

SUMMARY
To further our understanding of the genetic etiology of autism, we generated and analyzed genome sequence data from 516 idiopathic autism families (2,064 individuals). This resource includes >59 million single-nucleotide variants (SNVs) and 9,212 private copy number variants (CNVs), of which 133,992 and 88 are de novo mutations (DNMs), respectively. We estimate a mutation rate of ~1.5×10−8 SNVs per site per generation with a significantly higher mutation rate in repetitive DNA. Comparing probands and unaffected siblings, we observe several DNM trends. Probands carry more gene-disruptive CNVs and SNVs resulting in severe missense mutations and mapping to predicted fetal brain promoters and embryonic stem cell enhancers. These differences become more pronounced for autism genes (p=1.8×10−3, OR=2.2). Patients are more likely to carry multiple coding and noncoding DNMs in different genes, which are enriched for expression in striatal neurons (p=3×10−3), suggesting a path forward for characterizing genetically more complex cases of autism.
ETOC
Genomic analysis of 516 families with an autistic child and an unaffected sibling suggests that simplex autism results from de novo mutation and is oligogenic.
INTRODUCTION
While the heritability of autism is high (~40% (Hallmayer et al., 2011) to 80% (Bailey et al., 1995; Steffenburg et al., 1989)), large copy number variants (CNVs) and de novo mutations (DNMs) in genes account for only a fraction of cases. Estimates for their contribution range from ~10%–30% of simplex autism cases (Gratten et al., 2016; Iossifov et al., 2014; Krumm et al., 2015). Common genetic variants (Gaugler et al., 2014), inherited gene-disruptive mutations (Krumm et al., 2015), as well as rare variants of large effect outside of the coding sequence (Turner et al., 2016) likely play an important role in autism disease etiology. Our understanding of the genetic properties of these other sources of genetic variation is currently limited. Most published studies, to date, have focused on single-nucleotide polymorphism (SNP) microarray analysis, targeted sequencing of genes, or whole-exome sequencing (WES) of parent–child trios designed to predict large CNVs and variants within the coding portions of the genome, respectively. There are relatively few publicly available whole-genome sequencing (WGS) datasets (Turner et al., 2016; Yuen et al., 2017) to begin to address other forms of DNM with respect to autism etiology.
In order to assess the contribution of variants in the noncoding, putative regulatory portions of the genome, we selected 476 autism families from the Simons Simplex Collection (SSC) for deep WGS (30-fold coverage). We specifically selected families where blood DNA was available from both parents, a proband, and one unaffected child (termed quads) facilitating DNM comparisons for various classes of mutation within the context of each family. The unaffected sibling in this context serves as a genetic control to estimate rates of DNM. Probands were selected that were negative for known pathogenic mutations, including large CNVs and de novo loss-of-function DNMs as determined by WES and SNP microarray analyses (see STAR Methods, Figure S1). Thus, most high-impact DNMs in coding sequence were eliminated, allowing us to focus exclusively on more difficult cases of the disease where no genetic etiology had yet been determined. We combined these data with 40 SSC quad families that were previously genome sequenced and negative for any known large CNVs or likely gene-disrupting (LGD) de novo variants (Turner et al., 2016). In total, we analyzed the pattern of DNM in 516 autism families (2,064 genomes) to investigate the combined effect of genic and noncoding mutations underlying autism in addition to generating one of the largest genome-wide resource for the study of DNMs.
RESULTS
de novo SNVs/indels
We applied two different single-nucleotide variant (SNV) callers—FreeBayes and GATK—and identified >59 million SNV and small insertion and deletion (indel) events (STAR Methods). Comparing parental genome sequence data and combining with previous WES results (Iossifov et al., 2014; Krumm et al., 2015), we classified 133,992 de novo variants, of which 127,543 were autosomal de novo variants in the 1,032 children (64,060 in probands and 63,483 in siblings; Table 1). It should be noted that families were selected to minimize birth-order effects associated with older fathers and autism probands (Turner et al., 2011), and as a result, the total number of SNVs and indels in probands does not differ significantly from that in siblings in this study (SNV Mann-Whitney two-sided p=0.27, indel Mann-Whitney two-sided p=0.36; Figure 1). Among children for whom we had information on father’s age at birth (n=986), we observe the expected strong correlation between the number of de novo SNVs and indels and paternal age (SNV=0.50, p=1.17×10−64, indel r=0.27, p=1.75×10−17, Pearson’s correlation coefficient). The linear model (SNV adjusted r2=0.25, indel adjusted r2=0.07) is identical to the exponential model (SNV adjusted r2=0.25, indel adjusted r2=0.07). We estimate an increase by 1.49 [1.32, 1.65] SNVs and 0.16 [0.12, 0.19] indels for each additional year of father’s age at birth (Figure 1). Focusing on noncoding de novo SNVs and indels also reveals the significant correlation of de novo events and paternal age (Figure S2).
