7) |
| Gender, females (%) | 57.6% | 59.1% | 62.0% |
| Ethnicity, Caucasians (%) | 97.1% | 97.7% | 97.7% |
| APOE ε4 carriers (%) | 33.6% | 33.3% | 31.0% |
| MMSE, mean score (SD) | 29.5 (0.9) | 29.6 (0.8) | 29.6 (0.7) |
| Education, mean years (SD) | 17.0 (2.4) | 17.0 (2.4) | 17.2 (2.3) |
| NART, mean (SD) | 119.6 (7.9) | 120.1 (7.6) | 120.6 (7.2) |
| WAIS vocabulary, mean (SD) | 14.2 (2.3) | 14.3 (2.3) | 14.5 (2.2) |
| CR Composite, mean (SD) | 0.0 (0.8) | 0.0 (0.8) | 0.1 (0.7) |
| Paired Associates Immediate (SD) | 20.2 (3.4) | 20.5 (3.0) | 20.5 (2.9) |
| Logical Memory Delayed (SD) | 12.3 (4.0) | 12.6 (3.9) | 13.0 (3.8) |
| Boston Naming, % Correct (SD) | 96.0 (5.3) | 96.2 (5.3) | 96.6 (5.1) |
| Digit Symbol Substitution (SD) | 52.2 (11.7) | 52.8 (11.8) | 53.2 (11.4) |
| Cognitive Composite, mean (SD) | −0.1 (0.6) | −0.1 (0.6) | −0.0 (0.6) |
Table 2.
Participant Characteristics at Baseline by Follow-up Diagnosis
| Variable | Remained Normal (N = 237) |
Progressed to MCI/Dementia (N = 66) |
|---|---|---|
| Age in years, mean (SD) | 55.7 (9.4) | 63.0 (10.8)** |
| Follow-up years, mean (SD) | 12.1 (4.1) | 12.0 (4.4) |
| Gender, females (%) | 62.0% | 51.5% |
| Ethnicity, Caucasians (%) | 98.7% | 93.9% |
| APOE ε4 carriers (%) | 32.5% | 36.4% |
| MMSE, mean score (SD) | 29.6 (0.7) | 29.3 (1.0)* |
| Education, mean years (SD) | 17.2 (2.3) | 16.6 (2.5) |
| NART, mean (SD) | 121.1 (7.0) | 116.7 (8.8)** |
| WAIS-R vocabulary, mean (SD) | 14.6 (2.1) | 13.1 (2.5)** |
| CR Composite, mean (SD) | 0.1 (0.8) | −0.3 (0.9)** |
| Paired Associates Immediate (SD) | 20.9 (2.8) | 19.1 (3.0)** |
| Logical Memory Delayed (SD) | 13.1 (3.8) | 11.0 (3.7)** |
| Boston Naming, % Correct (SD) | 96.7 (4.8) | 94.1 (6.2)** |
| Digit Symbol Substitution (SD) | 54.8 (12.0) | 46.1 (7.8)** |
| Cognitive Composite, mean (SD) | 0.1 (0.6) | −0.5 (0.5)** |
| Biomarker Composite, mean (SD) | −0.1 (0.4) (N=136) |
0.3 (0.7)** (N=34) |
Abbreviations: NART = National Adult Reading Test; WAIS-R = Wechsler Adult Intelligence Scale - Revised. Significant differences between the group who remained normal and the group who progressed are indicated by asterisks:
p < 0.05;
p < 0.005.
3.1. Cross-sectional associations at baseline
For both Models 1 and 2, there was no interaction between the biomarker composite score and follow-up diagnosis [both p > 0.8], thus both models were rerun without these interaction terms. The results showed that at baseline, the biomarker composite score was not associated with either the cognitive composite score [Model 1, β (SE) = −0.10 (0.11), p=0.28] or the CR composite score [Model 2, β (SE) = −0.04 (0.11), p=0.72]. However, for both models there was a main effect of follow-up diagnosis, indicating that individuals who progressed to MCI at follow-up had lower baseline cognitive scores [Model 1, β (SE) = −0.45 (0.11), p<0.0001] and lower baseline CR scores [Model 2, β (SE) = −0.54 (0.15), p=0.0003]. In addition, older age was associated with lower cognitive composite scores at baseline [Model 1, β (SE) = −0.01 (0.004), p=0.03] and higher CR scores [Model 2, β (SE) = 0.02 (0.006), p = 0.0003]. Model 3 showed that the group who progressed to MCI had higher biomarker composite scores at baseline than the group who remained normal [β (SE) = 0.37 (0.10), p=0.0003].
3.2. Cognitive reserve, rate of change in cognition, and relationship to follow-up diagnosis
The results of the model testing the association between the baseline CR composite score and longitudinal cognitive trajectories are summarized in Table 3. The most important finding was the three-way interaction between the baseline CR score, follow-up diagnosis, and time [p=0.0016], indicating that the relationship between the CR score and cognitive change over time differs for individuals who remain cognitively normal compared to individuals who developed MCI or dementia at follow-up.
Table 3.
