

Summary
Cellular tropism during persistent viral infection is commonly conferred by the interaction of a viral surface protein with a host receptor complex. Norovirus, the leading global cause of gastroenteritis, can be persistently shed during infection, but its in vivo cellular tropism and tropism determinants remain unidentified. Using murine norovirus (MNoV), we determine that a small number of intestinal epithelial cells (IECs) serve as the reservoir for fecal shedding and persistence. The viral non-structural protein NS1, rather than a viral surface protein, determines IEC tropism. Expression of NS1 from a persistent MNoV strain is sufficient for an acute MNoV strain to target IECs and persist. Additionally, interferon-lambda (IFN-λ) is a key host determinant blocking MNoV infection in IECs. The inability of acute MNoV to shed and persist is rescued in Ifnlr1−/− mice, suggesting that NS1 evades IFN-λ-mediated antiviral immunity. Thus, NS1 and IFN-λ interactions govern IEC tropism and persistence of MNoV.
Keywords: Norovirus, interferon-lambda, persistence, reservoir, tropism
eTOC Blurb
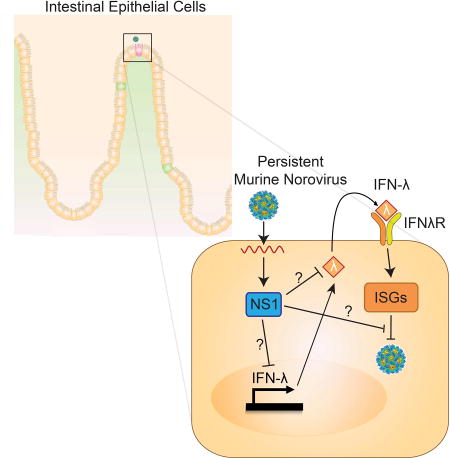
The in vivo tropism of norovirus, a gastrointestinal pathogen, is unknown. Lee et al. discover that a small number of intestinal epithelial cells are a reservoir for persistent murine norovirus. This tropism is regulated by viral protein NS1 and host IFN-lambda, findings that provide critical insight into norovirus pathogenesis.
Introduction
Noroviruses (NoVs) are non-enveloped, positive-sense, single-stranded RNA viruses comprising one genus of the family Caliciviridae, which contains six genogroups (GI-GVI). Genogroups I, II and IV primarily contain human noroviruses (HNoVs) and genogroup V contains murine noroviruses (MNoV) (Karst et al., 2014). HNoVs are the leading cause of gastroenteritis outbreaks across the globe, and are responsible for severe childhood diarrhea and foodborne disease outbreaks (Ahmed et al., 2014). During the acute phase of infection, HNoV causes vomiting and diarrhea, which typically resolves within one to two days (Atmar et al., 2008; Rockx et al., 2002). Viral shedding into the stool, however, can continue for weeks to months in asymptomatic patients (Gustavsson et al., 2017; Kaufman et al., 2014; Saito et al., 2013; Teunis et al., 2015), and for years in immunocompromised patients (Bok and Green, 2012). This phenomenon is referred to as persistence herein. Norovirus is transmitted by the fecal-oral route, predominantly from contaminated food, water, and person-to-person contact (de Graaf et al., 2016). Given that asymptomatic HNoV-infected patients can continue to persistently shed a substantial quantity of infectious virus into stool for prolonged periods, a more complete understanding of the mechanisms underlying viral shedding may lead to strategies to interrupt viral transmission and/or cure chronically infected patients (de Graaf et al., 2016; de Graaf et al., 2017).
Since HNoV has not had an in vitro cultivation system until recently (Ettayebi et al., 2016; Jones et al., 2014) or a genetically manipulable physiologically relevant animal model, murine norovirus (MNoV), first identified in 2003 (Karst et al., 2003), has been widely used as a model system to understand HNoV biology (Karst et al., 2014). MNoV can be efficiently cultivated in vitro and readily infects mice to provide a tractable in vivo model for NoV studies (Karst et al., 2014; Wobus et al., 2016). MNoV infection recapitulates many aspects of HNoV infection in vivo, including fecal-oral transmission, intestinal viral replication and prolonged viral shedding after acute infection, thus effectively modeling chronic asymptomatic HNoV infection (Karst et al., 2014; Wobus et al., 2016). The MNoV model has facilitated identification of many host and viral factors regulating NoV replication and pathogenesis in vivo (Baldridge et al., 2016). These findings include identifying a proteinaceous NoV receptor that can confer permissiveness for MNoV replication upon human cells (Haga et al., 2016; Orchard et al., 2016), uncovering NoV as a trigger for intestinal pathology in models for inflammatory bowel disease (Basic et al., 2014; Cadwell et al., 2010), discovering viral interactions with the commensal microbiota (Baldridge et al., 2015; Jones et al., 2014), and identifying the molecular basis for an interaction between the NS1/2 protein and the host protein VAPA that is shared between MNoV and HNoV proteins (McCune et al., 2017).
The cellular tropism of NoVs is critical to understanding the pathogenesis of intestinal infection. Multiple studies have provided experimental evidence for the cellular tropism of HNoV and MNoV, but the cell type responsible for viral transmission and disease symptoms has not yet been elucidated. Exploration of HNoV tropism has mostly relied on immunodeficient human patients (Karandikar et al., 2016) and non-native animal models. HNoV infection in immunocompromised mice showed macrophage-like cell tropism in the liver and spleen (Taube et al., 2013). HNoV infection in gnotobiotic pigs showed viral replication in small intestinal enterocytes (Cheetham et al., 2006). In chimpanzees, HNoVs were primarily detected in DC-SIGN+ phagocytes and B-cells in the lamina propria (Bok et al., 2011). Recent in vitro studies of HNoV showed successful cultivation of HNoV in B-cells and human intestinal enteroids (Ettayebi et al., 2016; Jones et al., 2014). However, all previous experimental evidence from in vivo models was from immunodeficient mice or other animal hosts. Studies of MNoV cellular tropism have shown that dendritic cells, macrophages and B-cells support MNoV growth in vitro (Jones et al., 2014; Wobus et al., 2004), while MNoV infection of immunodeficient mice suggested intestinal epithelial cell (IEC) tropism (Mumphrey et al., 2007; Ward et al., 2006). M-cells in Peyer’s patches are reported to transcytose MNoV into the lamina propria (Gonzalez-Hernandez et al., 2013; Gonzalez-Hernandez et al., 2014), but the role of M-cells in persistent infection and viral shedding is not clear. Importantly, there has been very limited data concerning HNoV and MNoV cellular tropism in immunocompetent hosts. Thus, the cellular tropism contributing to NoV disease symptoms, persistent intestinal infection, or viral transmission has not been fully elucidated.
Innate and adaptive immune responses are both important to control MNoV infection. Type III interferon (also known as IFN-lambda or IFN-λ) has specialized antiviral activities at mucosal barriers, such as the intestine (Hernandez et al., 2015; Mahlakoiv et al., 2015; Nice et al., 2015; Pott et al., 2011), lung (Crotta et al., 2013; Mordstein et al., 2008; Mordstein et al., 2010) and genital tract (Ank et al., 2006), and is thought to be a primary innate barrier against mucosally-transmitted viruses (Lazear et al., 2015). Intestinal growth and shedding of persistent MNoV strain CR6 is controlled by IFN-λ-mediated innate immunity but not type I or II interferons (Nice et al., 2015), and indeed exogenous IFN-λ can prevent and cure persistent MNoV infection in the absence of adaptive immunity (Nice et al., 2015). Persistent MNoV induces functionally suboptimal virus-specific CD8+ T cell responses (Tomov et al., 2013), but even with highly functional CD8+ T cell responses, persistent MNoV is generally unaffected and persists in the intestine (Tomov et al., In press). These findings suggest a critical role for innate immunity and a less important role for adaptive immunity in control of persistent MNoV infection. In contrast, an acute strain of MNoV, CW3, which infects mice systemically and is cleared within 2 weeks in immunocompetent mice, is known to be controlled by type I and II interferons (Hwang et al., 2012; Maloney et al., 2012; Nice et al., 2016; Thackray et al., 2012) but a role for type III interferon in regulation of acute MNoV infection has not been reported. Both T-cell and B-cell responses are required to clear acute MNoV infection (Chachu et al., 2008a; Chachu et al., 2008b; Tomov et al., 2013). These phenotypic and regulatory differences between persistent and acute MNoV strains suggest that specific viral genetic differences, influencing host responses, may contribute to MNoV persistence.
