

Summary
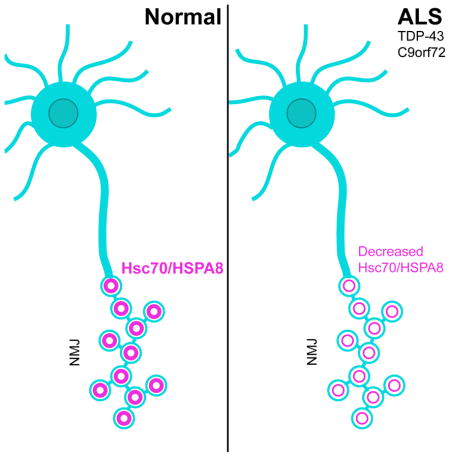
Amyotrophic Lateral Sclerosis (ALS) is a synaptopathy accompanied by the presence of cytoplasmic aggregates containing TDP-43, an RNA binding protein linked to ~97% of ALS cases. Using a Drosophila model of ALS, we show that TDP-43 OE in motor neurons results in decreased expression of the Hsc70-4 chaperone at the neuromuscular junction (NMJ). Mechanistically, mutant TDP-43 sequesters hsc70-4 mRNA and impairs its translation. Expression of Hsc70-4’s ortholog, HSPA8 is also reduced in primary motor neurons and NMJs of mice expressing mutant TDP-43. Electrophysiology, imaging, and genetic interaction experiments reveal TDP-43 dependent defects in synaptic vesicle endocytosis. These deficits can be partially restored by OE of Hsc70-4, cysteine-string protein (Csp) or dynamin. This suggests that TDP-43 toxicity results in part from impaired activity of the synaptic CSP/Hsc70 chaperone complex impacting dynamin function. Finally, Hsc70-4/HSPA8 expression is also post-transcriptionally reduced in fly and human iPSC C9orf72 models, suggesting a common disease pathomechanism.
Graphical Abstract
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a progressive neurodegenerative disease linked to several including SOD1 (Rosen et al., 1993), TARDBP (TDP-43) (Kabashi et al., 2008; Sreedharan et al., 2008), FUS (Kwiatkowski et al., 2009; Vance et al., 2009), and C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011), suggesting the presence of multiple disease mechanisms (Robberecht and Philips, 2013). Synaptic degeneration is a culminating point in ALS, however the molecular mechanisms that trigger this remain poorly understood (Gillingwater and Wishart, 2013).
With 2–3% of ALS patients harboring mutations in TDP-43 (Kabashi et al., 2008; Neumann, 2009; Sreedharan et al., 2008; Van Deerlin et al., 2008) and ~97 % of all ALS cases exhibiting TDP-43 pathology (Ling et al., 2013), TDP-43 is a common denominator in ALS. Thus, elucidating the mechanisms by which both wild-type and mutant TDP-43 cause neurodegeneration is essential to understanding of ALS pathogenesis.
TDP-43 is a nucleo-cytoplasmic shuttling, DNA/RNA binding protein containing a prion-like C-terminal domain where the majority of ALS causing mutations cluster causing an increase in TDP-43’s intrinsic propensity for aggregation (Johnson et al., 2009). TDP-43 was shown to bind UG-rich sequences and regulate the splicing of several transcripts encoding synaptic proteins (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). In addition, TDP-43 regulates the localization and translation of specific mRNAs (Alami et al., 2014; Coyne et al., 2014; Fallini et al., 2012). TDP-43 also associates with RNA stress granules (SG), which sequester specific mRNAs resulting in translation inhibition (Dewey et al., 2011; Kim et al., 2014; Liu-Yesucevitz et al., 2010; McDonald et al., 2011).
TDP-43 has previously been shown to associate with human Hsc70 (HSPA8) in mammalian cells (Freibaum et al., 2010). Hsc70 proteins comprise a family of constitutive, ubiquitous molecular chaperones with roles in protein folding and degradation, stress response, endosomal microautophagy, and chaperone-mediated autophagy (Liu et al., 2012). Consistent with its role in proteostasis, HSPA8 has been implicated in neurodegeneration as it colocalizes with ubiquitin-positive inclusions in sporadic ALS (Watanabe et al., 2001). Among the multiple Hsc70 proteins in Drosophila (Palter et al., 1986; Perkins et al., 1990), the clathrin-uncoating ATPase Hsc70-4 has roles in synaptic vesicle (SV) exo- and endocytosis, and is highly expressed in neurons and at the larval NMJ (Bronk et al., 2001; Chang et al., 2002; Zinsmaier and Bronk, 2001).
The critical role of Hsc70-4 in synaptic vesicle cycling (SVC) led us to hypothesize that abnormal interactions between TDP-43 and Hsc70-4 may lead to synaptic impairments prior to degeneration. Here we show that motor neuron overexpression (OE) of TDP-43 causes a reduction in synaptic Hsc70-4 levels and impairments in SVC at the Drosophila NMJ. Using electrophysiology and FM1-43 dye uptake experiments we show that defects in SVC are due to deficits in SV endocytosis. Similar to our findings in Drosophila, we show that HSPA8 protein is reduced post-transcriptionally at the NMJ in TDP-43A315T mice and in TDP-43G298S, patient derived iPSC motor neurons. Furthermore, flies harboring 36 G4C2 repeats (Mizielinska et al., 2014) and C9orf72 patient derived iPSC motor neurons (Zhang et al., 2015) exhibit a post-transcriptional reduction in Hsc70-4/HSPA8 protein. Together, our findings uncover defects in SV endocytosis as a source of synaptic dysfunction in TDP-43 and C9orf72 mediated ALS.
Results
Hsc70-4 protein and mRNA form a complex with TDP-43 in motor neurons in vivo
To test whether TDP-43 associates with Hsc70-4 we performed immunoprecipitations (IPs) from Drosophila adults expressing TDP-43 in motor neurons (Figure 1A) followed by mass spectrometry (data not shown). We found that Hsc70-4 associates with both wild-type TDP-43 (TDP-43WT) and ALS-associated mutant TDP-43 (TDP-43G298S; 22.9 and 30.1% sequence coverage respectively), albeit its interaction with TDP-43G298S is significantly stronger (Figure 1A). These results confirm that endogenous Hsc70-4 associates with human wild-type and mutant TDP-43 in Drosophila motor neurons.
Figure 1. hsc70-4 mRNA is a translation target of TDP-43G298S and Hsc70-4 protein expression is reduced at synaptic terminals of TDP-43 expressing animals.