Table 1.
Summary of DNMs.
| de novo (n=516 families) | ||
|---|---|---|
| Category | Proband | Sibling |
| Autosomal | 64060 | 63483 |
| SNVs/indels | ||
| X SNVs/indels | 3276 | 2914 |
| SNV/indel ratio | 12.7 | 12.7 |
| CpG to TpG (SNVs) | 10412 | 10401 |
| Autosomal deletion* | 41 | 36 |
| Autosomal duplication* | 6 | 5 |
| X deletion* | 1 | 2 |
| X duplication* | 0 | 0 |
n=476 phase 1 families only
Figure 1. Patterns of DNM.

(A) There was a strong correlation between the number of de novo SNVs and paternal age (SNV Pearson’s r=0.50, p=1.17×10−64) with an estimated increase of 1.49 [1.32, 1.65] SNVs for each additional year of father’s age. (B) There was a strong correlation between the number of de novo indels and paternal age (indel Pearson’s r=0.27, p=1.75×10−17) with an increase of 0.16 [0.12, 0.19] indels for each additional year of father’s age. (C) Histogram of de novo SNVs per individual (red=proband, blue=sibling). (D) Histogram of de novo indels per individual (red=proband, blue=sibling). (E) Mutation rate estimates comparing unique and ancient repeat portions of the genome. The overall mutation rate based on experimental validation was 1.7×10−8 substitutions per site per generation with a mutation rate of 1.3×10−8 in unique regions (blue arrow) and 1.5×10−8 in ancient repetitive DNA (red arrow). VRs were comparable between unique regions (97.6%, n=1,640) and ancient repetitive DNA (95.8%, n=216). The average paternal age was 33.4 ± 5.9 years for the 1,032 genomes analyzed here.
We established an overall 96.4% validation rate (VR) based on PCR amplification and Sanger or single-molecule, real-time sequencing of a subset of 687 autosomal variants. Combining this with previous validation data (Iossifov et al., 2014; Krumm et al., 2015; Turner et al., 2016) for all chromosomes (n=1,964) we estimate a VR of 95.4% or 95.5% if restricting to autosomes (n=1,932). Of these, 1,640 mapped to unique regions (1200 Mbp) of the genome (97.6% VR) and 292 mapped to repetitive regions (1484 Mbp) (83.6% VR). Because of the difficulties associated with validating SNVs in repetitive regions, we further stratified the repeat regions into evolutionarily recent (<10% divergence from the consensus [see STAR Methods], 362 Mbp) and more ancient repetitive DNA (1121 Mbp). The VR for DNMs in ancient repeats was substantially higher (95.8%; n=216 DNM) when compared to recently retrotransposed or duplicated DNA (48.7%; n=76 DNM).
Based on these validation results, we estimated ~89.4 SNV and ~4.8 indel DNMs per individual (Figure 1) and an overall mutation rate of 1.7×10−8 SNVs per site per generation, including recent and ancient repeats. If we restrict our analysis to only those regions with the highest validation (>95%), we calculate the mutation rate as 1.3×10−8 in unique regions of the genome and 1.5×10−8 in ancient repeats (Figure 1). These two mutation rates are significantly different from one another (chi-squared test p=1.7×10−74). It is interesting that the total number of validated de novo SNVs per individual was higher than previous studies (Kong et al., 2012a) and concomitantly we estimate a higher human mutation rate. We note, however, that the average age at birth for fathers in our study was 33.4 ± 5.9 years—3.7 years higher than the previous study (Kong et al., 2012a). If we adjust our estimates for an average paternal age of 29.7 years, we still predict a higher overall higher mutation rate (1.6×10−8) but this difference is driven by DNM in repetitive DNA as opposed to unique DNA, which is comparable between Kong and this study (1.2 ×10−8).