In this study, using a highly sensitive flow cytometry assay to simultaneously detect two non-structural proteins of MNoV, we discovered that a small number of IECs are infected by persistent MNoV, and that IEC infection is responsible for persistence. Furthermore, we identified the viral protein NS1 as the determinant of IEC tropism, IFN-λ to be a key host determinant of IEC tropism, and suggest an antagonistic relationship between these viral and host components as a basis for persistence and shedding in the intestine. Our findings help clarify the role of intestinal antiviral innate immune responses in controlling NoV infection and also point to a potentially key immune evasion strategy utilized by persistent MNoV.
Results
Intestinal epithelial cells are infected by persistent murine norovirus in vivo
Detecting MNoV-infected cells from the tissues of an immunocompetent host has been a substantial challenge, due to the rarity of MNoV-infected cells and high background signals in intestinal tissues. To overcome this limitation, we developed a sensitive flow cytometry assay to detect two non-structural proteins, NS1/2 and NS6/7, also known respectively as N-terminal protein and Protease/Polymerase (Thorne and Goodfellow, 2014). Since many enteric pathogens infect IECs and recent evidence from HNoV cultivation in enteroids suggested IEC tropism (Ettayebi et al., 2016), we hypothesized that MNoV infects IECs in vivo. Because Ifnlr1−/− mice exhibit increased viral shedding and replication in the intestine during persistent MNoV infection (Figure S1) (Baldridge et al., 2017; Nice et al., 2015), Ifnlr1−/− mice were included in this assay. We infected wild-type and Ifnlr1−/− mice with CR6, a persistent strain of MNoV, and collected epithelial cell fractions from the proximal colon 7 days post-infection (dpi). Among EpCam+/CD45- cells (pre-gated as in Figure 1A), we detected a very small proportion of IECs from wild-type and Ifnlr1−/− mice that were NS1/2 and NS6/7 double positive (here referred to as “MNoV+ IECs”) in the flow cytometry assay (Figure 1A). This NS1/2 and NS6/7 double positive population was not detected in either control sera-stained IECs (Figure 1A) or in EpCam-/CD45+ cells (Figure S1). Both control-stained and MNoV-stained IECs displayed comparable levels of NS1/2 single positive and NS6/7 single positive cells, indicating that double staining of different non-structural proteins efficiently decreases background signal (Figure 1A and S1). We also examined IECs from uninfected wild-type mice using this assay, and found that NS1/2 or NS6/7 single staining exhibits significant background signals at ~10–1000 events/million IECs but that NS1/2 and NS6/7 double staining shows background signal below 1 event/million IECs (Figure S1).
Figure 1. Detection of intestinal epithelial cells infected by persistent MNoV in vivo.

(A) Flow cytometry of NS1/2 and NS6/7 double staining. Colonic IECs from wild-type and Ifnlr1−/− mice infected with CR6 were stained with control sera or anti-NS1/2 and anti-NS6/7 and analyzed. EpCam+/CD45− cells were pre-gated as IECs. (B) MNoV genome quantification by quantitative RT-PCR of RNA from sorted NS1/2 and NS6/7 double positive cells. N = 4 to 8 mice per group, combined from two independent experiments. (C–E) Quantification of MNoV+ IECs during CR6 infection in wild-type and Ifnlr1−/− mice at (C) 7 dpi, (D) 14 dpi, and (E) 35 dpi. N = 5 to 13 mice per group, combined from two to four independent experiments. Dashed lines represent limit of detection. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparisons test (B) and Mann Whitney test (C–E). **P < 0.01, ***P < 0.001. See also Figure S1.
To independently confirm that MNoV+ IECs are MNoV-infected, we sorted NS1/2 and NS6/7 double positive cells and double negative cells and assessed for the presence of the MNoV RNA genome. MNoV+ IECs were >1000 fold-enriched for MNoV genomes compared to NS1/2 and NS6/7 double negative cells or unsorted cells (Figure 1B). We also stained IECs with an antibody recognizing the capsid protein of MNoV, VP1, and found that 67.9% of MNoV+ IECs were positive for VP1 (Figure S1). These data indicate that the double positive IECs are a site of MNoV gene and protein expression.
Quantification of the MNoV+ IECs from infected Ifnlr1−/− mice at day 7 post-infection indicated that they are more abundant than those observed in wild-type mice (Figure 1C), consistent with previous reports indicating enhanced CR6 replication in the intestines of Ifnlr1−/− mice (Baldridge et al., 2017; Nice et al., 2015). Fourteen and 35 dpi represent persistent timepoints for CR6 infection, as acute MNoV infection is fully cleared by 14 dpi (Nice et al., 2013). During persistent infection, we found that wild-type mice maintain about 21–23 MNoV+ cells/million IECs (95% confidence interval [CI], 0.0017%–0.0026% at 14 dpi and 0.0021%–0.0027% at 35 dpi). In contrast, Ifnlr1−/− mice maintain 38–61 MNoV+ cells/million IECs (95% CI, 0.0034%–0.0087% at 14 dpi and 0.0029%–0.0046% at 35 dpi) in the colon during persistent infection (Figure 1D and 1E). The ileum is also a site of viral infection by CR6 (Nice et al., 2013); we analyzed ileal IECs by flow cytometry at 7, 14, and 35 dpi, and found a similar percentage (95% CI, 0.0010%–0.0038% at 7 dpi and 0.0012%–0.0044% at 14 dpi) of MNoV+ cells at 7 and 14 dpi which decreased by 35 dpi (Figure S1). Therefore, a persistent strain of MNoV productively infects very low numbers of IECs at a level maintained over 35 days post-infection, and the level of this infection is controlled by endogenous IFN-λ signaling.
MNoV-infected IECs can be visualized microscopically
To confirm MNoV infection in IECs in the intestine and assess for infection-associated pathological changes in nearby tissue, we stained colonic sections for MNoV non-structural proteins and an IEC marker. Wild-type and Ifnlr1−/− mice were infected with CR6 and sacrificed at 14 dpi. Proximal colons were fixed and stained with antibodies recognizing NS1/2, NS6/7, and E-Cadherin. Consistent with flow cytometric results, infrequent NS1/2 and NS6/7 double positive cells were visible in the colonic epithelium of both wild-type and Ifnlr1−/− mice as E-Cadherin-positive (Figure 2A and 2B). There was no detectable disruption of the epithelial barrier or infiltration of inflammatory cells around infected IECs. Staining indicated that intracellular NS1/2 and NS6/7 localize in the perinuclear region, consistent with previous observations in macrophage cell lines (Hyde et al., 2009). We confirmed colocalization of NS6/7 with double-stranded RNA (dsRNA), which represents the MNoV replication complex, in wild-type mice (Figure 2C) (Hyde et al., 2009). Therefore, using both flow cytometry and an immunofluorescence assay, we validated that the in vivo tropism of persistent MNoV is for IECs.
Figure 2. Immunofluorescence microscopy to visualize MNoV-infected IECs.