(A) Immunoprecipitation (IP) of TDP-43 expressed in motor neurons. Genotypes and antibodies used for IP indicated on top. Antibodies used for western blot (WB) indicated on right. (B) qPCR for hsc70-4 mRNA in TDP-43 complexes. (C) qPCR for hsc70-4 mRNA in soluble and insoluble TDP-43 complexes. (D–F) qPCR for hsc70-4 mRNA in input (D), RNP (E), and polysomes (F) from flies expressing TDP-43WT and TDP-43G298S in motor neurons compared to controls. (G) WB for Hsc70-4 levels in whole larvae expressing TDP-43 in motor neurons. Genotypes indicated on bottom. Actin was used as loading control. (H) Quantification of Hsc70-4 protein levels from WBs represented as a ratio to w1118 controls. (I) qPCR for hsc70-4 mRNA in whole larvae of animals expressing TDP-43WT or TDP-43G298S in motor neurons versus controls. (J) WB for Hsc70-4 levels in VNCs of TDP-43 expressing larvae. Genotypes indicated on bottom. Actin was used as loading control. (K) Quantification of Hsc70-4 protein levels from WBs represented as a ratio to w1118 controls. (L) qPCR for hsc70-4 mRNA in VNCs of animals expressing TDP-43WT or TDP-43G298S in motor neurons versus controls. (M–O) Single confocal sections of synaptic boutons in NMJ preparations immunostained for Hsc70-4 and the neuronal membrane marker Hrp from larvae expressing TDP-43WT (N), and TDP-43G298S (O) compared to w1118 controls (M). Antibodies as indicated on left. (P) Quantification of Hsc70-4 intensity in synaptic boutons normalized to bouton area. Note p<0.001 for TDP-43WT vs TDP-43G298S. (Q) qPCR for hsc70-4 mRNA in NMJ preparations of animals expressing TDP-43WT or TDP-43G298S in motor neurons versus controls. (R) Quantification of Hsc70-4 intensity in muscle normalized to muscle area. Scale bars (M–O) 5 μm, 1 μm.
To determine whether hsc70-4 mRNA is present in TDP-43 complexes, we performed RNA IPs (RIP) from Drosophila adults expressing TDP-43WT or TDP-43G298S in motor neurons followed by quantitative real-time PCR (qRT-PCR). hsc70-4 mRNA was strongly enriched in TDP-43G298S while less was present in TDP-43WT complexes (Figure 1B).
It has been proposed that under prolonged stress TDP-43 may sequester its mRNA targets into insoluble complexes thereby preventing their translation and normal function (Ramaswami et al., 2013). To test whether TDP-43 sequesters hsc70-4 mRNA, we performed subcellular fractionations from Drosophila larvae expressing TDP-43 in motor neurons. Using qRT-PCR we found an enrichment of hsc70-4 mRNA in the insoluble fraction of animals expressing TDP-43G298S, but not TDP-43WT (Figure 1C), consistent with hsc70-4 mRNA sequestration specifically by TDP-43G298S. These data show that both Hsc70-4 protein and its cognate mRNA associate with TDP-43, albeit preferentially with TDP-43G298S.
OE of TDP-43G298S inhibits the translation of hsc70-4 mRNA
The sequestration of hsc70-4 mRNA by TDP-43G298S may impair its translation. To test this possibility, we performed polysome fractionations from adult Drosophila overexpressing TDP-43WT or TDP-43G298S in motor neurons. Using qRT-PCR we found that TDP-43G298S OE resulted in a shift for hsc70-4 mRNA from actively translating polysomes to non-translated, (ribonucleoprotein particle, RNP) fractions, while TDP-43WT had no effect (Figure 1E–F). Given that no changes in overall hsc70-4 mRNA levels were detected in whole animals (Figure 1D), these data suggest that TDP-43G298S impairs the translation of hsc70-4 mRNA by sequestering it into insoluble RNPs. In contrast, TDP-43WT did not affect hsc70-4 mRNA translation, at least within the limits of sensitivity provided by this assay.
TDP-43 expression reduces Hsc70-4 levels at the larval NMJ in Drosophila
The translation inhibition of hsc70-4 mRNA by TDP-43G298S predicts that Hsc70-4 levels are reduced, which in turn may impair Hsc70-4’s function (Stricher et al., 2013; Uytterhoeven et al., 2015; Zinsmaier and Bronk, 2001). No changes in protein expression were found in extracts of whole larvae or dissected ventral nerve cords (VNCs) from animals overexpressing TDP-43WT or TDP-43G298S in motor neurons (Figure 1G–H, J–K). In contrast, immunostainings at NMJs revealed a significant decrease in Hsc70-4 within synaptic boutons in animals overexpressing TDP-43WT and TDP-43G298S compared to controls (14% and 43% decrease respectively, p<0.001, Figure 1M–P). Notably, muscle Hsc70-4 levels remained unchanged (Figure 1R, Figure S1D–F). Since we found no changes in transcript levels in extracts from whole larvae, VNCs, or NMJ preparations (Figure 1I, L, Q), our data suggest that hsc70-4 mRNA is regulated by TDP-43 post-transcriptionally.
Although we cannot eliminate the possibility that Hsc70-4 protein transport or stability at the NMJ may be altered by TDP-43 OE, together with the polysome fractionation results, our findings suggest that TDP-43G298S OE reduces Hsc70-4 levels cell autonomously by inhibiting hsc70-4 mRNA translation. While TDP-43WT also decreases Hsc70-4 levels at synaptic terminals, this appears to arise from a distinct mechanism.
OE of disease-associated mutant TDP-43 reduces HSPA8 levels in mouse primary motor neurons
Having identified reduced synaptic expression of Hsc70-4 in the fly, we next asked if HSPA8 levels were also altered in a mammalian model of ALS. To evaluate HSPA8 levels, we transfected mouse primary motor neurons with GFP-tagged TDP-43WT, TDP-43Q331K, TDP-43M337V or GFP alone and performed immunostainings for HSPA8. OE of TDP-43WT had no effect on HSPA8 levels in growth cones or cell bodies (p>0.05, Figure 2B, F, I, J). In contrast, TDP-43Q331K or TDP-43M337V OE significantly reduced HSPA8 levels in growth cones (by 39% and 41%, respectively; p<0.01, Figure 2G, H, J) and cell bodies (12% decrease for TDP-43Q331K, p<0.05, Figure 2C, I, and 33% decrease for TDP-43M337V, p<0.001, Figure 2D, I). These data indicate that, similar to what has been observed in the fly model, HSPA8 levels are reduced in mammalian primary motor neurons overexpressing disease-associated mutant TDP-43.
Figure 2. Hsc70/HSPA8 expression is reduced in mutant TDP-43 expressing mouse primary motor neurons, at synaptic terminals of mouse NMJs, and in TDP-43G298S human iPSC neurons.

(A–D) Representative fluorescence images of cell bodies from primary motor neurons transfected with expression constructs for GFP (A–A‴) or GFP-tagged TDP-43WT (B–B‴), TDP-43Q331K (C–C‴), or TDP-43M337V (D–D‴) indicated on left. Antibodies and stains indicated on top. (E–H) Representative fluorescence images of growth cones from primary motor neurons expressing GFP (E–E′) or GFP-tagged TDP-43WT (F–F′), TDP-43Q331K (G–G′), or TDP-43M337V (H–H′) as indicated on left. Antibodies indicated on top. (I–J) Quantification of fluorescent intensity (a.u.) of Hsc70 in the cell body (I) and growth cones (J). (K–L) Epifluorescent images of mouse NMJs immunostained for Hsc70/HSPA8 and AChR. Genotypes indicated on left and antibodies indicated on top. (M) Quantification of Hsc70/HSPA8 intensity from NMJs. (N) Confocal images of control and TDP-43G298S human iPSC motor neurons labeled with DAPI, the dendritic marker Map2, and Hsc70/HSPA8. Genotypes indicated on left and antibodies indicated on top. (O) Quantification of Hsc70/HSPA8 intensity in the soma and dendrites of control and TDP-43G298S human iPSC motor neurons normalized to control. Symbols represent mean of 25–30 neurons from each line (controls) or differentiation (TDP-43) and indicate differentiation pairs. (P) qPCR for Hsc70/HSPA8 mRNA in control and TDP-43G298S human iPSC motor neurons. Scale bars (A, N) 10 μm, (K) 20 μm.