Results of linear mixed effects model including follow-up diagnosis as a predictor
| Model Predictors | Estimate | SE | p-value |
|---|---|---|---|
| Time | 0.0875 | 0.0180 | <0.0001 |
| Baseline age | −0.0143 | 0.0031 | <0.0001 |
| Gender (male) | −0.2828 | 0.0520 | <0.0001 |
| CR composite | 0.2500 | 0.0422 | <0.0001 |
| Follow-up diagnosis (MCI) | −0.2554 | 0.0756 | 0.0008 |
| Baseline age × time | −0.0013 | 0.0003 | 0.0001 |
| CR composite × time | 0.0013 | 0.0043 | 0.7601 |
| Follow-up diagnosis × time | −0.0551 | 0.0076 | <0.0001 |
| CR composite × follow-up diagnosis | 0.1299 | 0.0786 | 0.0992 |
| CR composite × follow-up diagnosis × time | −0.250 | 0.0078 | 0.0016 |
As shown in Figure 2, participants who remained cognitively normal showed improvements in cognitive performance and the degree of improvement did not differ for high and low CR individuals. In contrast, among individuals who became cognitively impaired (i.e., those in the preclinical phase when first evaluated), cognitive performance declined over time and the rate of decline was greater among those with higher baseline CR composite scores. The main effect of age and the age by time interaction were also significant [p≤0.0001], signifying lower cognitive performance and less improvement in performance over time with increasing age. Lastly, there was a main effect of CR [p<0.0001], signifying higher mean cognitive performance with increasing CR. The results were unchanged when APOE ε4 status and its interaction with time were added to the model, and neither APOE ε4 status nor its interaction with time was significant [both p>0.3].
Figure 2. Estimates of longitudinal cognitive change by follow-up diagnosis and baseline cognitive reserve composite score.
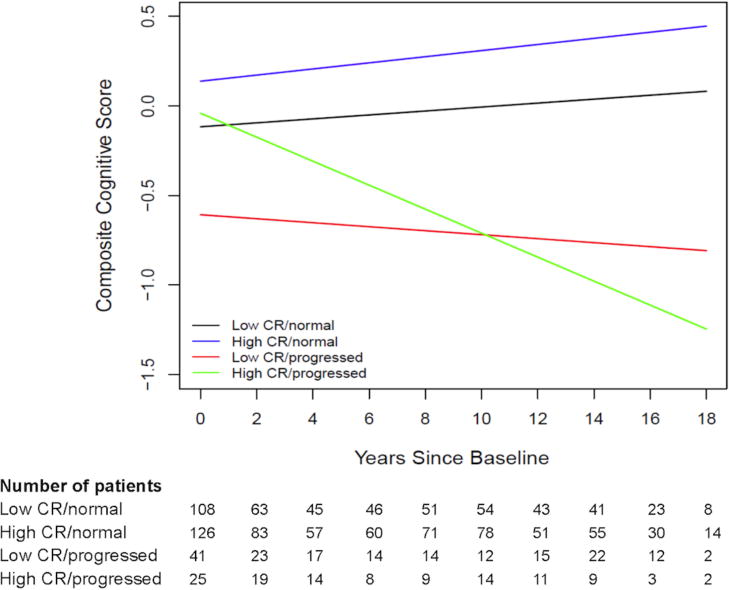
Estimates from linear mixed effects model predicting longitudinal cognitive composite scores over time among four groups: (1) low CR/normal (black line) are individuals with CR scores below the median who remained cognitively normal over time; (2) high CR/normal (blue line) are individuals with CR scores at or above the median who remained cognitively normal; (3) low CR/progressed (red line) are individuals with CR scores below the median who progressed to MCI at follow-up; and (4) high CR/progressed (green line) are individuals with CR scores at or above the median who progressed to MCI at follow-up. Estimates are adjusted for baseline age, gender, and the age × time interaction. The number of participants contributing data at each time point is shown in the table below the figure.
Of note, when the diagnosis variable and its interactions with time and CR were removed from the model, the interaction between CR and time was not significant [p=0.29, see Supplementary Materials 5], underscoring the importance of taking into account follow-up diagnosis when evaluating CR-cognition relationships.
3.2.1. Follow-up models in individuals who developed MCI or dementia
Among those who progressed, higher CR was associated with higher baseline cognitive scores and a faster rate of cognitive decline (p=0.0046), see Supplementary Materials 6 for full model results. The analysis including scores obtained prior to the onset of symptoms associated with MCI revealed a main effect of CR [β (SE) = 0.27 (0.08), p=0.0006] but no CR by time interaction [β (SE) = 0.017 (0.08), p = 0.27]. By comparison, the analysis including only scores obtained after the onset of symptoms of MCI showed both a main effect of CR [β (SE) = 0.69 (0.23), p=0.0044] and a CR × time interaction [β (SE) = −0.06 (0.02), p=0.0032]. This suggests a greater rate of cognitive decline among individuals with higher levels of CR after the onset of symptoms, but not before. A follow-up linear regression analysis furthermore showed that higher baseline CR scores were strongly associated with an older age of symptom onset [co-varying gender, t=4.29, p<0.0001].