(A–C) Proximal colons from wild-type (A, C) and Ifnlr1−/−(B) mice infected with CR6 were analyzed at 14 dpi by immunofluorescence microscopy for detection of (A, B) NS1/2, NS6/7, DAPI and E-cadherin staining or (C) dsRNA (J2 antibody) and NS6/7, and Hoechst staining. Low frequency E-cadherin positive epithelial cells expressed the viral non-structural proteins NS1/2 and NS6/7 which were tightly co-localized in a punctated pattern throughout the cytoplasm. Below is a magnified inset represented by the white box. Dashed line represents the luminal interface. Scale bars represent 10 µm.
Adaptive immunity is not sufficient to control MNoV infection in IEC
While MNoV CR6 establishes persistent infection in immunocompetent mice, MNoV CW3 infects mice but is cleared by adaptive immune responses by 14 dpi (Chachu et al., 2008a; Karst et al., 2003; Nice et al., 2013). However, CW3 can persist for prolonged periods in both intestinal and extra-intestinal tissues in Rag1−/− mice (Chachu et al., 2008a; Tomov et al., 2013). Interestingly, while CR6-infected Rag1−/− mice shed virus into stool over 35 dpi, virus was barely detectable in the stool of CW3-infected Rag1−/−mice over the same time course (Figure 3A). In contrast, CW3 genomes in the colon of Rag1−/− mice were readily detected, at a level comparable to that observed for CR6 (~105 copies per ug RNA), but at a 10-fold lower level than observed for CR6-infected Rag1−/− mice (Figure 3B). We examined wild-type and Rag1−/− mice at 14 and 35 dpi with CW3, and found that CW3 did not infect IECs in either mouse strain (Figure 3C and 3D). CR6-infected IECs are 4–5 fold enriched in Rag1−/− mice over wild-type mice at 14 and 35 dpi, indicating that adaptive immune responses can suppress CR6 infection in IECs but cannot eliminate persistence in these cells (Figure 3C and 3D). To rule out differential recognition of CW3 NS1/2 and NS6/7 proteins in our flow cytometric assay, we compared antibody staining for CW3 and CR6 in BV2 cells and found them to be equivalent (Figure S2). Thus, in the absence of adaptive immunity, MNoV CW3 can chronically infect the intestine of the murine host but doesn’t recapitulate IEC tropism or viral shedding into stool.
Figure 3. Regulation of IEC infection by adaptive immunity.

(A) Time-course of MNoV genome copies shed into fecal pellets with time points at 7, 14, 21, 24, 28, and 35 days after CR6 or CW3 infection in Rag1−/− mice. N = 9 to 31 mice per group, combined from three independent experiments. (B) MNoV genome copies in colon from wild-type or Rag1−/− mice at 35 dpi. (C, D) Quantification of MNoV+ IECs from wild-type and Rag1−/− mice at 14 dpi (C) and at 35 dpi (D). Dashed lines represent limit of detection. Statistical significance was determined by two-way ANOVA (A), one-way ANOVA followed by Tukey’s multiple-comparisons test (B) and Mann Whitney test (C, D). **P < 0.01, ***P < 0.001. See also Figure S2.
NS1 confers IEC tropism and persistence of MNoV
Since NS1 is a viral determinant of MNoV persistent infection (Nice et al., 2013), we hypothesized that NS1 may also confer IEC tropism. To test this hypothesis, we employed mutant NS1-chimeric viruses (Figure 4A). CR6 efficiently infected IECs, shed virus into stool and persisted in the colon (Figure 4B–4E). However, CR6NS1-CW3, which carries NS1-CW3 in the CR6 genome, lost these CR6 phenotypes. CR6NS1-CW3 failed to infect IECs, was not shed into stool, and didn’t persist in the intestine (Figure 4B–4E). Conversely, the acute strain CW3 did not infect IECs, get shed into stool or persist in the intestine. But with the reciprocal exchange of NS1-CR6 into the CW3 genome (CW3NS1-CR6), the virus substantially but not completely recapitulated the phenotypes of CR6 (Figure 4B–4E). We confirmed CW3NS1-CR6 infection of IECs, and maintenance of the NS1-CR6 sequence in vivo, by immunofluorescence and sequencing (Figure S3). Thus, NS1 of the persistent MNoV strain CR6 was both necessary and sufficient for IEC tropism, viral shedding and persistence.
Figure 4. IEC tropism of persistent MNoV is NS1-dependent.

(A) Schematic depicting the chimeric viruses used in this assay. (B–E) Wild-type mice were infected with the indicated viruses and analyzed at 14 dpi. (B, C) MNoV+ IECs from the proximal colon of infected mice were analyzed (B) and quantified (C) by flow cytometry. N = 5 mice per group, combined from two independent experiments. (D–E) MNoV genome copies in (D) stool and (E) colon. Dashed lines represent limit of detection. N = 5 to 13 mice per group, combined from two independent experiments. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparisons test. **P < 0.01, ***P < 0.001, NS = not significant. See also Figure S3.
NS1-mediated IEC tropism determines IFN-λ sensitivity of MNoV
We previously showed that exogenous IFN-λ prevents and cures persistent MNoV infection in mice (Nice et al., 2015) and that IFNLR1 expression in IECs is critical for the antiviral activity of IFN-λ (Baldridge et al., 2017). Since CW3 infection in wild-type and Rag1−/− mice did not show IEC infection in the intestine (Figure 3C, 3D, and 4C), we hypothesized that CR6 and CW3 may be differentially affected by IFN-λ. To test this hypothesis, we injected recombinant IFN-λ 1 day before CR6 or CW3 infection in wild-type mice, and assessed tissue and stool infection 7 dpi (Figure 5A–5C). IFN-λ completely blocked CR6 viral shedding and replication in the colon (Figure 5B and 5C), consistent with previous reports (Baldridge et al., 2017; Nice et al., 2015). Interestingly, IFN-λ had no effect on CW3 infection in the spleen or colon (Figure 5A and 5C). The NS1-chimeric MNoVs (i.e., CR6NS1-CW3 and CW3NS1-CR6) were examined in the same way. IFN-λ prevented CW3NS1-CR6 infection in the intestine and viral shedding into the stool (Figure 5B and 5C), correlating with CW3NS1-CR6 IEC infection (Figure 4C). However, the action of IFN-λ was restricted to the intestine with no effect on replication of CW3NS1-CR6 in the spleen (Figure 5A). CR6NS1-CW3 failed to grow in the intestine or other tissues with or without IFN-λ pre-treatment (Figure 5A–5C), consistent with its lack of IEC tropism (Figure 4C). Therefore, the NS1 of the persistent MNoV strain CR6 determined IEC tropism, which correlated with the preventative effect of IFN-λ on different viral strains. These results suggest that the antiviral effect of IFN-λ on MNoV infection is due to cell intrinsic effects in IECs.
Figure 5. Differential IFN-λ sensitivity of chimeric MNoVs.

(A–C) MNoV genome copies in tissues and fecal pellets. Wild-type mice were pre-treated with PBS or 3 µg of IFN-λ 1 day before infection, then infected with the indicated MNoV strains. MNoV genomes from spleen (A), stool (B), and colon (C) were quantified by qRT-PCR. N = 7 to 10 mice per group, combined from two independent experiments. Dashed lines represent limit of detection. Statistical significance was determined by Mann Whitney test. *P < 0.05, **P < 0.01, ***P < 0.001, NS = not significant. See also Figure S4.