Coding de novo SNVs/indels
Since the DNA of most families had been previously exome sequenced, we compared the efficacy of detecting DNMs by WES and WGS. As expected, 98.8% (111,655/113,027) of DNMs were unique to the WGS data when comparing samples where both WES and WGS were available (n=479 probands and 436 siblings) because most mutations map outside the coding region. We then compared WES and WGS events mapping specifically to the coding portion of genes, including splice donors and acceptors. This analysis revealed 1,105 WGS-only events, 188 WES-only events, and 850 events discovered both by WES and WGS (Figure S3). The majority of genic mutations discovered only by WGS (54%, n=596) were not present on the WES capture design (NimbleGen EZ-SeqCap v2.0 targets, ~36 Mbp). While most portions of the capture regions were covered well by WES and WGS (>=10× depth, n=189,069 regions), there is a 30-fold difference in the number of genic regions covered well by WGS (n=3,818) compared to WES (n=105). Moreover, the de novo VR of the WES-only events is lower (64.0%, n=75 tested) when compared to WGS-only mutations identified in protein-coding sequence (84.4%, n=64).
We identified and validated 33 de novo LGD events mapping to protein-coding genes. Three additional events were identified and attempted by Sanger but failed sequencing by multiple attempts thus resulting in unknown validation status. This leads to a total of 36 novel de novo LGD events with 15 in probands and 21 in siblings (Figure 2, Table S1, Table S2). The difference between proband and sibling LGD is not statistically significant—an unsurprising result because these families were supposed to have been pre-filtered for LGD by WES. Of these 36 DNMs, one was detected only by WES, five by both WES and WGS, and 25 by WGS alone (five additional events occurred in samples not previously WES). An examination of these missed LGD DNMs indicates that most of the gain in sensitivity is due to the more uniform sequence coverage provided by WGS when compared to WES (Figure S3). To calculate a false negative rate for LGD discovery in WES data, we assessed the new LGD DNMs identified in individuals who were previously exome sequenced (n=479 probands and 436 siblings). There were 12 novel LGD events in probands (false negative rate=12/479 [2.5%]) and 13 novel events in siblings (false negative rate=13/436 [3.0%]) detected only by WGS. Among these were events in genes previously associated with autism, including ARID1B, PHIP, and CNTNAP3, highlighting the benefit of WGS over WES.
Figure 2. Proband–sibling DNM differences by functional annotation.

Number of autosomal de novo variants by functional category by (A) coding variants (LGD=likely gene-disrupting, MIS30=missense with CADD score >30, DEL=exonic deletion); (B) putative noncoding regulatory variants (UTR=untranslated region, 3' UTR=3' untranslated region, TFBS=putative noncoding regulatory with a transcription factor binding site); and (C) ENCODE/ChromHMM putative regulatory variants (fetal promoters=within a TFBS in a fetal brain transcription start site and embryonic enhancers=within a TFBS in a human embryonic stem cell strong enhancer). * Indicates nominal significance (p<0.05) by FET. (D) One of the three de novo sequence variants identified in autism probands that was tested for in vivo enhancer activity in the CNS (reference allele from enhancer.lbl.gov) (Visel et al., 2007). We extended the previous assessment for the reference allele in our current study by also testing the variant allele. For the locus, we show, from top to bottom, the human genome reference allele, the patient variant (red text), the location of a conserved TFBS near the variant, the VISTA enhancer with hs number (blue bar), and representative transgenic embryonic day 11.5 mouse embryos for the reference and variant alleles, respectively, displaying the enhancer activity pattern (blue staining). Whole embryos are shown on left, with enlarged images of the forebrain on the right. FB: forebrain, MB: midbrain, HB: hindbrain. (E) Results of the enhancer assay identified a novel forebrain enhancer activity pattern being driven by the allele containing the de novo patient variant. The expression in both the midbrain and hindbrain were unaffected. P-values by FET.