HSPA8 expression is reduced at the NMJ in TDP-43A315T mutant mice
We next asked whether levels of HSPA8 are also reduced at NMJs of TDP-43 mutant mice. To address this, dissected mouse NMJs were immunostained for HSPA8 and postsynaptic acetylcholine receptors (AChR). In comparison to controls, synaptic HSPA8 immunoreactivity at NMJs expressing TDP-43A315T (Wegorzewska et al., 2009) was decreased by 21% (p<0.05, Figure 2K–M). Taken together, these data indicate that disease-associated mutant TDP-43 reduces levels of HSPA8 at NMJs of both flies and mice.
HSPA8 expression is reduced in TDP-43G298S human iPSC motor neurons
Next, we sought to determine if expression of HSPA8 was also altered in human iPSC derived motor neurons harboring the TDP-43G298S mutation. Immunofluorescence experiments show a significant decrease in HSPA8 expression in the soma and dendrites of TDP-43G298S iPSC motor neurons compared to controls (Figure 2N–O). Notably, hspa8 mRNA expression is unchanged (Figure 2P), consistent with post-transcriptional regulation of HSPA8 expression, as in the fly.
Hsc70-4 OE mitigates TDP-43 mediated toxicity in flies
To determine whether the reduced levels of Hsc70-4 at NMJs contribute to the locomotor and lifespan defects induced by TDP-43 OE in flies (Coyne et al., 2014; Coyne et al., 2015; Estes et al., 2011; Estes et al., 2013) we co-overexpressed Hsc70-4 with either TDP-43WT or TDP-43G298S in motor neurons and found that it significantly improved locomotor function, as indicated by a faster turning time (see Experimental Procedures, Figure 3A). Co-OE of Hsc70-4 also significantly increased lifespan from 33 to 45 days and from 28 to 55 days for TDP-43 WT and TDP-43G298S animals, respectively (p<0.001, Figure 3B–C). Notably, suppression of TDP-43 toxicity by Hsc70-4 was not due to a reduction in TDP-43 protein or mRNA levels (Figure S2). As with TDP-43G298S, Hsc70-4 also suppressed the locomotor dysfunction induced by OE of disease-associated TDP-43Q331K or TDP-43M337V in Drosophila motor neurons (Figure S3C), which supports the notion that different C-terminus mutations in TDP-43 employ similar mechanisms. Importantly, co-OE of the Hsc70-4 related protein Hsc70-3 or the Hsc70 interacting protein Hsp90 (Hsp83) had no effect on TDP-43 induced locomotor dysfunction (Figure S3D–E). Taken together, these findings suggest that a specific loss of synaptic Hsc70-4 activity underlies, at least in part, the toxic effects of wild-type or mutant TDP-43 OE in Drosophila motor neurons.
Figure 3. TDP-43 expression results in defects in SV endocytosis that are suppressed by Hsc70-4 in a variant dependent manner.

(A) Motor neuron expression of TDP-43WT or TDP-43G298S results in increased larval turning time, which is mitigated by OE of Hsc70-4. (B–C) Motor neuron expression of TDP-43 variants WT (B) or G298S (C) leads to reduced lifespan. OE of Hsc70-4 increases lifespan for both TDP-43WT (B) and TDP-43G298S (C). (D) Representative electrophysiology traces of EJPs. Genotypes indicated on top. (E–G) EJP amplitude (E), mEJP amplitude (F), and quantal content (G) measurements from electrophysiology recordings. Genotypes indicated on bottom. (H–P) Confocal images of FM1-43 dye uptake in synaptic boutons of Drosophila larvae after 5 min of stimulation in HL-3 saline containing 90 mM KCl and 2 mM Ca2+. Genotypes indicated on top and left. (Q) Quantification of FM1-43 dye uptake normalized to total FM1-43 uptake area. Note an 18 ± 1.8% reduction in FM1-43 dye uptake for TDP-43G298S, Hsc70-4 OE compared to w1118 controls (p<0.001). Scale bar (D) 10 μm.
Hsc70-4 is known to act as a molecular chaperone and is also involved in endosomal microautophagy (Uytterhoeven et al., 2015; Zinsmaier, 2010). To determine whether OE of TDP-43WT or TDP-43G298S affects these functions we co-overexpressed TDP-43 with chaperone-dead (Hsc70-4D10N) or microautophagy-dead (Hsc70-43KA) Hsc70-4 (Uytterhoeven et al., 2015). In contrast to the mitigating effect of Hsc70-4WT, co-OE of either Hsc70-4D10N or Hsc70-43KA enhanced TDP-43WT (Figure S3A) and had no effect on TDP-43G298S induced locomotor dysfunction (Figure S3B). Since Hsc70-4D10N or Hsc70-43KA OE alone had no effect on locomotor activity (Figure S3A–B), these results suggest that TDP-43 compromises both the molecular chaperone and the endosomal microautophagy activities of Hsc70-4.
OE of TDP-43 in motor neurons impairs SV endocytosis
Given the various critical synaptic roles of Hsc70-4 in SVC such as the uncoating of clathrin-coated SVs in cooperation with auxilin, maintaining SNARE complex assembly and dynamin oligomerization in cooperation with cysteine string protein (Csp), and degradation of synaptic proteins by endosomal microautophagy (Bronk et al., 2001; Burgoyne and Morgan, 2015; Chang et al., 2002; Eisenberg and Greene, 2007; Uytterhoeven et al., 2015; Zhang et al., 2012), we hypothesized that the reduced synaptic levels of Hsc70-4 induced by wild-type and mutant TDP-43 may impair synaptic function.
We have previously shown that OE of wild-type or mutant TDP-43 decreases the number of synaptic boutons at larval NMJs (Coyne et al., 2014; Estes et al., 2013) and increases bouton size (Figure S4Q–R), similar to mutations that affect SV endocytosis (Verstreken et al., 2002). We do not observe a reduction in the number of active zones for neurotransmitter release per bouton area when TDP-43 is expressed in motor neurons (Figure S4A–H). Furthermore, TDP-43 OE in motor neurons does not alter the intensity or area occupied by Csp within synaptic boutons (Figure S4I–P), suggesting that the total number of SVs is within normal range.
To test whether wild-type or mutant TDP-43 OE in motor neurons affects synaptic transmission, we recorded spontaneous and evoked excitatory postsynaptic potentials (EPSPs) from larval NMJs of muscle 6. In comparison to controls, neither TDP-43 WT nor TDP-43G298S OE had an effect on spontaneous or stimulus-evoked EPSP amplitudes (Figure 3D–F). Accordingly, quantal content of evoked neurotransmitter release was normal (Figure 3G).
Next, we examined SVC by performing FM1-43 dye uptake assays at the larval NMJ (see Experimental Procedures, (Kuromi and Kidokoro, 2005)). In comparison to controls, TDP-43WT and TDP-43G298S OE significantly reduced K+-induced FM1-43 dye uptake by 33 ± 1.8% and 39 ± 2.1%, respectively (p<0.001, Figure 3H–J, Q). Subsequent K+ stimulation to unload FM1-43 dye from boutons showed no difference between TDP-43 mutant NMJs and controls (Figure S5A–C). Taken together, these results indicate that TDP-43 OE does not affect exocytosis but causes defects in SV endocytosis.