IFN-λ does not cure parenterally-transmitted MNoV
While CW3 infects the intestine and MLN and also spreads systemically to extra-intestinal tissues such as spleen and liver, CR6 is restricted to the intestine and MLN in immunocompetent mice (Chachu et al., 2008a; Karst et al., 2003; Nice et al., 2013; Strong et al., 2012). To determine whether extra-intestinal infection of CR6 was affected by exogenous IFN-λ treatment, we pretreated wild-type mice with IFN-λ 1 day before infection, and then inoculated them with CR6 perorally (PO) or intraperitoneally (IP) for systemic infection (Figure 6A–6C). At 3 dpi, viral shedding into stool and growth in the colon was completely prevented by IFN-λ in orally-infected mice (Figure 6B and 6C). In contrast, IP-administered CR6 was rarely shed, exhibited lower viral replication in the colon, and was detected in the spleen (Figure 6A–6C). IFN-λ had a significant but minor effect on CR6 replication in the colon and no effect on splenic virus in mice infected by the IP route (Figure 6A–6C). Interestingly, the IP-administered CR6 phenocopied CW3 (i.e., rare shedding into the stool, lower replication in the intestine, systemic spread to the spleen, and insensitivity to exogenous IFN-λ). In sum, these results suggest systemically-introduced CR6 may share a non-IEC tropism with CW3 in IFN-λ-insensitive cells.
Figure 6. IFN-λ does not control parenterally-introduced MNoV infection.

(A–C) Wild-type mice were pre-treated with PBS or 3 µg of IFN-λ 1 day before infection, then infected with CR6 perorally (PO) or intraperitoneally (IP), and analyzed at 3 dpi. MNoV genomes from spleen (A), stool (B), and colon (C) were quantified by qRT-PCR. N = 9 to 10 mice per group, combined from two independent experiments. Dashed lines represent limit of detection. Statistical significance was determined by Mann Whitney test. *P < 0.05, ***P < 0.001, NS = not significant.
Exogenous IFN-λ rapidly clears MNoV-infected IECs
We previously showed that exogenous IFN-λ cures persistent CR6 infection in mice (Nice et al., 2015) and that IFN-λ signaling in IECs is critical for CR6 clearance in the intestine (Baldridge et al., 2017). To unveil the sterilizing activity of IFN-λ, we dissected the kinetics of MNoV clearance in IECs, stool, colon and MLN after IFN-λ treatment. Wild-type mice were infected with CR6 and then treated at 7 dpi with exogenous IFN-λ. MNoV+ IECs were rapidly cleared within the first day after IFN-λ treatment (Figure S4). Viral shedding into the stool was also quickly reduced by IFN-λ treatment, decreasing ~1 log after one day, and being cleared by 3 days after IFN-λ treatment (Figure S4). Viral genomes in intestinal tissue exhibited a similar clearance pattern to stool (Figure S4). In contrast, viral genomes in the MLN exhibited delayed clearance, with viral levels appearing unaffected at 1 or 3 days after IFN-λ treatment, decreasing ~1 log at 7 days post-treatment, then only being cleared 14 days after IFN-λ treatment (Figure S4). These data demonstrate that MNoV clearance in intestinal and lymphoid tissues was delayed relative to clearance from IECs.
NS1 evades IFN-λ-mediated antiviral immunity
From our NS1-chimera studies, we found that CR6NS1-CW3 was unable to infect IECs or grow in the intestine in wild-type mice (Figure 4 and 5), and that sensitivity to the antiviral activity of IFN-λ was NS1-dependent (Figure 5). Therefore, we hypothesized that NS1-CR6 may be able to antagonize IFN-λ immunity in IECs while NS1-CW3 lacks this capability. To further explore interactions of NS1 and IFN-λ signaling, we tested IFN-λ induction and responses in multiple cell lines in vitro. BV2 cells, which are permissive for CR6 and CW3 (Orchard et al., 2016), were insensitive to IFN-λ treatment (Figure S5). Since IEC cell lines we tested were not permissive for MNoV, we generated an MNoV-permissive IEC line, M2C-CD300lf, by lentiviral transduction of the immortalized colonic epithelial cell line M2C with the MNoV receptor (Orchard et al., 2016; Padilla-Nash et al., 2012) (Figure S5). CR6 infection of M2C-CD300lf cells induced IFN-λ expression and was sensitive to exogenous IFN-λ treatment, consistent with our in vivo findings. However, CW3 was phenotypically indistinguishable from CR6 in these cells, indicating the existence of critical differences between this in vitro model and our observed in vivo CR6-specific IEC tropism. Further, replication of CW3, CR6, and the NS1-chimeras was not affected by knock-down of Ifnlr1 in these cells, indicating that cell-intrinsic functions of IFN-λ do not control differences between CW3 and CR6 infection in these cells (Figure S5).
In the absence of an in vitro system that effectively recapitulated our in vivo observations, we elected to perform further studies in vivo. CW3 does not persist in Ifnlr1−/− mice (Nice et al., 2015), nor does it persist in Ifnlr1f/f-Villincre mice, which were previously demonstrated to be specifically deficient for Ifnlr1 only in IECs (Baldridge et al., 2017) (Figure S6). However, we hypothesized that growth of the CR6NS1-CW3 virus could be rescued in IFN-λ-signaling-deficient mice. Indeed, we found that CR6NS1-CW3 established persistent infection only in Ifnlr1−/− mice but not in wild-type, Ifnar1−/− or Rag1−/− mice (Figure 7A and 7B). Ifnlr1−/− mice infected with CR6NS1-CW3 exhibited high levels of viral shedding in stool (Figure 7A) and viral replication in the colon (Figure 7B). Importantly, persistence and fecal shedding of CR6NS1-CW3 were also rescued in Ifnlr1f/f-Villincre mice (Figure 7C and 7D). These results indicate that the NS1 of the persistent MNoV strain CR6 was necessary to escape IFN-λ-mediated antiviral immunity in IECs, a requirement that is relieved when IFN-λ responses are absent in the host. Future illumination of the mechanisms of NS1 activity will demand finer resolution of the differences governing in vivo tropism of the MNoV strains.
Figure 7. Complemented viral persistence of CR6NS1-CW3 in Ifnlr1 knockout mice.

(A–D) Wild type, Ifnlr1−/−, Ifnar1−/−, Rag1−/−, Ifnlr1f/f and Ifnlr1f/f-Villincre mice were infected with CR6NS1-CW3, and MNoV genomes from stool (A, C) and colon (B, D) were analyzed at 14 dpi by qRT-PCR. N = 6 to 13 mice per group, combined from two or three independent experiments. Dashed lines represent limit of detection. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple-comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001. See also Figures S5 and S6.
Discussion
Here, we present the identification of IECs as a reservoir of persistent MNoV infection in an immunocompetent murine host, and make the surprising observation that tropism for these cells as well as viral persistence and shedding is determined by a viral nonstructural protein, NS1, in combination with a host cytokine, IFN-λ. To detect this low frequency population, we developed a highly sensitive flow cytometry assay relying on simultaneous detection of two different viral non-structural proteins. We found that only ~20 cells per million IECs are infected by MNoV strain CR6 in wild-type mice at persistent time points. It is remarkable that this small number of cells is responsible for shedding approximately one million copies of MNoV into each fecal pellet, which is the major source of viral transmission of norovirus. HNoV cellular tropism in the intestine has not yet been elucidated in an immunocompetent host in vivo, which has been a barrier to understanding the pathogenesis of HNoV infection. Previous evidence for HNoV cellular tropism has come from immunodeficient human patients (Karandikar et al., 2016), non-human primates (Bok et al., 2011), gnotobiotic pigs (Cheetham et al., 2006), and immunodeficient animals (Taube et al., 2013). Cumulatively these data have suggested that macrophages, dendritic cells, B-cells and IECs are all potential target cells for HNoV (Karst and Wobus, 2015). Since persistent viral shedding of MNoV in mice resembles the prolonged viral shedding seen in asymptomatic HNoV-infected patients after resolution of acute infection, the IEC tropism of MNoV may also apply to HNoV. This would be consistent with studies identifying IECs as an important cell type for HNoV replication (Ettayebi et al., 2016; Karandikar et al., 2016). Bile acids have been identified as an important factor for HNoV cultivation in enterocytes (Ettayebi et al., 2016), which may correlate with a requirement for intestinal microbes for infection by MNoV strain CR6 (Baldridge et al., 2015); the exploration of the role of host and microbial metabolites in IEC infection will be important for future study. Although infection of other cell types may contribute to virus production and disease, we speculate that IEC tropism is likely to be a common feature for all noroviruses during viral shedding and fecal-oral transmission phases of the pathogenetic process.