3.2.2. Follow-up models in individuals who remained cognitively normal
Among individuals who remained cognitively normal over time, higher CR was associated with higher levels of cognitive performance (p<0.0001), but not with the rate of change in cognition (p=0.51), see Supplementary Materials 6 for full model results. In a post-hoc analysis, we also explored whether baseline CR modifies the age-related decline in the improvement in performance over time (presumed to be a practice effect). The interaction between CR, age, and time was not significant [β (SE)= −0.0004 (0.0004), p=0.28] suggesting that CR does not modify the age-related decrease in the practice effect among those who remain cognitively normal.
3.4. Cognitive reserve, rate of change in cognition and relationship to baseline biomarker composite scores
The results from the analysis examining whether baseline biomarker levels modify the association between CR and cognitive change are summarized in Table 4 (both full and reduced model excluding non-significant interaction terms). Both the main effects of CR and the biomarker composite score were significant, indicating better cognitive performance among individuals with higher CR scores and lower biomarker levels at baseline (Figure 2). The three-way interaction between CR, biomarker composite score and time was not significant (p=0.11), suggesting that baseline biomarker levels do not modify the association between CR and cognitive change. The CR score × biomarker score × time interaction was also not significant when the model was run separately among progressors and those who remained cognitively normal (both p>0.18, see Supplementary Materials 7 for full model results), but this may reflect the reduced sample size, particularly for progressors (N=34). Separate analyses including scores only before or after symptom onset for progressors are presented in Supplementary Materials 8.
Table 4.
Results of linear mixed effects model including baseline biomarker composite score as predictor
| Full Model | Reduced Model | |||||
|---|---|---|---|---|---|---|
| Model Predictors | Estimate | SE | p-value | Estimate | SE | p-value |
| Time | 0.0829 | 0.0230 | 0.0004 | 0.0957 | 0.0219 | <0.0001 |
| Baseline age | −0.0195 | 0.0041 | <0.0001 | −0.0189 | 0.0042 | <0.0001 |
| Gender (male) | −0.3431 | 0.0697 | <0.0001 | −0.3391 | 0.0690 | <0.0001 |
| CR composite | 0.3083 | 0.0503 | <0.0001 | 0.2975 | 0.0465 | <0.0001 |
| Biomarker composite | −0.2248 | 0.0716 | 0.0020 | −0.2506 | 0.0674 | 0.0003 |
| Baseline age × time | −0.0013 | 0.0004 | 0.0017 | −0.0015 | 0.0004 | 0.0001 |
| CR composite × time | −0.0022 | 0.0049 | 0.6464 | – | – | – |
| Biomarker composite × time | −0.0075 | 0.0075 | 0.3189 | – | – | – |
| CR composite × biomarker composite | 0.0290 | 0.0857 | 0.7357 | – | – | – |
| CR composite × biomarker composite × time | −0.0144 | 0.0091 | 0.1136 | – | – | – |
Note: The Reduced model excludes all non-significant interaction terms.
4. Discussion
This study investigated the relationship between level of CR, long-term diagnostic outcomes, a composite score composed of measures related to the underlying pathological processes associated with AD and long-term cognitive trajectories among middle-aged and older individuals with normal cognition at baseline. The main finding was that the relationship between level of CR and longitudinal change in cognition differs as a function of diagnostic outcome. Among individuals, who remained cognitively normal (who also had lower biomarker composite scores), higher levels of CR were associated with better cognitive performance, but CR had no impact on the rate of change in cognition. By comparison, among individuals who progressed to MCI and had higher biomarker scores at baseline, a higher level of CR was associated with higher baseline cognitive performance prior to the onset of clinical symptoms, and a faster rate of cognitive decline after the onset of symptoms of MCI.
As discussed in the introduction, prior studies have produced inconsistent results regarding the relationship between CR and the rate of change in cognition among middle aged and older adults. The current study suggests that this discrepancy can be attributed, at least in part, to differences in the proportion of individuals in each cohort who progress to MCI or dementia. For example, our results suggest that in studies of individuals with normal cognition at baseline, higher CR is more likely to be associated with greater cognitive decline (or less practice-related improvement) if the proportion of individuals who progress to MCI over the follow-up time is high. By comparison, within a group where few individuals develop clinical symptoms over time, a null association between CR and the rate of change in cognition is more likely to be found. Moreover, lower CR is more likely to be associated with greater cognitive decline over time when studies enroll individuals with a mixed diagnostic status (e.g., cognitively normal and MCI participants) and do not take this baseline clinical status into account. This is because individuals with MCI are more likely to harbor greater AD related and other pathology, to show cognitive decline over time and to have lower CR relative to age-matched cognitively normal individuals. Consequently, in studies with a mix of cognitively normal and MCI participants at baseline, the effects of CR on baseline cognition and change in cognitive performance may be confounded with the effects of differences in baseline diagnosis (normal vs. MCI) as well as level of underlying pathology. Additionally, as reviewed by Zahodne et al. (2011, 2015), many older studies reporting reduced rates of cognitive decline for individuals with higher CR had methodological limitations that biased them in favor of finding associations between higher CR and reduced cognitive decline (including many studies included in an earlier meta-analysis by Valenzuela & Sachdev (2016b)). Inconsistencies in prior findings may also be attributable to differences in educational attainment across cohorts (Zahodne, et al., 2015) as well as to differences in measures of CR and cognitive outcome variables, as may be the case if different cognitive and brain processes are differentially sensitive to different components of CR.