We also considered de novo missense mutations and focused on those representing the top 0.1% of deleterious mutations (Combined Annotation Dependent Depletion [CADD] (Kircher et al., 2014) score ≥30; Table S2). In total, we discovered 28 severe de novo missense mutations in probands and 13 in siblings. Mindful of potential biases that may affect statistical testing due to paternal age, we applied a Fisher’s exact test (FET) for the number of events as opposed to the number of individuals demonstrating a significant enrichment of severe de novo missense mutations in cases compared to controls (nominal p=0.01, OR=2.1) (Figure 2, Table S1).
de novo structural variants
We also applied a suite of structural variant (SV) callers (STAR Methods) to maximize sensitivity for de novo SV mutation detection of various size ranges. To eliminate false positives, we assessed sequence read-depth for all putative events, requiring evidence for increase (duplication) or decrease (deletion) in read-depth in the child when compared to parental genomes (STAR Methods). In total, we detected 9,212 private autosomal CNVs. Of these, 9,124 events showed evidence of transmission while 88 were predicted to be de novo in the 476 phase 1 families analyzed in the present study. We did not include the pilot 40 families in the SV analysis due to technical differences in WGS and a different complement of callers used previously (Turner et al. 2016). Using SNP microarray data from the same samples (STAR Methods), we estimate a VR of 87.5% for de novo CNVs (median size 18 kbp). The set included a total of 77 deletions (median 500 bp, mean 3,363 bp) and 11 duplications (median 13,512 bp, mean 48,894 bp). In probands we identified 41 deletions and 6 duplications and in siblings we identified 36 deletions and 5 duplications. We also identified three de novo SVs on the X chromosome: two noncoding events in siblings (6.6 and 2.1 kbp) and one 50 kbp DMD coding deletion in a proband. In addition to similar autosomal rates of CNVs, a comparison of the largest CNV per individual in probands and siblings yielded no significant difference (nominal p=0.77, deletion nominal p=0.53, duplication nominal p=0.79 log rank test), likely due to the ascertainment criteria of this cohort, which excluded probands with large CNV events. However, when we restricted the analysis to autosomal CNVs that intersect a RefSeq exon, we observed a significant increase of exon-intersecting deletions amongst proband CNVs (20 in probands, 9 in siblings, FET, one-sided, nominal p=0.03 OR=2.8; Figure 2, Table S1). Three of these genic CNVs in probands are predicted to disrupt genes (CHD2, UBE3B, ZNF462) and correspond to autism risk genes (SFARI845, https://gene.sfari.org) (Basu et al., 2009). No such events are observed in unaffected siblings.
Noncoding de novo SNVs/indels
For the purpose of this study, we limited our analysis to the two most well-known functional classes of noncoding DNA, namely untranslated regions (UTRs) of genes and putative noncoding regulatory DNA corresponding to promoters and enhancers. Because of the association of autism gene networks expressed early in brain development (Hormozdiari et al., 2015; Konopka et al., 2012; Pinto et al., 2014; Voineagu et al., 2011), we defined putative noncoding regulatory (pNCR) DNA as conserved transcription factor binding sites (TFBS) mapping to regions of fetal brain DNase I hypersensitivity.
Combining annotated 5' and 3' UTR sequence, we identified 640 DNMs in probands versus 568 in siblings (Figure 2, Table S1). We observed an enrichment of DNMs in UTRs in probands (nominal p=0.03, OR=1.1, FET, one-sided; Figure 2, Table S1). This effect appeared to be driven by the 3' UTR events (nominal p=0.04, OR=1.1, FET, one-sided; Figure 2, Table S1) and not the 5' UTR events (nominal p=0.29, OR=1.1, FET, one-sided) but this likely reflects differences in length and, thus, reduced power for 5' UTR.
We initially assessed events within all putative noncoding regulatory regions of the fetal central nervous system (CNS) as previously described (Turner et al., 2016)). Although no significant difference was observed overall comparing DNMs in probands (n=2,360 events) and siblings (n=2,313 events), we observed a difference if we restricted the analysis to evolutionarily conserved regions (mean GERP++ (Davydov et al., 2010) score >2) (254 proband variants, 203 sibling variants; FET, two-sided, nominal p=0.02, OR=1.3). Using the ChromHMM classification from ENCODE (Ernst and Kellis, 2012) and Epigenomics Roadmap projects (Kundaje et al., 2015), we assessed the potential functional categories represented in the proband CNS DNase I TFBS. We found enrichment of DNM variants corresponding to transcriptional start sites/promoter regions in the fetal brain (nominal p=0.03, OR=1.8, FET, one-sided; Figure 2, Table S1) and strong enhancers in human embryonic stem cells (nominal p=0.02, OR=8.0, FET, one-sided; Figure 2, Table S1).