The persistent and acute strains of MNoV exhibited differential infectivity of IECs in both wild-type and Rag1−/− mice, facilitating our identification of the viral determinant of IEC tropism to be the viral non-structural protein NS1. NS1-CR6 was both necessary and sufficient for IEC tropism, for persistence, and for fecal-oral viral transmission. Most interestingly, defective growth of a viral strain lacking NS1-CR6 (CR6NS1-CW3) in wild-type mice was rescued in Ifnlr1−/− mice, suggesting that persistent MNoV evades IFN-λ-mediated antiviral immunity in IECs via NS1-mediated activity. The combination of a host IFN-λ-mediated immune response in IECs, and the counteracting viral immune evasion by NS1-CR6, confers the exclusive IEC tropism of MNoV strain CR6 for persistence and fecal-oral viral transmission. The molecular mechanism of NS1, while not identified in this study, will be key to understanding its role in conferring IEC tropism. Determining whether NS1 acts through direct antagonism of IFN-λ immunity, and whether this activity is conserved in HNoV, also requires additional exploration. As differences between persistent and acute MNoV strains were not recapitulated in an MNoV-permissive colonic IEC line, either mechanistic studies in vivo or new systems for analysis of NoV tropism in vitro will be required to perform such studies.
The acute MNoV strain CW3 elicits a different set of host immune responses from CR6. CW3 infection is not under the control of IFN-λ, even in the colon, but is known to be naturally cleared by T-cell and B-cell responses within 2 weeks after infection (Chachu et al., 2008a; Chachu et al., 2008b). Given that CW3 did not infect IECs in wild-type or Rag1−/− mice and was not IFN-λ sensitive, CW3 may primarily infect non-IEC cells that are effectively IFN-λ unresponsive in the intestine (i.e., dendritic cells, macrophages and B-cells). When inoculated via a parenteral route, CR6 likely shares this tropism for IFN-λ-insensitive cells with CW3, making IEC infection after oral CR6 infection unique. A recent study showed that persistent CR6 ignores highly functional MNoV-specific CD8+ T cell responses in the intestine (Tomov et al., In press), suggesting that MNoV+ IECs could be an immune-privileged site for escaping T-cell responses. Therefore, differential cell tropism between persistent and acute strains of MNoV in the intestine may confer differential host immune sensitivities. In this scenario, persistent infection of CR6 in IECs is controlled by IFN-λ but escapes CD8+ T-cell responses, even though the CD8+ T-cell response is highly functional. Conversely, acute infection of CW3 in non-IEC cells is not controlled by IFN-λ but is targeted by CD8+ T-cells and other adaptive immune responses, and ultimately cleared in an immunocompetent murine host.
One of the interesting features of IFN-λ-mediated immunity is its sterilizing activity against persistent MNoV infection in the absence of adaptive immunity (Nice et al., 2015). We recently showed that IFN-λ signaling in IECs is critical for MNoV clearance (Baldridge et al., 2017). Since exogenous IFN-λ treatment of mice persistently infected with CR6 immediately cleared MNoV+ IECs within a day, IFN-λ-mediated cell intrinsic MNoV clearance in IECs is likely the initial mechanism of sterilizing immunity by IFN-λ.
Rapid MNoV clearance in IECs also correlates with rapid clearance of stool, corroborating our supposition that IECs are responsible for viral shedding and transmission. MNoV clearance in intestinal tissues was delayed compared to clearance in the IECs, and even more dramatically delayed in the MLN. The differential kinetics of MNoV clearance in tissues suggests that MNoV in the intestine and MLN depends on the presence of infected IECs and is gradually cleared by immune mechanisms other than IFN-λ when infected IECs are absent. Thus, sterilizing immunity by IFN-λ may require at least two steps. Firstly, IFN-λ triggers rapid clearance of the IEC reservoir of persistent MNoV infection. Secondly, other components of the immune system clear MNoV virions from non-IECs in tissues. Further studies will be necessary to elucidate the secondary steps of IFN-λ-mediated sterilizing innate immunity.
In summary, we identified IECs as a reservoir of persistent MNoV infection by showing, through cellular and genetic analysis, that they are responsible for viral shedding and persistence of MNoV. This study also clarifies how the host innate and adaptive immune systems control persistent MNoV infection, an effect mediated predominantly by the IFN-λ antiviral immune response. This IFN-λ-mediated antiviral response and the counteracting viral antagonism by NS1 determine the IEC tropism of persistent MNoV. This work provides a unique example of control of viral tropism by a non-structural protein in combination with a host cytokine, since both MNoV CW3 and CR6 utilize the same cellular receptor (Orchard et al., 2016). This unveiling of a reservoir of MNoV and the host-virus interactions that govern IEC tropism provide us with critical insight into norovirus pathogenesis and infection. We speculate that these findings will facilitate better understanding of HNoV pathology and transmission, and may provide us with important therapeutic targets to prevent HNoV outbreaks and cure chronically infected immunocompromised patients.
STAR Methods
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Cells
Viruses
METHOD DETAILS
MNoV infections and IFN-λ treatment in mice
MNoV infection in cell lines
Knockdown of Ifnlr1 in M2C-CD300lf cells
MNoV plaque assay
Quantitative reverse transcription-PCR
Flow cytometry for MNoV+ intestinal epithelial cells (IECs)
Sorting of MNoV+ IECs
Fluorescence microscopy
Sequencing of MNoV NS1 gene
QUANTIFICATION AND STATISTICAL ANALYSIS
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Megan Baldridge (mbaldridge@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild-type C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) (stock number 000664) and housed at the Washington University School of Medicine under specific-pathogen-free conditions in an MNoV-free facility according to university guidelines. Animal protocols were approved by the Washington University Animal Studies Committee. Knock-out mice on the C57BL/6J background were maintained in the same conditions and included the following strains: Ifnar1−/− (B6.129.Ifnar1tm1) (Muller et al., 1994), Ifnlr1−/− (B6.Cg-Ifnlr1tm1Palu) (Ank et al., 2008; Baldridge et al., 2017), and Rag1−/− (B6.129S7-Rag1tm1Mom/J) (Mombaerts et al., 1992). Ifnlr1f/f mice were generated from Ifnlr1tm1a(EUCOMM)Wtsi ES cells as described previously (Baldridge et al., 2017). Ifnlr1f/f-Villincre were generated by crossing Ifnlr1f/f to Villin-Cre mice for selective disruption of Ifnlr1 in intestinal epithelial cells (Madison et al., 2002). Equal ratios of adult male and female mice, aged 6 to 12 weeks, were used in all experiments for all strains. Experimental mice were cohoused with up to 5 mice of the same sex per cage with autoclaved standard chow pellets and water provided ad libitum.
Cells
M2C and M2K cells, both derived from male mice, and F1K and F3K cells, both derived from female mice, were cultured in DMEM/F-12 medium (Gibco, #11039-047) with 15% fetal bovine serum (FBS). mICCL2 cells (originally isolated from 20-day old mouse fetuses of unreported sex) and BV2 cells (derived from female mice) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% HEPES. M2C-CD300lf cells were generated by lentiviral transduction of the M2C cells with a pCDH-CMV-MCS-T2A–Puro vector (System Biosciences, Inc) containing CD300lf. Two days after transduction, cells were selected with puromycin (2 µg/ml) for 2 weeks. Cell lines were authenticated by microscopic morphologic evaluation and screening for mycoplasma, and BV2 cells were also authenticated by karyotyping.