The chaperone activity of Hsc70-4 is required to mitigate SVC defects caused by TDP-43G298S
We next asked whether Hsc70-4 OE can mitigate TDP-43 induced defects in FM1-43 dye uptake. When OE in a wild-type background or together with TDP-43WT, Hsc70-4 had no effect on FM1-43 dye uptake at the NMJ (p>0.05, Figure 3K–L, Q). In contrast, co-OE of Hsc70-4 with TDP-43G298S strongly suppressed the defects in FM1-43 dye uptake induced by TDP-43G298S OE (p<0.001, Figure 3M, Q).
To determine whether the chaperone activity of Hsc70-4 is required for its protective effect, we co-overexpressed chaperone-dead Hsc70-4 (Hsc70-4D10N; (Uytterhoeven et al., 2015)) and found that it does not alter FM1-43 dye uptake deficits induced by TDP-43 (p>0.05, Figure 3O–Q). It also has no effect on its own, when OE in a wild-type background (p>0.05, Figure 3N, Q). These data suggest that TDP-43WT OE induces SV endocytosis defects that are at least in part independent of Hsc70-4. In contrast, mutant TDP-43G298S induces defects in SV endocytosis that are mechanistically linked to the chaperone activity of Hsc70-4.
Auxilin OE or RNAi-mediated knock down does not alter TDP-43 locomotor phenotypes
Hsc70’s molecular chaperone activity critically requires DNAJ proteins, which transition Hsc70-ATP to the ADP-bound state to stabilize client interactions (Jiang et al., 2007; Kampinga and Craig, 2010). At synaptic terminals, Hsc70’s chaperone function in SV exo- and endocytosis is facilitated by the DNAJ proteins auxilin and Csp on SVs (Kampinga and Craig, 2010; Liu et al., 2012; Zinsmaier and Bronk, 2001). To determine the mechanisms underlying Hsc70-4 dependent phenotypes of TDP-43 OE, we used larval turning assays to examine the effects of mutations affecting critical steps of SVC (Figure 4A).
Figure 4. Factors involved in Hsc70-4 independent and dependent steps of SVC modulate TDP-43 toxicity.

(A) Diagram of the SV cycle. (B–F) Motor neuron expression of TDP-43 WT or G298S results in increased larval turning time, which is enhanced upon OE of the Csp J domain mutant CspH45Q (E), and suppressed upon genomic expression of Csp (D) or OE of dynamin (shibire, F). OE (B) or reduction in auxilin (Auxilin RNAi, C) does not alter TDP-43 mediated locomotor defects. (G–O) Confocal images of FM1-43 dye uptake after 5 min of stimulation in HL-3 saline containing 90 mM KCl and 2 mM Ca2+. Genotypes indicated on top and left. (P) Quantification of FM1-43 dye uptake normalized to total FM1-43 uptake area. Scale bar (G) 10 μm.
We first tested whether TDP-43 OE impairs auxilin/Hsc70-mediated clathrin-uncoating of SV. Auxilin OE or RNAi-mediated knock down (55% decrease in expression by qPCR, data not shown) had no effect on larval turning on its own or in combination with wild-type and mutant TDP-43 (Figure 4B, C). This suggests that the synaptic defects induced by wild-type or mutant TDP-43 OE are unlikely due to a compromised auxilin/Hsc70 clathrin-uncoating activity.
Csp levels and chaperone activity modulate TDP-43 mediated locomotor dysfunction
Next, we tested for genetic interactions with Csp, which recruits Hsc70-4 to a chaperone complex on SVs ensuring proper assembly of SNARE and dynamin complexes during SV exo- and endocytosis, respectively (Chandra et al., 2005; Tobaben et al., 2001; Zhang et al., 2012; Zinsmaier, 2010). OE of Csp in a wild-type background from a genomic transgene (increasing Csp gene dosage by one copy) (Zinsmaier et al., 1994) had no effect on larval turning time (Figure 4D) while co-OE with TDP-43WT or TDP-43G298S restored larval turning time to control levels (Figure 4D). OE of Csp with a UAS transgene had a significant effect on larval turning time on its own (data not shown), likely due to high levels of Csp causing neurodegeneration (Nie et al., 1999), therefore we did not pursue this line for further genetic interactions with TDP-43.
Csp’s J domain promotes the ATPase activity of both Hsc70 and Hsp70, which is abolished by mutations in the HPD motif and J domain (Braun et al., 1996; Bronk et al., 2005; Chamberlain and Burgoyne, 1997; Zhang et al., 1999). OE of J domain mutant CspH45Q alone (Bronk et al., 2005) had no effect on larval turning (Figure 4E), however, co-OE with TDP-43WT or TDP-43G298S strongly enhanced locomotor dysfunction (Figure 4E). Taken together, these results suggest that OE of wild-type or mutant TDP-43 impairs the activity of the Csp/Hsc70-4 chaperone complex.
OE of Csp/Hsc70-4’s client dynamin mitigates TDP-43 induced locomotor dysfunction
Csp/Hsc70-4 chaperone activity maintains SV endocytosis by ensuring proper oligomerization of the GTPase dynamin (Rozas et al., 2012; Zhang et al., 2012). Once a critical mass is reached, oligomerization activates dynamin’s GTPase activity, which in turn pinches or pops off newly formed SVs from the plasma membrane (Haucke et al., 2011; Jahn and Fasshauer, 2012; Ramachandran, 2011). To determine whether TDP-43 OE affects Csp/Hsc70-4’s chaperone activity on dynamin, we tested whether OE of Drosophila dynamin (shibire) may restore TDP-43 induced locomotor defects and found that while OE of shibire alone had no effect on larval turning (Figure 4F), co-OE with TDP-43WT or TDP-43G298S in motor neurons restored locomotor function (Figure 4F). Notably, none of the mRNAs encoding the examined SVC proteins were enriched in wild-type or mutant TDP-43 complexes, or were translationally dysregulated, as suggested by polysome fractionations (Figure S6).
OE of the Hsc70-4’s co-chaperone Csp or its client dynamin suppresses SVC defects induced by TDP-43 in motor neurons
To determine whether OE of Hsc70-4’s co-chaperone Csp or their client dynamin can mitigate TDP-43 dependent defects in SVC, we tested if the genomic Csp construct or dynamin OE can restore FM1-43 dye uptake defects induced by TDP-43. We found that OE of either Csp from a genomic transgene or dynamin alone had no effect on K+-induced FM1-43 dye uptake in comparison to control (p>0.05, Figure 4J, M, P). Co-OE of Csp fully restored the defect in FM1-43 dye uptake induced by TDP-43WT or TDP-43G298S OE (Figure 4G–I) to control levels (Figure 4K–L, P). Similarly, co-OE of dynamin also fully suppressed the defect in FM1-43 dye uptake to control levels (Figure 4N–P). Taken together, these data suggest that OE of wild-type or mutant TDP-43 impairs the activity of the Csp/Hsc70 complex in maintaining normal dynamin function for SV endocytosis.