In a series of prior investigations, we reported associations between higher CR and a reduced risk of progressing from normal cognition to MCI, even after accounting for levels of individual AD biomarkers at baseline (Pettigrew et al., 2016; Soldan et al., 2013; Soldan et al., 2015). The current results suggest that this reduction in risk of progression is not due to a direct effect of CR on the rate of cognitive decline prior to symptom onset (as reflected by the absence of a CR × time interaction), but rather a delay in the onset of symptoms and a corresponding delay in the onset of cognitive decline, in the presence of underlying pathology. This interpretation derives from the observation that older age of symptom onset was strongly associated with higher baseline CR scores. Specifically, individuals with CR scores above the median had a mean age of symptom onset that was approximately 7 years later than for those with CR scores below the median of the group, despite similar pathology levels at baseline. As illustrated in Supplementary Materials 9, we hypothesize that individuals with higher CR remain asymptomatic for a longer period of time and maintain their cognitive performance for a longer duration, while individuals with lower CR become symptomatic earlier and begin to show cognitive decline when they are younger.
The results of the current study provide support for the predictions of Stern’s (2009) hypothetical model of CR: among individuals with similar biomarker levels at baseline, higher CR is associated with a) better cognitive performance prior to the onset of symptoms, b) a later age of onset of cognitive decline, and c) a faster rate of decline after the onset of clinical symptoms. To our knowledge, the current study provides the first direct empirical support for these components of the model. The Stern model further hypothesizes that the more rapid cognitive decline among high CR individuals reflects the presence of greater levels of pathology at the time of symptom onset. In the current study, biomarker measures of pathology were not available for the time that coincides with the onset of symptoms for most individuals; therefore, this aspect of the model cannot be tested with the current data. However, cross-sectional studies of individuals with MCI or dementia have reported that, when equating patients for clinical severity, those with higher CR tend to harbor more pathology (e.g., Roe et al., 2008; Vemuri et al., 2011), as predicted by the model.
The present results also suggest that the levels of pathology present in this cognitively normal sample have little impact on the association between CR and cognitive change. Of note, it is difficult to compare the present findings to studies that have primarily focused on the impact of amyloid on cognition, since the biomarker composite used here combined multiple measures of AD-related pathology together. Future studies are needed to test if biomarker levels at the time of symptom onset modify the relationship between CR and subsequent cognitive change, as Stern’s model would predict.
Related to the Stern model, a number of previous studies of individuals with dementia have reported greater rates of cognitive decline (e.g., Roselli et al., 2009; Scarmeas et al., 2006; Stern et al., 1999) or functional decline (de Oliveira et al., 2015) among those with more education compared to those with low levels of education. Our study supports and extends these findings by suggesting that this acceleration in cognitive decline is present not only in the dementia phase of AD, but may begin in the early symptomatic phase.
In addition to informing theoretical models of CR, the current study provides evidence that higher levels of CR protect against the clinical manifestations of AD by delaying the onset of symptoms associated with the disease. In the absence of medications that reduce underlying AD pathology, this delay in the onset of symptoms allows individuals to maximize daily functioning as pathology levels increase. The results further suggest that interventions aimed at increasing CR could provide significant clinical benefits by delaying the symptomatic phase of the disease.
In the present study, we found no cross-sectional association between biomarker levels and CR scores at baseline, even among the group who progressed, suggesting baseline biomarker levels are not responsible for the more rapid cognitive decline among participants with high CR. One possible explanation may be related to participant age. In the total study sample, as well as among individuals who progressed, higher CR scores were associated with older age at baseline and at symptom onset. Importantly, older age has also been associated with greater accumulation of amyloid and tau, as measured by CSF and PET imaging (Resnick et al., 2015; Sutphen et al., 2015), and with higher levels of other brain pathologies, including cerebrovascular disease (Elobeid et al., 2016; Jeerakathil et al., 2004; P. T. Nelson et al., 2011). It is therefore possible that the greater cognitive decline observed among individuals with high CR who progressed is due to a greater rate of AD pathology accumulation after the baseline evaluation, as well as to higher levels of pathologies not measured by our biomarker composite score. Future studies with concurrent measures of CR, AD biomarkers, and cerebrovascular disease before and after symptom onset are necessary to examine this issue.
Another finding of the current study was that among individuals who remained cognitively normal over time, there was no relationship between the CR composite score and the age-related decrease in the practice effect. This suggests that level of CR, at least as measured in the current study, is associated with baseline rates of cognitive performance, but does not alter the trajectories of normal cognitive aging. Taken together, the present results suggest that the mechanism by which CR benefits cognitive functioning among older individuals is not by reducing the rate of cognitive decline associated with aging or preclinical AD. Rather, CR appears to delay the onset of clinical symptoms associated with underlying AD pathology.