Viruses
Stocks of MNoV strains CR6, CW3, CR6NS1-CW3 and CW3NS1-CR6 were generated from molecular clones as previously described (Baldridge et al., 2017; Strong et al., 2012). Briefly, a plasmid encoding the viral genome was transfected into 293T cells to generate infectious virus, which was subsequently passaged on BV2 cells. After two passages, cell cultures were frozen and thawed to liberate virions, then cultures were cleared of cellular debris and virus was concentrated by ultracentrifugation through a 30% sucrose cushion. Titers of all virus stocks were determined by plaque assay on BV2 cells.
METHOD DETAILS
MNoV infections and IFN-λ treatment in mice
For MNoV infections, mice were inoculated with a dose of 106 PFU of the indicated viral strain at 6 to 8 weeks of age by the oral route in a volume of 25 µl or the intraperitoneal route in a volume of 200 µl. Recombinant IFN-λ was provided by Bristol-Myers Squibb (Seattle, WA) as a monomeric conjugate comprised of 20-kDa linear polyethylene glycol (PEG) attached to the amino terminus of murine IFN-λ, as previously reported (Baldridge et al., 2017). For treatment of mice, 3 µg of IFN-λ diluted in PBS was injected intraperitoneally. Stool and tissues were harvested into 2-ml tubes (Sarstedt, Germany) with 1-mm-diameter zirconia/silica beads (Biospec, Bartlesville, OK). Tissues were flash frozen in a bath of ethanol and dry ice and either processed on the same day or stored at −80°C.
MNoV infection in cell lines
For MNoV infection, IEC lines and BV2 cells were seeded at 1 × 105 cells/well of a 24-well plate. After 24 hours, cells were infected with MNoV in 200 µl volume for 1 hour with gentle shaking at room temperature. Viral inoculum was removed and 500 µl of media was added to the cells. At 12, 24 or 48 hpi, the culture supernatant was harvested for plaque assay, and the cells were washed once with PBS and used for RNA prep. For the IFN-sensitivity assay, IFN-β (R&D systems, #12405-1) and IFN-λ (Bristol-Myers Squibb) were added to the cells at the time of seeding to the culture plate. The cells were incubated with IFNs for 24 hours, then infected with MNoV as described above.
Knockdown of Ifnlr1 in M2C-CD300lf cells
M2C-CD300lf cells were seeded at 2 × 104 cells/well of a 24-well plate 16 hours before siRNA transfection. The siRNAs targeting Ifnlr1 (Dharmacon, #L-041660-01–0005) or a control sequence (Dharmacon, #D-001810-01–05) were transfected using Dharmafect 2 reagent (Dharmacon) at final concentration of 20 nM, according to the manufacturer’s instructions. The M2C-CD300lf cells were incubated with the transfection mixture for 48 hours, then cells were infected with CR6, CW3, CR6NS1-CW3 or CW3NS1-CR6. At 12 hpi, the culture supernatant was harvested for plaque assay. Mock-infected cells were used for Ifnlr1 qRT-PCR to confirm knockdown efficiency.
MNoV plaque assay
The culture supernatant from infected cells was harvested and frozen at −80°C. BV2 cells were seeded at 1 × 106 cells/well of a six-well plate. 16 hours after seeding, 10-fold serially diluted sample was applied to each well for 1 hour with gentle rocking. The viral inoculum was aspirated and 2 ml of methylcellulose media (MEM, 10% FBS, 2mM L-Gluatmine, 10 mM HEPES, and 1% methylcellulose) was added. Plates were incubated for 60–72 hours prior to visualization of plaques with crystal violet solution (0.2% crystal violet and 20% ethanol).
Quantitative reverse transcription-PCR
As previously described (Baldridge et al., 2017), RNA was isolated from stool using a ZR-96 viral RNA kit (Zymo Research, Irvine, CA). RNA from tissues or cells was isolated using TRI Reagent (Invitrogen) with a Direct-zol-96 RNA kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. 5 µl of RNA from stool or 1 µg of RNA from tissue was used for cDNA synthesis with the ImPromII reverse transcriptase system (Promega, Madison, WI). DNA contamination was removed using the DNAfree kit (Life Technologies). MNoV TaqMan assays were performed, using a standard curve for determination of absolute viral genome copies, as described previously (Baert et al., 2008). PrimeTime qPCR assays for Ifna4 (Mm.PT.58.7678281.g), Ifnb (Mm.PT.58.30132453.g), Ifnlr1 (Mm.PT.58.32691011), Ifit1 (Mm.PT.58.32674307) and Ifi44 (Mm.PT.58.12162024) (IDT, Coralville, Iowa), and a Taqman assay for Ifnl2/3 (Mm04204156_gH) (Thermo Fisher Scientific) were performed using the same protocol. Quantitative PCR for housekeeping gene Rps29 was performed as previously described (Baldridge et al., 2017). Standard curves for quantitative PCR assays were used to facilitate absolute quantification of transcript copy numbers.
Flow cytometry for MNoV+ IECs
The epithelial fraction of the proximal colon was isolated as previously described (Lefrancois and Lycke, 2001). In brief, after mice were euthanized, proximal colons and ileums were collected. Tissues were washed with cold PBS twice, then chopped and transferred to new tubes. The tissues were incubated with stripping buffer (for proximal colon, 10% fetal bovine serum, 15 mM HEPES, 5 mM EDTA, 5 mM DTT in 1x HBSS; for ileum, 10% fetal bovine serum, 15 mM HEPES, 5 mM EDTA, in 1x HBSS) for 20 min in 37°C and vortexed for 15 seconds at maximum sett ing. The dissociated cells were passed through 100 µm and 40 µm filter mesh and washed with cold PBS. The collected cells were stained with a live/dead staining kit (live/dead fixable blue dead cell stain kit, Life Technology) according to the manufacturer’s protocol. After live/dead staining, cells were stained with anti-EpCam-APC-Cy7 (clone G8.8, Biolegend) and anti-CD45-PacBlue (clone 30-F11, Biolegend) for 20 minutes on ice, then washed with wash buffer (2% FBS, 1 mM EDTA in PBS). For intracellular staining, cells were fixed with Cytofix/Cytoperm buffer (BD Bioscience) for 20 minutes in room temperature. Cells were washed with Perm/Wash buffer (BD Bioscience) twice, and stained with anti-NS1/2 (Rabbit polyclonal antibody, provided by Vernon Ward) and anti-NS6/7 (Guinea-pig polyclonal antibody, provided by Kim Green) for 1 hour at room temperature. VP1 (A6.2) antibody was used for MNoV capsid staining. Cells were stained with anti-Rabbit-PE (Jackson) and anti-Guinea-pig-AF647 (Invitrogen) for one hour at room temperature. Cells were analyzed using a BD LSR II flow cytometer (BD Bioscience). Doublets were excluded by SSC-A × SSC-H analysis, and dead cells were excluded by live/dead staining. IECs were defined as EpCAM+CD45- cells, consistent with previous publications (Mahlakoiv et al., 2015). Data were processed using FACSDiva software (BD) and FlowJo (FlowJo).
Sorting of MNoV+ IECs
Cells were sorted using the BD ARIA II (BD Biosciences) using FACSDiva software. 100–1000 cells from NS1/2+NS6/7+ or NS1/2-NS6/7- gates were sorted into a microtube containing 100 µl of PKD buffer (Qiagen) with a 1:16 proteinase K solution (Qiagen). To lyse cells and purify RNA, the collected cells were heat-inactivated at 56°C for 1 hour. Total RNA was extracted from the cells by TRIzol-LS (Life technology) according to the manufacturer’s protocol with a few alterations. For example, Isopropanol precipitation was carried out overnight, and 0.5 µl of glycogen was used to facilitate RNA precipitation. Prepped total RNAs were used for reverse-transcriptase reactions as described.