Although all of these trends are currently nominally significant and do not withstand multiple test correction, it is interesting that the TFBS effects become more pronounced as further functional constraint is applied. Interestingly, a small subset (n=11) of these putative regulatory sites had previously been tested using mouse transgenic enhancer assays (Pennacchio et al., 2006) in mouse embryos (E11.5). Of the eleven sites, eight corresponded to regions with DNM in probands (seven confirmed as positive enhancers by the VISTA assay) with reporter expression in the midbrain, hindbrain, forebrain, and neural tube (Figure 2). Three corresponded to regions with DNM in siblings (one confirmed as an enhancer by the VISTA assay (Visel et al., 2007)). To assess whether there were any functional differences between reference and variant alleles identified in patients, we focused on three proband variants (Figure 2, Figure S5) within sites that previously showed enhancer activity based on VISTA. Using the reporter assay described previously (Visel et al., 2007), we show that one of the three variants, hs737 (Figure 2, Figure S5), demonstrated a reproducible expression difference. The reference allele showed enhancer activity in the midbrain and hindbrain, while the single de novo substitution mutation maintained expression in midbrain and hindbrain but also showed reproducible expression in the forebrain (Figure 2).
Gene ontology enrichment analysis of the genes closest to the variants in TFBS (Table S3) was performed for both proband and sibling variants (Table S3) considering biological process (BP), molecular function (MF), and cellular component (CC). Only proband DNMs showed MF enrichment, namely, beta-catenin binding. The top five significant (binomial test, Bonferroni corrected) BP proband enrichments, based on fold enrichment (FE), included positive regulation of nervous system development, developmental cell growth, positive regulation of neuron differentiation, developmental growth involved in morphogenesis, and regulation of cell morphogenesis involved in differentiation.
Oligogenic DNM burden
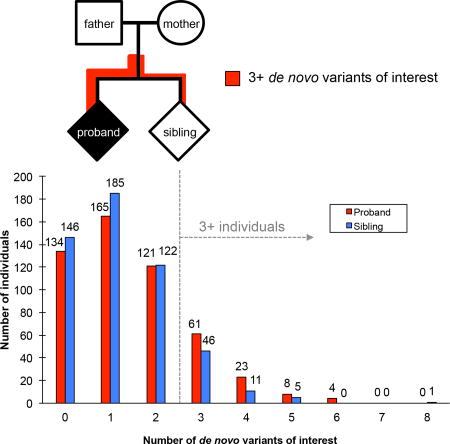
Several studies have reported multiple de novo or private deleterious mutation events (Girirajan et al., 2010; Jiang et al., 2004; Schaaf et al., 2011; Turner et al., 2016) in risk genes among children with autism suggesting that two or more loci (oligogenic) might contribute to a fraction of autism cases. Based on the functional classes established above, we specifically tested if two or more potentially deleterious DNMs were likely to occur in probands compared to their unaffected siblings. We compared the total number of de novo variants of interest (VOI) between probands and siblings (see STAR Methods) defined here as the total number of LGD, severe missense (CADD score >30), noncoding pNCR TFBS variants, 3' UTRs and deletions that disrupted an exon. Comparing probands and siblings, we observe an excess of multiple VOI in individuals with autism (nominal p=0.01, Mann-Whitney test) (Figure 3A, Figure S4). This trend remains even after correcting for the father’s age at birth (FET p=0.05).
Figure 3. Oligogenic mutation burden.

(A) The number of individuals carrying 0 or more de novo variants of interest (see Methods). (B) TSEA of genes in probands with 3 or more DNMs shows enrichment in the brain. (C) TSEA of genes in siblings with 3 or more DNMs shows no enrichment for any tissue. (D) CSEA) analysis of genes in probands with 3 or more DNMs shows an enrichment for striatal D2 + spiny neurons as well as striatal D1 + spiny neurons. (E) CSEA of genes in siblings with three or more DNMs shows no enrichments. For each plot in B-E, hierarchical clustering is shown for tissues (TSEA) or for cell types (CSEA). For each bullseye image, different stringency thresholds are shown by each hexagon and the darker the color the more significant the p-value.