Additionally, we found that when follow-up diagnosis is taken into account, APOE ε4 status did not modify the cognitive trajectories, nor the relationship between CR and cognitive change. Although APOE ε4 status is sometimes associated with greater cognitive decline among older adults, this tends to reflect the fact that a greater proportion of the e4 carriers develop AD-related cognitive impairment over time (Yu et al., 2013). We interpret the lack of a main effect of APOE ε4 status on the rate of cognitive change in our analysis as an indication that after accounting for the fact that APOE-e4 carriers are more likely to progress to MCI (captured by the follow-up diagnosis indicator), the rates of change in cognition are similar for e4 carriers and non-carriers. This interpretation is consistent with the observation that the ε4 allele primarily results in an earlier age of amyloid accumulation and thereby an earlier age of onset, but has less impact on the rate of cognitive decline among those who develop symptoms of MCI (Albert et al., 2014). It is also consistent with the observation that ε4 carriers and non-carriers benefit similarly from education (et al., 2013) and from CR more broadly (e.g., Pettigrew et al., 2013) with respect to risk of progression.
Our study has several limitations. First, participants were primarily Caucasian and have a strong family history of dementia, so the results may not generalize to the US population at large. Second, the level of CR is relatively high in this study, with all individuals having at least high school education. Our results may therefore underestimate the impact of CR on cognitive change and it is possible that the relationship between CR and cognitive change differs for individuals with very low education. Additionally, our measure of CR was limited to education, vocabulary, and verbal intelligence. It remains unclear if our results generalize to other proxy measures of CR, such as occupational complexity and participation in leisure activities. We are presently in the process of studying these issues in this cohort. However, it is noteworthy that in all analyses conducted to date education, vocabulary, and reading ability tend to have similar relationships between cognition and AD biomarkers, with education generally showing somewhat weaker associations than vocabulary and reading measures, and the composite score showing the most robust associations (Pettigrew et al., 2013, 2017). Third, the sample size of the current study was not sufficient to reliably examine whether the presence of one or two APOE ε4 alleles alters the associations between measures of CR, AD biomarkers, and cognitive change among cognitively normal or symptomatic individuals, as has been suggested by some studies (e.g., Vemuri et al., 2016). Fourth, it is worth noting that these analyses focused on cognitive measures. This approach was selected since participants were cognitively normal at baseline, and functional outcome measures have limited variability among such participants who tend to perform at ceiling on these types of measures. By contrast, cognitive measures are likely more sensitive to subtle cognitive change that may be evident during the preclinical phase of AD, particularly among middle-aged, cognitively normal participants. Lastly, although the biomarker composite score includes the major categories of biomarkers (measures of amyloid, tau, and neurodegeneration), it does not take into account the fact that some component measures (e.g., amyloid plus tau abnormality) may be more strongly associated with cognitive change and underlying pathology than others (e.g., abnormal levels of neurodegeneration in the absence of amyloid pathology). Consequently, two individuals with the same biomarker composite scores may differ in their underlying pathology, and future studies are therefore needed to test if different types of pathology profiles are differentially related to CR and cognitive change.
Supplementary Material
Figure 3. Estimates of longitudinal cognitive change by level of baseline biomarker and cognitive reserve composite scores.
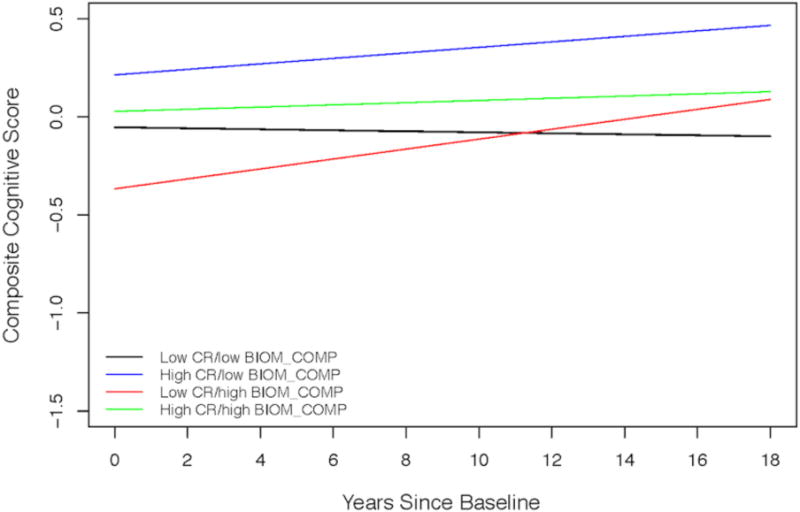
Estimates from linear mixed effects model predicting longitudinal cognitive composite scores over time among four groups: (1) low CR/low biomarker (black line) are individuals with CR scores below the median and biomarker composite scores below the median; (2) high CR/low biomarker (blue line) are individuals with CR scores at or above the median and biomarker scores below the median; (3) low CR/high biomarker (red line) are individuals with CR scores below the median and biomarker scores at or above the median; and (4) high CR/high biomarker (green line) are individuals with CR scores at or above the median and biomarker scores at or above the median. Estimates are adjusted for baseline age, gender, and the age × time interaction.