Fluorescence microscopy
Wild-type or Ifnlr1−/− mice were sacrificed at 14 dpi. The colon was harvested, flushed with PBS followed by 10% neutral buffered formalin and then fixed overnight. Tissue was then blocked in 1% agar and embedded in paraffin for sectioning. Slides were then washed in xylene (3 washes × 5 min), isopropanol (3 washed × 5 min) and diH20 (5 min). Antigen retrieval was performed by boiling for 20 min in Dako antigen retrieval solution. Blocking was performed in TBST+10% goat serum+1%BSA. Primary antibodies were then applied in TBST overnight at 4 °C (E-cadherin 1:400, NS1/2 1:1000, NS6/7 1:1000, dsRNA(J2, Scicons) 1:100) followed by washing in TBST and incubation with secondary antibodies (1:500). Images were collected on a Zeiss LSM 880 Confocal Laser Scanning Microscope and analyzed on FIJI software. A maximum projection Z-stack was generated for each channel prior to merging.
Sequencing of MNoV NS1 gene
Phusion High-Fidelity DNA Polymerase (Thermo Scientific) was used to PCR-amplify the MNoV NS1 gene with primers MNoV-F1 5’-GTGAAATGAGGATGGCAACGC-3’ and MNoV-R718 CTAGAGATCTTCGCCCTCTTC from cDNA isolated from stool. PCR products were Sanger sequenced and aligned to reference genomes using Geneious 9.1.2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with Prism 7 software (GraphPad Software, San Diego, CA). In all graphs, three asterisks indicate a P value of <0.001, two asterisks indicate a P value of <0.01, one asterisk indicates a P value of <0.05, and ns indicates not significant (P > 0.05) as determined by Mann-Whitney test, one-way analysis of variance (ANOVA), or two-way ANOVA with Tukey’s multiple-comparison test, as specified in the relevant figure legends. The number of mice and/or replicates used in experiments is specified in the relevant figure legends.
Supplementary Material
Highlights.
Intestinal epithelial cells (IECs) are the persistent reservoir of murine norovirus (MNoV).
MNoV infects a very small number of IECs in immunocompetent hosts.
NS1, rather than a viral surface protein, is a viral determinant of IEC tropism.
NS1 and IFN-λ interactions govern IEC tropism and persistence of MNoV.
Acknowledgments
The authors would like to thank D. Kreamalmeyer for animal care and breeding, the members of the Virgin lab for manuscript review and discussion, and the Flow Cytometry & Fluorescence Activated Cell Sorting Core Facility, the Molecular Microbiology Imaging Facility and the Elvie L. Taylor Histology Core Facility at Washington University School of Medicine for assistance with cell sorting and immunohistochemistry. The authors would also like to thank Thomas Ried for providing colonic IEC lines. H.W.V. was supported by National Institutes of Health (NIH) grant U19 AI109725, and the Crohn’s and Colitis Foundation grant #326556. S.L. was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2016R1A6A3A03012352). M.T.B. was supported by National Institutes of Health (NIH) grant K22 AI127846-01 and DDRCC grant P30 DK052574. T.J.N. was supported by NIH training grant 5T32A100716334 and postdoctoral fellowships from the Cancer Research Institute and American Cancer Society. C.B.W. was supported by NIH grant 1K08AI128043-01. B.T.M. was supported by NCI-NIH award F31CA177194-01. K-W.K. was supported by NIH R37 AI049653 to G. J. Randolph. A.O. was supported by Pediatric Infectious Diseases Society-St. Jude Children’s Research Hospital Fellowship Program in Basic and Translational Research. Washington University holds patents related to murine norovirus. The University and H.W.V. receive income based on licenses for this MNV technology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, S.L., M.T.B.; Methodology, S.L., C.B.W., B.T.C., K-W.K., R.C.O., T.J.N.; Validation, S.L., C.B.W., S.T.P., M.T.B.; Investigation, S.L., M.T.B.; Resources, B.T.C., T.J.N.; Writing – Original Draft, S.L.; Writing – Review & Editing, C.B.W., A.O., B.T.C., K-W.K., R.C.O., T.J.N., M.T.B.; Funding Acquisition, H.W.V.; Supervision, H.W.V.
References
- Ahmed SM, Hall AJ, Robinson AE, Verhoef L, Premkumar P, Parashar UD, Koopmans M, Lopman BA. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. The Lancet Infectious diseases. 2014;14:725–730. doi: 10.1016/S1473-3099(14)70767-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. Journal of immunology. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. Journal of virology. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. Norwalk virus shedding after experimental human infection. Emerging infectious diseases. 2008;14:1553–1557. doi: 10.3201/eid1410.080117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baert L, Wobus CE, Van Coillie E, Thackray LB, Debevere J, Uyttendaele M. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Applied and environmental microbiology. 2008;74:543–546. doi: 10.1128/AEM.01039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, Virgin HW. Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda against Norovirus and Reovirus. Journal of virology. 2017;91 doi: 10.1128/JVI.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. 2015;347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, Turula H, Wobus CE. Norovirus Regulation by Host and Microbe. Trends in molecular medicine. 2016;22:1047–1059. doi: 10.1016/j.molmed.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basic M, Keubler LM, Buettner M, Achard M, Breves G, Schroder B, Smoczek A, Jorns A, Wedekind D, Zschemisch NH, et al. Norovirus Triggered Microbiota-driven Mucosal Inflammation in Interleukin 10-deficient Mice. Inflammatory bowel diseases. 2014;20:431–443. doi: 10.1097/01.MIB.0000441346.86827.ed. [DOI] [PubMed] [Google Scholar]
- Bens M, Bogdanova A, Cluzeaud F, Miquerol L, Kerneis S, Kraehenbuhl JP, Kahn A, Pringault E, Vandewalle A. Transimmortalized mouse intestinal cells (m-ICc12) that maintain a crypt phenotype. The American journal of physiology. 1996;270:C1666–1674. doi: 10.1152/ajpcell.1996.270.6.C1666. [DOI] [PubMed] [Google Scholar]
- Bok K, Green KY. Norovirus gastroenteritis in immunocompromised patients. The New England journal of medicine. 2012;367:2126–2132. doi: 10.1056/NEJMra1207742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok K, Parra GI, Mitra T, Abente E, Shaver CK, Boon D, Engle R, Yu C, Kapikian AZ, Sosnovtsev SV, et al. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:325–330. doi: 10.1073/pnas.1014577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachu KA, LoBue AD, Strong DW, Baric RS, Virgin HW. Immune mechanisms responsible for vaccination against and clearance of mucosal and lymphatic norovirus infection. PLoS pathogens. 2008a;4:e1000236. doi: 10.1371/journal.ppat.1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachu KA, Strong DW, LoBue AD, Wobus CE, Baric RS, Virgin HW. Antibody is critical for the clearance of murine norovirus infection. Journal of virology. 2008b;82:6610–6617. doi: 10.1128/JVI.00141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham S, Souza M, Meulia T, Grimes S, Han MG, Saif LJ. Pathogenesis of a genogroup II human norovirus in gnotobiotic pigs. JVirol. 2006;80:10372–10381. doi: 10.1128/JVI.00809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, Wack A. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS pathogens. 2013;9:e1003773. doi: 10.1371/journal.ppat.1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf M, van Beek J, Koopmans MP. Human norovirus transmission and evolution in a changing world. Nature reviews Microbiology. 2016;14:421–433. doi: 10.1038/nrmicro.2016.48. [DOI] [PubMed] [Google Scholar]