OE of Hsc70-4 attenuates the aggregation of TDP-43WT but not TDP-43G298S
Given the general role of Hsc70-4 in clearance of misfolded or aggregated proteins (Liu et al., 2012; Stricher et al., 2013; Zetterstrom et al., 2011), we hypothesized that Hsc70-4 OE may influence TDP-43 aggregation. To examine this, we performed subcellular fractionations from Drosophila larvae co-overexpressing wild-type or mutant TDP-43 with Hsc70-4 in motor neurons and quantified the amount of TDP-43 in the soluble (low salt, LS), sarkosyl (Sark), and urea (Urea) fractions. Interestingly, Hsc70-4 co-OE significantly reduced the amount of TDP-43WT in the Urea fraction and increased the amount of TDP-43WT in the Sark fraction (p<0.01 and p<0.05 respectively, Figure 5A, E). Notably, Hsc70-4 OE had no significant effect on the solubility of ALS-associated mutant TDP-43G298S (Figure 5B, E), despite its neuroprotective effects on TDP-43G298S induced functional phenotypes (Figure 3A–C, H–M).
Figure 5. TDP-43WT insolubility is reduced by OE of Hsc70-4 and Hsc70-4 insolubility is increased in an age dependent manner.

(A–B) Solubility studies of third instar larvae show the distribution of TDP-43WT (A) and TDP-43G298S (B) in low salt (LS), sarkosyl (Sark), and Urea containing fractions alone and in the context of Hsc70-4 OE. (C) Solubility studies of third instar larvae show Hsc70-4 distribution in LS, Sark, and Urea Fractions. (D) Solubility studies of 7 day old adults show Hsc70-4 distribution in LS, Sark, and Urea Fractions. (E) Quantification of TDP-43WT and TDP-43G298S levels in LS, Sark, and Urea fractions normalized to input. (F) Quantification of Hsc70-4 levels in LS, Sark, and Urea fractions from larval fractionations normalized to input. (G) Quantification of Hsc70-4 levels in LS, Sark, and Urea fractions from adult fractionations normalized to input.
Conversely, TDP-43WT or TDP-43G298S OE in motor neurons did not affect the solubility of endogenous Hsc70-4 at the larval stage (Figure 5C, F). However, adult flies (7 days) exhibited a significant increase in insoluble Hsc70-4 protein for both wild-type and mutant TDP-43 (p<0.001, Figure 5D, G). Adults expressing TDP-43WT also showed a corresponding decrease of Hsc70-4 protein in the Sark fraction in comparison to controls (p<0.001, Figure 5D, G). These data suggest an age-dependent sequestration of Hsc70-4 by insoluble cytoplasmic RNA/protein complexes, as previously observed sporadic ALS patient samples (Watanabe et al., 2001).
Co-OE of Hsc70-4 with TDP-43WT significantly decreased the amount of insoluble Hsc70-4 in the urea fraction (p<0.001, Figure 5D, G) and increased the amount of Hsc70-4 in the Sark fraction compared to TDP-43WT expression alone (p<0.001, Figure 5D, G), mirroring the effects Hsc70-4 OE on TDP-43WT solubility. Thus, these data suggest that OE of Hsc70-4 may mitigate TDP-43WT toxicity by improving the solubility of both TDP-43 and Hsc70.
C9orf72 repeat expansions reduce Hsc70-4/HSPA8 levels and induce defects in SVC
We next sought to determine whether similar SVC defects were present in C9orf72 (C9) repeat expansion models (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Using a fly model of C9 ALS (Mizielinska et al., 2014) containing 36 G4C2 repeats, we detected no alterations in Hsc70-4 expression in dissected VNCs (Figure 6A–B). However, at synaptic boutons of larval NMJs, Hsc70-4 levels were significantly decreased compared to controls (21%, p<0.001, Figure 6E–G). There was no change in hsc70-4 mRNA transcript levels in VNCs or NMJ preparations (Figure 6C–D) and Hsc70-4 protein levels in muscles were normal (Figure 6H). Together, these data indicate that OE of G4C2 repeats impairs Hsc70-4 protein expression post-transcriptionally and cell autonomously in motor neurons.
Figure 6. C9orf72 repeat expansions cause reduced Hsc70-4/HSPA8 expression and defects in SVC.

(A) WB for Hsc70-4 levels in VNCs of G4C2 expressing larvae. Genotypes indicated on bottom. Actin was used as loading control. (B) Quantification of Hsc70-4 protein levels from WBs represented as a ratio to G4C2 3X controls. (C) qPCR for hsc70-4 mRNA in VNCs from animals expressing G4C2 36X in motor neurons versus G4C2 3X controls. (D) qPCR for hsc70-4 mRNA in NMJ preparations from animals expressing G4C2 36X in motor neurons versus G4C2 3X controls. (E–F) Single confocal sections of synaptic boutons in NMJ preparations immunostained for Hsc70-4 and the neuronal membrane marker Hrp from larvae expressing G4C2 36X (F) compared to G4C2 3X controls (E). Antibodies indicated on left. (G) Quantification of Hsc70-4 intensity in synaptic boutons normalized to bouton area. (H) Quantification of Hsc70-4 intensity in muscle normalized to muscle area. (I) Confocal images of control and C9 ALS human iPSC motor neurons immunostained for DAPI, the dendritic marker Map2, and Hsc70/HSPA8. Genotypes indicated on left and antibodies indicated on top. (J) Quantification of Hsc70/HSPA8 intensity in the soma and dendrites of control and C9 ALS human iPS motor neurons. (K) qPCR for Hsc70/HSPA8 mRNA in control and C9 ALS human iPS motor neurons. (L) Confocal images of FM1-43 dye uptake at Drosophila NMJs after 5 min of stimulation in HL-3 saline containing 90 mM KCl and 2 mM Ca2+. Genotypes indicated on top and left. (M) Quantification of FM1-43 dye uptake normalized to total FM1-43 uptake area. Scale bars (E–F) 5 μm, 1 μm, (I, L) 10 μm.
To determine if HSPA8 expression was altered in a human C9 model of ALS, we used induced pluripotent stem cells (iPSCs) derived motor neurons. In comparison to controls, HSPA8 levels were reduced in the soma and dendrites of C9 iPSC motor neurons by 57% and 32%, respectively (p<0.001, Figure 6I–J). Similar to the fly model, there was no significant change in hsc70-4 mRNA in C9 iPSC motor neurons compared to controls (Figure 6K). This suggests that Hsc70 expression is post-transcriptionally down-regulated in C9 ALS human motor neurons.
Since the reduction in Hsc70-4 levels in C9 models of ALS is reminiscent of that seen in the TDP-43 fly model, we hypothesized that C9 repeat expansions may also disrupt SVC. To test this possibility, we performed FM1-43 dye uptake experiments at the fly NMJ. OE of 36 G4C2 repeats in motor neurons significantly decreased FM1-43 dye uptake compared to G4C2 3X controls (53 ± 4.4%, p<0.001, Figure 6L–M). To determine the contribution of reduced synaptic expression of Hsc70-4 to SVC deficits we co-overexpressed Hsc70-4 and found that it restores FM1-43 dye uptake in flies expressing expanded G4C2 repeats back to control levels (Figure 6L–M). These data indicate that C9 mediated FM1-43 dye uptake deficits are at least in part the result of decreased Hsc70-4 expression at the NMJ. Furthermore, these findings suggest that both TDP-43 and C9orf72 models of ALS share common features of post-transcriptionally reduced Hsc70 expression levels that are accompanied by defects in SVC.