Highlights.
Studied association between cognitive reserve (CR), AD biomarkers, and cognition.
Association between CR and cognitive change differed by follow-up diagnosis.
CR unrelated to cognitive change among those who remained cognitively normal.
Higher CR associated greater cognitive decline after onset of symptoms of MCI.
AD biomarkers did not modify association between CR and cognitive change.
Acknowledgments
The BIOCARD Study consists of 7 Cores with the following members: (1) the Administrative Core (Marilyn Albert, Rostislav Brichko); (2) the Clinical Core (Ola Selnes, Marilyn Albert, Anja Soldan, Corinne Pettigrew, Rebecca Gottesman, Ned Sacktor, Scott Turner, Leonie Farrington, Maura Grega, Gay Rudow, Daniel D’Agostino, Scott Rudow); (3) the Imaging Core (Michael Miller, Susumu Mori, Tilak Ratnanather, Timothy Brown, Hayan Chi, Anthony Kolasny, Kenichi Oishi, Laurent Younes); (4) the Biospecimen Core (Abhay Moghekar, Richard O’Brien); (5) the Informatics Core (Roberta Scherer, David Shade, Ann Ervin, Jennifer Jones, Matt Toepfner, Alicia Wentz, April Patterson, Aisha Mohammed); (6) the Biostatistics Core (Mei-Cheng Wang, Qing Cai, Daisy Zhu); and (7) the Neuropathology Core (Juan Troncoso, Barbara Crain, Olga Pletnikova, Gay Rudow, and Karen Fisher). The authors are grateful to the members of the BIOCARD Scientific Advisory Board who provide continued oversight and guidance regarding the conduct of the study including: Drs John Cernansky, David Holtzman, David Knopman, Walter Kukull, and Kevin Grimm, and Drs John Hsiao and Laurie Ryan, who provide oversight on behalf of the National Institute on Aging. The authors thank the members of the BIOCARD Resource Allocation Committee who provide ongoing guidance regarding the use of the biospecimens collected as part of the study, including: Drs Constantine Lyketsos, Carlos Pardo, Gerard Schellenberg, Leslie Shaw, Madhav Thambisetty, and John Trojanowski.
The authors acknowledge the contributions of the Geriatric Psychiatry Branch of the intramural program of NIMH who initiated the study (Principle investigator: Dr. Trey Sunderland). The authors are particularly indebted to Dr. Karen Putnam, who has provided ongoing documentation of the Geriatric Psychiatry Branch study procedures and the data files received from NIMH.
Study funding:
This study was supported in part by grants from the National Institutes of Health (U19-AG03365, P50-AG005146, and T32-AG027668).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures:
Dr. Soldan reports no disclosures.
Dr. Pettigrew reports no disclosures.
Dr. Cai reports no disclosures.
Dr. J. Wang reports no disclosures.
Dr. M.C. Wang reports no disclosures.
Dr. Moghekar reports no disclosures.
Dr. Miller owns a significant equity share in “Anatomy Works”. This arrangement is being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
Dr. Albert is an advisor to Eli Lilly.
Original submission
The data contained in this manuscript has not been published previously, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
IRB approval:
Participants in this study were treated in accordance with the ethical standards of the Johns Hopkins University IRB and provided written consent for participation and use of their data.
Authors’ approval:
All authors have reviewed the contents of the manuscript being submitted, approve of its contents and validate the accuracy of the data.
References
- Albert M, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2011;7(3):270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M, Soldan A, Gottesman R, McKhann G, Sacktor N, Farrington L, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Current Alzheimer Research. 2014;11(8):773–84. doi: 10.2174/156720501108140910121920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva H, Mokri H, Le Goff M, Meillon C, Jacqmin-Gadda H, Foubert-Samier A, et al. Compensatory mechanisms in higher-educated subjects with Alzheimer’s disease: a study of 20 years of cognitive decline. Brain: a journal of neurology. 2014;137(Pt 4):1167–75. doi: 10.1093/brain/awu035. [DOI] [PubMed] [Google Scholar]
- Clouston SA, Glymour M, Terrera GM. Educational inequalities in aging-related declines in fluid cognition and the onset of cognitive pathology. Alzheimers Dement (Amst) 2015;1(3):303–10. doi: 10.1016/j.dadm.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7(2):180–4. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I. Altered Proteins in the Aging Brain. Journal of neuropathology and experimental neurology. 2016;75(4):316–25. doi: 10.1093/jnen/nlw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari C, Xu WL, Wang HX, Winblad B, Sorbi S, Qiu C, Fratiglioni L. How can elderly apolipoprotein E ε4 carriers remain free from dementia? Neurobiol Aging. 2013;34(1):13–21. doi: 10.1016/j.neurobiolaging.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Glymour MM, Tzourio C, Dufouil C. Is cognitive aging predicted by one’s own or one’s parents’ educational level? results from the three-city study. Am J Epidemiol. 2012;175(8):750–9. doi: 10.1093/aje/kwr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, Rawlings AM, Sharrett AR, Albert M, Alonso A, Bandeen-Roche K, et al. Impact of differential attrition on the association of education with cognitive change over 20 years of follow-up: the ARIC neurocognitive study. Am J Epidemiol. 2014;179(8):956–66. doi: 10.1093/aje/kwu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CB, Derby C, LeValley A, Katz MJ, Verghese J, Lipton RB. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology. 2007;69(17):1657–64. doi: 10.1212/01.wnl.0000278163.82636.30. [DOI] [PubMed] [Google Scholar]