- de Graaf M, Villabruna N, Koopmans MP. Capturing norovirus transmission. Curr Opin Virol. 2017;22:64–70. doi: 10.1016/j.coviro.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016;353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez MB, Liu T, Blanco LP, Auble H, Payne HC, Wobus CE. Murine norovirus transcytosis across an in vitro polarized murine intestinal epithelial monolayer is mediated by M-like cells. Journal of virology. 2013;87:12685–12693. doi: 10.1128/JVI.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald JE, Ikizler M, Yagita H, Dermody TS, Williams IR, Wobus CE. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. Journal of virology. 2014;88:6934–6943. doi: 10.1128/JVI.00204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson L, Norden R, Westin J, Lindh M, Andersson LM. Slow clearance of norovirus following infection with emerging variants of genotype GII.4 strains. Journal of clinical microbiology. 2017 doi: 10.1128/JCM.00061-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, Yokoyama M, Murata K, Nakanishi A, Katayama K. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E6248–E6255. doi: 10.1073/pnas.1605575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, Gronke K, Ryffel B, Holscher C, Dumoutier L, et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nature immunology. 2015;16:698–707. doi: 10.1038/ni.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell host & microbe. 2012;11:397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JL, Sosnovtsev SV, Green KY, Wobus C, Virgin HW, Mackenzie JM. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. Journal of virology. 2009;83:9709–9719. doi: 10.1128/JVI.00600-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus C, Vinje J, et al. Enteric bacteria promote human and murine norovirus infection of B cells. Science. 2014;346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karandikar UC, Crawford SE, Ajami NJ, Murakami K, Kou B, Ettayebi K, Papanicolaou GA, Jongwutiwes U, Perales MA, Shia J, et al. Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. The Journal of general virology. 2016;97:2291–2300. doi: 10.1099/jgv.0.000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE. A working model of how noroviruses infect the intestine. PLoS pathogens. 2015;11:e1004626. doi: 10.1371/journal.ppat.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE, Goodfellow IG, Green KY, Virgin HW. Advances in norovirus biology. Cell host & microbe. 2014;15:668–680. doi: 10.1016/j.chom.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE, Lay M, Davidson J, Virgin HWt. STAT1-dependent innate immunity to a Norwalk-like virus. Science. 2003;299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- Kaufman SS, Green KY, Korba BE. Treatment of norovirus infections: moving antivirals from the bench to the bedside. Antiviral Res. 2014;105:80–91. doi: 10.1016/j.antiviral.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L, Lycke N. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im0319s17. Chapter 3, Unit 3 19. [DOI] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. JBiolChem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Mahlakoiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS pathogens. 2015;11:e1004782. doi: 10.1371/journal.ppat.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney NS, Thackray LB, Goel G, Hwang S, Duan E, Vachharajani P, Xavier R, Virgin HW. Essential cell-autonomous role for interferon (IFN) regulatory factor 1 in IFN-gamma-mediated inhibition of norovirus replication in macrophages. Journal of virology. 2012;86:12655–12664. doi: 10.1128/JVI.01564-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune BT, Tang W, Lu J, Eaglesham JB, Thorne L, Mayer AE, Condiff E, Nice TJ, Goodfellow I, Krezel AM, et al. Noroviruses Co-opt the Function of Host Proteins VAPA and VAPB for Replication via a Phenylalanine-Phenylalanine-Acidic-Tract-Motif Mimic in Nonstructural Viral Protein NS1/2. MBio. 2017;8 doi: 10.1128/mBio.00668-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS pathogens. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. Journal of virology. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Mumphrey SM, Changotra H, Moore TN, Heimann-Nichols ER, Wobus CE, Reilly MJ, Moghadamfalahi M, Shukla D, Karst SM. Murine Norovirus 1 Infection Is Associated with Histopathological Changes in Immunocompetent Hosts, but Clinical Disease Is Prevented by STAT1-Dependent Interferon Responses. JVirol. 2007;81:3251–3263. doi: 10.1128/JVI.02096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Osborne LC, Tomov VT, Artis D, Wherry EJ, Virgin HW. Type I Interferon Receptor Deficiency in Dendritic Cells Facilitates Systemic Murine Norovirus Persistence Despite Enhanced Adaptive Immunity. PLoS pathogens. 2016;12:e1005684. doi: 10.1371/journal.ppat.1005684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. Journal of virology. 2013;87:327–334. doi: 10.1128/JVI.01864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science. 2016;353:933–936. doi: 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Nash HM, Hathcock K, McNeil NE, Mack D, Hoeppner D, Ravin R, Knutsen T, Yonescu R, Wangsa D, Dorritie K, et al. Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes, chromosomes & cancer. 2012;51:353–374. doi: 10.1002/gcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. IFN-lambda determines the intestinal epithelial antiviral host defense. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockx B, de Wit M, Vennema H, Vinje J, De Bruin E, van Duynhoven Y, Koopmans M. Natural history of human calicivirus infection: a prospective cohort study. Clin InfectDis. 2002;35:246–253. doi: 10.1086/341408. [DOI] [PubMed] [Google Scholar]
- Saito M, Goel-Apaza S, Espetia S, Velasquez D, Cabrera L, Loli S, Crabtree JE, Black RE, Kosek M, Checkley W, et al. Multiple norovirus infections in a birth cohort in a Peruvian peri-urban community. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2013 doi: 10.1093/cid/cit763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong DW, Thackray LB, Smith TJ, Virgin HW. Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. Journal of virology. 2012;86:2950–2958. doi: 10.1128/JVI.07038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube S, Kolawole AO, Hohne M, Wilkinson JE, Handley SA, Perry JW, Thackray LB, Akkina R, Wobus CE. A mouse model for human norovirus. mBio. 2013;4:e00450–00413. doi: 10.1128/mBio.00450-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunis PF, Sukhrie FH, Vennema H, Bogerman J, Beersma MF, Koopmans MP. Shedding of norovirus in symptomatic and asymptomatic infections. Epidemiology and infection. 2015;143:1710–1717. doi: 10.1017/S095026881400274X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray LB, Duan E, Lazear HM, Kambal A, Schreiber RD, Diamond MS, Virgin HW. Critical role for interferon regulatory factor 3 (IRF-3) and IRF-7 in type I interferon-mediated control of murine norovirus replication. Journal of virology. 2012;86:13515–13523. doi: 10.1128/JVI.01824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne LG, Goodfellow IG. Norovirus gene expression and replication. The Journal of general virology. 2014;95:278–291. doi: 10.1099/vir.0.059634-0. [DOI] [PubMed] [Google Scholar]
- Tomov VT, Osborne LC, Dolfi DV, Sonnenberg GF, Monticelli LA, Mansfield K, Virgin HW, Artis D, Wherry EJ. Persistent enteric murine norovirus infection is associated with functionally suboptimal virus-specific CD8 T cell responses. Journal of virology. 2013;87:7015–7031. doi: 10.1128/JVI.03389-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomov VT, Palko O, Lau CW, Sun Y, Tacheva R, Bengsch B, Mane S, Cosma G, Eisenlohr L, Nice TJ, et al. Enteric MNV persistence is associated with a unique CD8 T cell differentiation state and rapid immune evasion. Immunity. doi: 10.1016/j.immuni.2017.09.017. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JM, Wobus CE, Thackray LB, Erexson CR, Faucette LJ, Belliot G, Barron EL, Sosnovtsev SV, Green KY. Pathology of immunodeficient mice with naturally occurring murine norovirus infection. Toxicologic pathology. 2006;34:708–715. doi: 10.1080/01926230600918876. [DOI] [PubMed] [Google Scholar]
- Wobus CE, Cunha JB, Elftman MD, Kolawole AO. Animal Models of Norovirus Infection. Viral Gastroenteritis. 2016:397–422. [Google Scholar]
- Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS biology. 2004;2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.