Discussion
ALS is a synaptopathy like other neurodegenerative disorders including Parkinson’s and Alzheimer’s disease (Gillingwater and Wishart, 2013). However, the mechanisms underlying synaptic dysfunction and neurodegeneration remain poorly understood. TDP-43 has been linked to a majority of ALS cases and is known to associate with RNA stress granules (SG), which sequester specific mRNAs and reduce their translation during stress (Dewey et al., 2011; Kim et al., 2014; Liu-Yesucevitz et al., 2010; McDonald et al., 2011). Collectively, these studies have suggested that TDP-43 association with RNA SG plays a critical role in maintaining both proteostasis and ribostasis by controlling the expression of target mRNAs and sequestration of protein partners. Because of its central role in ALS pathogenesis, understanding the molecular mechanisms underlying TDP-43 mediated neurodegeneration and defining its mRNA targets and their mode of regulation is expected to provide a deeper understanding of disease pathophysiology and identify novel therapeutic targets.
Distinct mechanisms govern TDP-43 dependent toxicity and regulation of Hsc70-4 expression
Using a Drosophila model of ALS based on TDP-43, we show that OE of ALS-associated mutant TDP-43 in motor neurons sequesters hsc70-4 mRNA and reduces synaptic Hsc70-4 protein levels at larval NMJs. HSPA8 expression is also reduced in cell bodies and growth cones of cultured primary motor neurons, at NMJs of mice expressing mutant TDP-43, and in human iPSC motor neurons, suggesting that the relationship between TDP-43 and Hsc70 is evolutionarily conserved.
TDP-43WT OE in fly motor neurons also leads to post-transcriptional reduction of synaptic Hsc70-4 levels at larval NMJs. However, immunoprecipitations and cellular fractionations suggest that this is unlikely due to hsc70-4 mRNA sequestration and reduced translation. It is possible that the sensitivity of this assay is limited, or that subtle defects in other aspects of RNA or protein processing result in reduced Hsc70-4 protein expression at the NMJ. Supporting this, bioinformatics analyses of published crosslinking immunoprecipitation (CLIP) data suggest that endogenous TDP-43 can bind HSPA8 introns and exons in mouse brains and human K562 cells (Polymenidou et al., 2011; Van Nostrand et al., 2016) (Figure S7).
Hsc70 belongs to a neuroprotective synaptic network that maintains SV exo- and endocytosis and prevents neurodegeneration (Bronk et al., 2001; Chang et al., 2002; Zinsmaier, 2010; Zinsmaier and Bronk, 2001). This led us to hypothesize that alterations in Hsc70-4 expression may be detrimental to synaptic function. Indeed, using a combination of FM1-43 dye loading and unloading experiments together with electrophysiology, we find that OE of TDP-43WT or TDP-43G298S in motor neurons leads to defects in SV endocytosis but not exocytosis. Although loss of endogenous fly TDP-43 (TBPH) has been shown to cause reduced expression of the synaptic proteins Syntaxin (Romano et al., 2014) and Cacophony (Chang et al., 2014) at the NMJ, and defects in SVC have been previously described in Parkinson’s disease (Xu et al., 2016), here we provide mechanistic insights into reduced SV endocytosis at the NMJ, in the context of TDP-43 toxicity.
In the case of mutant TDP-43, co-OE of Hsc70-4 provides significant rescue of the FM1-43 dye uptake defect although, we note that this is not a full restoration to the levels observed in controls. Co-OE of chaperone-dead Hsc70-4D10N fails to mitigate TDP-43G298S induced FM1-43 dye uptake defects suggesting that motor neuron expression of mutant TDP-43 reduces Hsc70-4 levels below a threshold that is necessary to maintain a molecular chaperone activity facilitating SV endocytosis/cycling.
In contrast, for TDP-43WT, co-OE of Hsc70-4, while not sufficient to rescue SVC, it appears to mitigate toxicity by generally improving proteostasis and specifically reducing the aggregation of TDP-43WT itself, which correlates with improved locomotor function and increased lifespan. This suggests that the mechanism by which TDP-43WT and TDP-43G298S cause defects in FM1-43 dye uptake are at least in part distinct but ultimately reduced Hsc70-4 levels at the NMJ impact the activity of its co-chaperone complexes and clients, many of which control critical steps in SVC.
Perturbations in the SV cycle highlight key TDP-43 dependent functional interactions between Hsc70-4, co-chaperone and client partners
Co-OE of Csp, an Hsc70-4 co-chaperone critical for stabilizing client binding (Jiang et al., 2007; Kampinga and Craig, 2010), restores FM1-43 dye uptake to control levels for both TDP-43WT and TDP-43G298S. This is surprising given that Hsc70-4 co-OE mitigated effects of TDP-43G298S but failed to rescue TDP-43WT mediated defects in endocytosis. A reasonable explanation for this result is that Csp has synaptic functions that are independent of Hsc70-4, as previously suggested (Bronk et al., 2005). These additional functions may contribute to improved SVC when Csp and TDP-43WT are co-overexpressed in motor neurons. Co-OE of the GTPase dynamin, a client of the Csp/Hsc70-4 chaperone complex (Zhang et al., 2012), also significantly restores FM1-43 dye uptake to control levels for wild-type and mutant TDP-43. Together, these findings are consistent with a scenario whereby TDP-43 impairs the activity of the Csp/Hsc70 chaperone complex to maintain normal dynamin function.
Towards a common, synaptic pathomechanism of disease
Similar to what we found for TDP-43, we show that HSPA8 expression is reduced at synaptic terminals of C9 repeat expressing flies, and in cell bodies and dendrites of human C9 iPSC motor neurons. Furthermore, we show that C9 repeat expansions in flies lead to defects in FM1-43 dye uptake. However, the precise mechanism by which C9 repeat expansions regulate Hsc70 expression and SVC remains to be determined. As previous studies have reported a decrease in EJP amplitude and quantal content in a fly model of C9 ALS (Zhang et al., 2015), it is possible that C9 mediated reduced synaptic expression of Hsc70 may elicit FM1-43 dye uptake defects that arise due to deficits in both SV exo- and endocytosis. Recent reports suggest that splicing and transcriptome alterations, including changes in synaptic gene expression, contribute to C9 ALS pathology (Cooper-Knock et al., 2015; Prudencio et al., 2015). Thus, it is likely that multiple mechanisms may be at play to alter SVC in C9 mediated ALS.
Although OE of either TDP-43WT or disease associated TDP-43G298S leads to similar phenotypes, our findings of hsc70-4 mRNA as a mutant specific sequestration and translation target suggests that the molecular mechanisms utilized by different TDP-43 variants are at least in part, distinct (see Figure 7). This knowledge could impact the development of therapeutic strategies that ought to take into consideration the specific mechanisms of toxicity associated with TDP-43WT, which is linked to 97% of ALS cases as opposed to ALS associated mutant TDP-43 that only represents 2–3% of ALS cases. Taken together, our data suggest that post-transcriptional dysregulation of Hsc70-4 expression connects defects in ribostasis and proteostasis at synapses in ALS across multiple models. Improving SVC through OE of Hsc70, its co-chaperone Csp or their client, dynamin highlights the SV cycle as a unifying therapeutic target in ALS and related neurodegenerative disorders such as FrontoTemporal Dementia (FTD) and Alzheimer’s disease in which TDP-43 pathology has been reported.
Figure 7. A model for TDP-43 and Hsc70-4 interactions.

(A) In controls, TDP-43 does not sequester mRNA targets or protein partners leading to normal levels of mRNA translation and synaptic proteins at the NMJ. As a result, SVC occurs as normal. (B) Motor neuron expression of TDP-43 results in decreased synaptic expression of Hsc70-4 and defects in SV endocytosis. Mutant TDP-43 sequesters hsc70-4 mRNA in insoluble complexes and inhibits its translation. Note: post-transcriptional reduction in Hsc70 expression and SVC deficits are also present in C9orf72 models of ALS.