- Hall CB, Lipton RB, Sliwinski M, Katz MJ, Derby CA, Verghese J. Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology. 2009;73(5):356–61. doi: 10.1212/WNL.0b013e3181b04ae3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–72. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539–47. doi: 10.1212/WNL.0000000000002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeerakathil T, Wolf PA, Beiser A, Hald JK, Au R, Kase CS, et al. Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke. 2004;35(8):1831–5. doi: 10.1161/01.STR.0000131809.35202.1b. [DOI] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Philadelphia: Lee & Febiger; 1983. [Google Scholar]
- Karlamangla AS, Miller-Martinez D, Aneshensel CS, Seeman TE, Wight RG, Chodosh J. Trajectories of cognitive function in late life in the United States: demographic and socioeconomic predictors. Am J Epidemiol. 2009;170(3):331–42. doi: 10.1093/aje/kwp154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly JJ, Schupf N, Tang MX, Stern Y. Cognitive decline and literacy among ethnically diverse elders. J Geriatr Psychiatry Neurol. 2005;18(4):213–7. doi: 10.1177/0891988705281868. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2011;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MI, Younes L, Ratnanather JT, Brown T, Trinh H, Postell E, et al. The diffeomorphometry of temporal lobe structures in preclinical Alzheimer’s disease. NeuroImage Clinical. 2013;3:352–60. doi: 10.1016/j.nicl.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghekar A, Goh J, Li M, Albert M, O’Brien RJ. Cerebrospinal fluid Abeta and tau level fluctuation in an older clinical cohort. Archives of neurology. 2012;69(2):246–50. doi: 10.1001/archneurol.2011.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghekar A, Li S, Lu Y, Li M, Wang MC, Albert M, et al. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology. 2013;81(20):1753–8. doi: 10.1212/01.wnl.0000435558.98447.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–4. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Muniz-Terrera G, Matthews F, Dening T, Huppert FA, Brayne C. Education and trajectories of cognitive decline over 9 years in very old people: methods and risk analysis. Age Ageing. 2009;38(3):277–82. doi: 10.1093/ageing/afp004. [DOI] [PubMed] [Google Scholar]
- Nelson HE. The National Adult Reading Test (NART): Test manual. Windsor: Nfer-Nelson; 1982. [Google Scholar]
- Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, et al. Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta neuropathologica. 2011;121(5):571–87. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira FF, Pivi GA, Chen ES, Smith MC, Bertolucci PH. Risk factors for cognitive and functional change in one year in patients with Alzheimer’s disease dementia from São Paulo, Brazil. J of Neurological Sci. 2015;359(1–2):127–32. doi: 10.1016/j.jns.2015.10.051. [DOI] [PubMed] [Google Scholar]
- Pettigrew C, Soldan A, Li S, Lu Y, Wang MC, Selnes OA, et al. Relationship of cognitive reserve and APOE status to the emergence of clinical symptoms in preclinical Alzheimer’s disease. Cogn Neurosci. 2013;4(3–4):136–42. doi: 10.1080/17588928.2013.831820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew C, Soldan A, Zhu Y, Wang MC, Moghekar A, Brown T, et al. Cortical thickness in relation to clinical symptom onset in preclinical AD. NeuroImage Clinical. 2016;12:116–22. doi: 10.1016/j.nicl.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinin AM, Muniz-Terrera G, Clouston S, Reynolds CA, Thorvaldsson V, Deary IJ, et al. Coordinated analysis of age, sex, and education effects on change in MMSE scores. J Gerontol B Psychol Sci Soc Sci. 2013;68(3):374–90. doi: 10.1093/geronb/gbs077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool LR, Weuve J, Wilson RS, Bultmann U, Evans DA, Mendes de Leon CF. Occupational cognitive requirements and late-life cognitive aging. Neurology. 2016;86(15):1386–92. doi: 10.1212/WNL.0000000000002569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proust-Lima C, Amieva H, Letenneur L, Orgogozo JM, Jacqmin-Gadda H, Dartigues JF. Gender and education impact on brain aging: a general cognitive factor approach. Psychol Aging. 2008;23(3):608–20. doi: 10.1037/a0012838. [DOI] [PubMed] [Google Scholar]
- Resnick SM, Bilgel M, Moghekar A, An Y, Cai Q, Wang MC, et al. Changes in Abeta biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiology of aging. 2015;36(8):2333–9. doi: 10.1016/j.neurobiolaging.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe CM, Mintun MA, D’Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Archives of neurology. 2008;65(11):1467–71. doi: 10.1001/archneur.65.11.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli F, Tartaglione B, Federico F, Lepore V, Defazio G, Livrea P. Rate of MMSE score change in Alzheimer’s disease: influence of education and vascular risk factors. Clinical neurology and neurosurgery. 2009;111(4):327–30. doi: 10.1016/j.clineuro.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, Albert SM, Manly JJ, Stern Y. Education and rates of cognitive decline in incident Alzheimer’s disease. Journal of neurology, neurosurgery, and psychiatry. 2006;77(3):308–16. doi: 10.1136/jnnp.2005.072306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Levy G, Tang MX, Manly J, Stern Y. Influence of leisure activity on the incidence of Alzheimer’s disease. Neurology. 2001;57(12):2236–42. doi: 10.1212/wnl.57.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Manoux A, Marmot MG, Glymour M, Sabia S, Kivimaki M, Dugravot A. Does cognitive reserve shape cognitive decline? Annals of neurology. 2011;70(2):296–304. doi: 10.1002/ana.22391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldan A, Pettigrew C, Li S, Wang MC, Moghekar A, Selnes OA, et al. Relationship of cognitive reserve and cerebrospinal fluid biomarkers to the emergence of clinical symptoms in preclinical Alzheimer’s disease. Neurobiology of aging. 2013;34(12):2827–34. doi: 10.1016/j.neurobiolaging.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldan A, Pettigrew C, Lu Y, Wang MC, Selnes O, Albert M, et al. Relationship of medial temporal lobe atrophy, APOE genotype, and cognitive reserve in preclinical Alzheimer’s disease. Human brain mapping. 2015;36(7):2826–41. doi: 10.1002/hbm.22810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y, Albert S, Tang MX, Tsai WY. Rate of memory decline in AD is related to education and occupation: cognitive reserve? Neurology. 1999;53(9):1942–7. doi: 10.1212/wnl.53.9.1942. [DOI] [PubMed] [Google Scholar]
- Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R. Influence of education and occupation on the incidence of Alzheimer’s disease. JAMA. 1994;271(13):1004–10. [PubMed] [Google Scholar]
- Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA neurology. 2015;72(9):1029–42. doi: 10.1001/jamaneurol.2015.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela MJ, Sachdev P. Brain reserve and dementia: A systematic review. Psychological Medicine. 2006a;36(4):441–454. doi: 10.1017/S0033291705006264. [DOI] [PubMed] [Google Scholar]
- Valenzuela MJ, Sachdev P. Brain reserve and cognitive decline: a non-parametric systematic review. Psychological Medicine. 2006b;36(8):1065–1073. doi: 10.1017/S0033291706007744. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain: a journal of neurology. 2015;138(Pt 3):761–71. doi: 10.1093/brain/awu393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Machulda M, Lowe VJ, Mielke MM, Roberts RO, Gunter JL, Senjem ML, Geda YE, Rocca WA, Petersen RC, Jack CR., Jr 2016;86(12):1128–35. doi: 10.1212/WNL.0000000000002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Weigand SD, Przybelski SA, Knopman DS, Smith GE, Trojanowski JQ, et al. Cognitive reserve and Alzheimer’s disease biomarkers are independent determinants of cognition. Brain: a journal of neurology. 2011;134(Pt 5):1479–92. doi: 10.1093/brain/awr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. Wechsler adult intelligence scale - Revised manual. New York, NY: The Psychological Corporation; 1981. [Google Scholar]
- Wechsler D. Wechsler Memory Scale - Revised Manual. San Antonio, TX: Psychological Corporation; 1987. [Google Scholar]
- Wilson RS, Hebert LE, Scherr PA, Barnes LL, Mendes de Leon CF, Evans DA. Educational attainment and cognitive decline in old age. Neurology. 2009;72(5):460–5. doi: 10.1212/01.wnl.0000341782.71418.6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Mendes De Leon CF, Barnes LL, Schneider JA, Bienias JL, Evans DA, et al. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA. 2002;287(6):742–8. doi: 10.1001/jama.287.6.742. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Weston A, Graff-Radford NR, Satterfield S, Simonsick EM, Younkin SG, et al. Association of plasma beta-amyloid level and cognitive reserve with subsequent cognitive decline. JAMA. 2011;305(3):261–6. doi: 10.1001/jama.2010.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Boyle P, Schneider JA, Segawa E, Wilson RS, Leurgans S, Bennett DA. APOE ε4, Alzheimer’s disease pathology, cerebrovascular disease, and cognitive change over the years prior to death. Psychol Aging. 2013;28(4):1015–23. doi: 10.1037/a0031642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahodne LB, Glymour MM, Sparks C, Bontempo D, Dixon RA, MacDonald SW, et al. Education does not slow cognitive decline with aging: 12-year evidence from the victoria longitudinal study. J Int Neuropsychol Soc. 2011;17(6):1039–46. doi: 10.1017/S1355617711001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahodne LB, Stern Y, Manly JJ. Differing effects of education on cognitive decline in diverse elders with low versus high educational attainment. Neuropsychology. 2015;29(4):649–57. doi: 10.1037/neu0000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.