Experimental Procedures
Drosophila Genetics
All Drosophila stocks and crosses were kept on standard yeast/cornmeal/molasses food at 22°C unless otherwise noted.
Larval Turning Assays
Wandering third instar larvae were placed on a grape juice plate. After 30 second acclimation, larvae were gently turned ventral side up and observed until they turned over and began making a forward motion.
Lifespan Analysis
Newly eclosed males and females (100 per genotype) were separated and placed in a new vial every 7 days. Survival plots were generated using R and RStudio.
Electrophysiology
Current-clamp recordings were made from muscle 6 of third-instar larvae using a modified Ca2+ free HL-3 saline, as described (Dawson-Scully et al., 2000).
FM1-43 Dye Uptake Assays
FM1-43 dye uptake assays using 4 μM FM1-43FX, 90 mM KCl, and 2 mM Ca2+ for stimulation were adapted from (Kuromi and Kidokoro, 2005; Verstreken et al., 2008).
Cellular Fractionations and Western Blotting
Cellular fractionations were previously described (Estes et al., 2013; Liachko et al., 2010). Western blots (WBs) were performed as described (Coyne et al., 2014; Coyne et al., 2015). Primary antibodies were 1:3000 rabbit anti-Hsc70-4 (from K. Zinsmaier), 1:6000 rabbit anti-GFP (Invitrogen), and 1:5000 rabbit anti-β Actin (Cell Signaling).
Immunoprecipitations (IPs)
IPs were performed using published protocols (Coyne et al., 2014).
Polysome Fractionations
Polysome fractionations were previously described (Coyne et al., 2014; Coyne et al., 2015).
qRT PCR
qRT PCR is described in Supplemental Experimental Procedures. An RNeasy kit (Qiagen) was used for RNA isolation. First strand cDNA synthesis was performed with a Superscript III cDNA synthesis kit (Invitrogen). qPCR reactions were carried out using SYBR Select Master Mix (Applied Biosystems) and an ABI 7300 Real Time PCR System (Applied Biosystems).
Immunohistochemistry and Confocal Imaging
Larval NMJs were prepared as described (Estes et al., 2011; Estes et al., 2013). Primary antibodies were 1:50 rabbit anti-Hsc70-4 (from K. Zinsmaier), 1:300 mouse anti-DCSP2 (DSHB), and 1:50 mouse anti-Bruchpilot, NC82 (DSHB).
Primary Neuron Cultures: Transfection, Staining, Image Acquisition, and Analysis
All procedures for animal experiments were approved by the Emory University IACUC and the Emory University Institutional Review Board. Primary motor neurons from spinal cords of mouse embryos were isolated, and plated at day 13.5 (Fallini et al., 2010).
Human iPSC Motor Neuron Differentiation and Immunocytochemistry for Hsc70/HSPA8
Control iPSC and C9 iPSC cells (>30 repeats) were differentiated to motor neurons as described by previous studies (Donnelly et al., 2013; Zhang et al., 2015).
Mouse NMJs
Animals
Lines and procedures are described in the Supplemental Experimental Procedures. All procedures were approved by the University of Kansas Medical Center IACUC. Immunohistochemistry, image analysis and statistics were described in (Chen et al., 2011; Chen et al., 2012; Nishimune et al., 2004).
Bioinformatics analyses
Statistical Analyses
Larval turning assays - 30 larvae/genotype. Lifespan analysis - 100 flies/genotype. At least 6 animals/genotype were used for electrophysiology. FM1-43 dye uptake experiments were performed on 12 animals and a total of 24 NMJs (2/animal) were used for analysis. Cellular fractionation, IPs, WBs, polysome fractionations, and qPCR were performed in triplicate. For analysis of mouse primary motor neurons, cell bodies and 10μm segments of the axonal growth cones were analyzed. Cell body: GFP (n = 57), TDP-43 WT (n = 49), TDP-43 Q331K (n = 42), TDP-43 M337V (n = 49); Growth cone: GFP (n = 30), TDP-43 WT (n = 24), TDP-43 Q331K (n = 35), TDP-43 M337V (n = 36). For immunocytochemistry in TDP-43 iPSC motor neurons, two control lines and two differentiations of one TDP-43 line were used. For immunocytochemistry in C9orf72 iPSC motor neurons, one control and one C9 lines were used. Statistical analyses were performed using the appropriate t test or ANOVA model with Tukey’s post hoc test. For cellular fractionations, Fisher’s test was used. Statistics were performed using GraphPad Prism software version 7.0. Lifespan statistical analysis was done using the log-rank test in R. All data shown are mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, n.s. not significant.
Additional details on fly lines, iPSC demographic information and experimental procedures can be found in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
This work was funded by NIH K-INBRE postdoctoral award P20 GM103418 (to RSR), NIH R00 NS082376 (to JCS), NIH R01NS078214 and R01AG051470 (to HN), NIH R21 NS094809 (to KZ), an Emory Medicine Catalyst Award (to WR), Muscular Dystrophy Association (to RS), ALS Association (to RS), Barrow Neurological Foundation (to RS), and NIH RO1 NS091299 (to DCZ). Additional funds were provided by the Himelic Family Foundation (to DCZ) and the Neuroscience Graduate Interdisciplinary Program at UA and ARCS (to ANC). We thank members of the Zarnescu laboratory for helpful feedback. We thank J Paul Taylor, Takeshi Iwatsubo, Fen-Biao Gao, Norbert Perrimon and Patrik Verstreken for sharing Drosophila lines (see Supplemental Experimental Procedures). We also acknowledge the Bloomington Drosophila Stock Center at Indiana University and the Developmental Studies Hybridoma Bank at University of Iowa for fly stocks and antibodies respectively.
Footnotes
Author Contributions
Conceived and designed the experiments: ANC, IL, CC, RSR, HN, KZ, WR, RS and DCZ. Performed the experiments: ANC, IL, CC, MT, RSR, AS, BLZ, JL and JJ. Analyzed the data: ANC, IL, CC, MT, RSR, AS, HN, KZ, WR, RS and DCZ. Contributed reagents/materials: ANC, IL, CC, RSR, HN, KZ, WR, RS, and DCZ. JCS performed bioinformatics analyses. Wrote the paper: ANC and DCZ with input from co-authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Wilbanks SM, Scheller RH. The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem. 1996;271:25989–25993. doi: 10.1074/jbc.271.42.25989. [DOI] [PubMed] [Google Scholar]
- Bronk P, Nie Z, Klose MK, Dawson-Scully K, Zhang J, Robertson RM, Atwood HL, Zinsmaier KE. The multiple functions of cysteine-string protein analyzed at Drosophila nerve terminals. J Neurosci. 2005;25:2204–2214. doi: 10.1523/JNEUROSCI.3610-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk P, Wenniger JJ, Dawson-Scully K, Guo X, Hong S, Atwood HL, Zinsmaier KE. Drosophila Hsc70-4 is critical for neurotransmitter exocytosis in vivo. Neuron. 2001;30:475–488. doi: 10.1016/s0896-6273(01)00292-6. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin Cell Dev Biol. 2015;40:153–159. doi: 10.1016/j.semcdb.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD. Activation of the ATPase activity of heat-shock proteins Hsc70/Hsp70 by cysteine-string protein. Biochem J. 1997;322(Pt 3):853–858. doi: 10.1042/bj3220853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Chang HC, Newmyer SL, Hull MJ, Ebersold M, Schmid SL, Mellman I. Hsc70 is required for endocytosis and clathrin function in Drosophila. J Cell Biol. 2002;159:477–487. doi: 10.1083/jcb.200205086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Hazelett DJ, Stewart JA, Morton DB. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res. 2014;1584:39–51. doi: 10.1016/j.brainres.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Billings SE, Nishimune H. Calcium channels link the muscle-derived synapse organizer laminin beta2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J Neurosci. 2011;31:512–525. doi: 10.1523/JNEUROSCI.3771-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Mizushige T, Nishimune H. Active zone density is conserved during synaptic growth but impaired in aged mice. J Comp Neurol. 2012;520:434–452. doi: 10.1002/cne.22764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS One. 2015;10:e0127376. doi: 10.1371/journal.pone.0127376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, Pearson A, Bowser R, Zarnescu DC. Futsch/MAP1B mRNA Is a Translational Target of TDP-43 and Is Neuroprotective in a Drosophila Model of Amyotrophic Lateral Sclerosis. J Neurosci. 2014;34:15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, Yamada SB, Siddegowda BB, Estes PS, Zaepfel BL, Johannesmeyer JS, Lockwood DB, Pham LT, Hart MP, Cassel JA, et al. Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Hum Mol Genet. 2015;24:6886–6898. doi: 10.1093/hmg/ddv389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson-Scully K, Bronk P, Atwood HL, Zinsmaier KE. Cysteine-string protein increases the calcium sensitivity of neurotransmitter exocytosis in Drosophila. J Neurosci. 2000;20:6039–6047. doi: 10.1523/JNEUROSCI.20-16-06039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P, 3rd, Good SK, Johnson BA, Herz J, Yu G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Molecular and cellular biology. 2011;31:1098–1108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8:640–646. doi: 10.1111/j.1600-0854.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- Estes PS, Boehringer A, Zwick R, Tang JE, Grigsby B, Zarnescu DC. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Human molecular genetics. 2011;20:2308–2321. doi: 10.1093/hmg/ddr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PS, Daniel SG, McCallum AP, Boehringer AV, Sukhina AS, Zwick RA, Zarnescu DC. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Disease models & mechanisms. 2013;6:721–733. doi: 10.1242/dmm.010710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Molecular neurodegeneration. 2010;5:17. doi: 10.1186/1750-1326-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21:3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. Journal of proteome research. 2010;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Wishart TM. Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol Appl Neurobiol. 2013;39:320–334. doi: 10.1111/nan.12014. [DOI] [PubMed] [Google Scholar]
- Haucke V, Neher E, Sigrist SJ. Protein scaffolds in the coupling of synaptic exocytosis and endocytosis. Nature reviews Neuroscience. 2011;12:127–138. doi: 10.1038/nrn2948. [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Maes EG, Taylor AB, Wang L, Hinck AP, Lafer EM, Sousa R. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–433. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Exocytosis and endocytosis of synaptic vesicles and functional roles of vesicle pools: lessons from the Drosophila neuromuscular junction. Neuroscientist. 2005;11:138–147. doi: 10.1177/1073858404271679. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Liachko NF, Guthrie CR, Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci. 2010;30:16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther. 2012;136:354–374. doi: 10.1016/j.pharmthera.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Human molecular genetics. 2011;20:1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M. Molecular Neuropathology of TDP-43 Proteinopathies. Int J Mol Sci. 2009;10:232–246. doi: 10.3390/ijms10010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, Zinsmaier KE. Overexpression of cysteine-string proteins in Drosophila reveals interactions with syntaxin. J Neurosci. 1999;19:10270–10279. doi: 10.1523/JNEUROSCI.19-23-10270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature. 2004;432:580–587. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- Palter KB, Watanabe M, Stinson L, Mahowald AP, Craig EA. Expression and localization of Drosophila melanogaster hsp70 cognate proteins. Mol Cell Biol. 1986;6:1187–1203. doi: 10.1128/mcb.6.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins LA, Doctor JS, Zhang K, Stinson L, Perrimon N, Craig EA. Molecular and developmental characterization of the heat shock cognate 4 gene of Drosophila melanogaster. Mol Cell Biol. 1990;10:3232–3238. doi: 10.1128/mcb.10.6.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Belzil VV, Batra R. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. 2015;18:1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R. Vesicle scission: dynamin. Semin Cell Dev Biol. 2011;22:10–17. doi: 10.1016/j.semcdb.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nature reviews Neuroscience. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Romano G, Klima R, Buratti E, Verstreken P, Baralle FE, Feiguin F. Chronological requirements of TDP-43 function in synaptic organization and locomotive control. Neurobiol Dis. 2014;71:95–109. doi: 10.1016/j.nbd.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Gomez-Sanchez L, Mircheski J, Linares-Clemente P, Nieto-Gonzalez JL, Vazquez ME, Lujan R, Fernandez-Chacon R. Motorneurons require cysteine string protein-alpha to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron. 2012;74:151–165. doi: 10.1016/j.neuron.2012.02.019. [DOI] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. Identification of Neuronal RNA Targets of TDP-43-containing Ribonucleoprotein Complexes. J Biol Chem. 2011;286:1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9:1937–1954. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B. A trimeric protein complex functions as a synaptic chaperone machine. Neuron. 2001;31:987–999. doi: 10.1016/s0896-6273(01)00427-5. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature neuroscience. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, Kuenen S, Wittocx R, Corthout N, Marrink SJ, et al. Hsc70-4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron. 2015;88:735–748. doi: 10.1016/j.neuron.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, Meinertzhagen IA, Bellen HJ. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 2002;109:101–112. doi: 10.1016/s0092-8674(02)00688-8. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ohyama T, Bellen HJ. FM 1-43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol Biol. 2008;440:349–369. doi: 10.1007/978-1-59745-178-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu XS, Sheng J, Zhang Z, Yue HY, Sun L, Sgobio C, Lin X, Peng S. alpha-Synuclein Mutation Inhibits Endocytosis at Mammalian Central Nerve Terminals. 2016;36:4408–4414. doi: 10.1523/JNEUROSCI.3627-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom P, Graffmo KS, Andersen PM, Brannstrom T, Marklund SL. Proteins that bind to misfolded mutant superoxide dismutase-1 in spinal cords from transgenic amyotrophic lateral sclerosis (ALS) model mice. J Biol Chem. 2011;286:20130–20136. doi: 10.1074/jbc.M111.218842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kelley WL, Chamberlain LH, Burgoyne RD, Lang J. Mutational analysis of cysteine-string protein function in insulin exocytosis. J Cell Sci. 1999;112(Pt 9):1345–1351. doi: 10.1242/jcs.112.9.1345. [DOI] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Henderson MX, Colangelo CM, Ginsberg SD, Bruce C, Wu T, Chandra SS. Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron. 2012;74:136–150. doi: 10.1016/j.neuron.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE. Cysteine-string protein’s neuroprotective role. J Neurogenet. 2010;24:120–132. doi: 10.3109/01677063.2010.489625. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Bronk P. Molecular chaperones and the regulation of neurotransmitter exocytosis. Biochem Pharmacol. 2001;62:1–11. doi: 10.1016/s0006-2952(01)00648-7. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.