This excess of multiple DNMs is observed if either 3' UTR events (nominal p=0.01, Mann-Whitney) or coding mutations (missense and LGD events, p=0.02, Mann-Whitney) were excluded from this calculation. Interestingly, the analysis showed the greatest distinction between probands and unaffected siblings when three or more DNMs were evaluated (Figure 3A). If we assume that this oligogenic signal contributes to autism risk, we estimate the attributable fraction for this group of idiopathic simplex autism, (i.e., without LGD mutations and large CNVs) as 7.28% [2.31, 12.01] (p=4×10−3). The largest differential between probands and unaffected siblings was observed between individuals with at least one pNCR TFBS and at least one 3' UTR event (58 probands, 31 siblings, attributable fraction=5.6% (1.94, 9.06), p=3×10−3, one-sided FET p=1.9×10−3, OR=2.0).
In order to gain insight into the biological relevance of variants in individuals with de novo oligogenic burden (three or more DNMs), we examined both Tissue Specific (TSEA) as well as Cell-type Specific Expression Analysis (CSEA) for the associated genes using recently developed tools (Dougherty et al., 2010; Xu et al., 2014). The tools attempt to identify molecular convergence of groups of genes with respect to tissue or cell types using recent transcriptomic profiling datasets. We defined candidate genes based on the location of the variants (UTR, missense, CNV) or the nearest mapped gene (in the case of noncoding regulatory DNA as defined by pNCR TFBS). Among probands with three or more DNMs, genes were enriched for brain expression (nominal p=3.8×10−4, Benjamini Hochberg (BH) corrected p=0.009; Figure 3B–E) and expression in the striatum. The latter was specifically enriched for striatal D2+ spiny neurons (nominal p=4.2×10−4, BH-corrected p=0.01) as well as striatal D1 + spiny neurons (nominal p=4.0×10−3, BH-corrected p=0.08) (Figure 3B–E). If we limit the analysis to individuals with three or more DNMs strictly in noncoding, regulatory DNA, the striatal signal becomes stronger in both the D2+ spiny neurons (nominal p=1.5×10−4, BH-corrected p=3×10−3) and the D1+ spiny neurons (nominal p=1.9×10−4, BH-corrected p=3×10−3). Unaffected siblings showed no significant enrichments by either TSEA or CSEA (Figure 3B–E).
Significant enrichment in autism genes
If the genomic trends with respect to DNM are relevant to disease, we would expect the signals to become more pronounced when we restrict our analysis to genes previously implicated in autism. We limited our assessment to two sets of genes specifically implicated in disease, namely, a set of 57 genes where an excess of de novo LGD/missense have been identified in cases but not in controls (Turner57) (Turner et al., 2016) and a more general list of manually curated autism risk genes (SFARI845) (Basu et al., 2009) (Figure 4). We first considered all DNMs of potential functional interest, including LGD mutations, missense mutations, UTRs, pNCR regions, and exonic deletions. We compared the number of events in probands versus the events in siblings, using a Fisher’s exact test, containing any variant within the gene set and found significant enrichments of DNMs in probands for both gene sets (Turner57 nominal p=1.76×10−3, OR=2.2; SFARI845 nominal p=1.36×10−3, OR=1.3; Figure 4A). We highlight the difference in counts for the Turner57 dataset between proband and sibling DNM events using a waterfall plot (Figure 4B).
Figure 4. Autism gene enrichment analysis.

(A) Number of DNMs in functional elements compared for two autism gene sets (Turner57 and SFARI845). (Turner57 nominal p=1.76×10−3, OR=2.2; SFARI845 nominal p=1.36×10−3, OR=1.3). (B) Waterfall plots compare DNMs in various classes of “functional elements” for genes implicated for DNM Turner57 (OR=2.2, p=1.8×10−3). Significance estimates are calculated using the FET and although nominal p-values are shown these are multiple test correction significant (n=2 tests). (C) Counts of individuals with two or more variants within SFARI845 genes.
Since we also observed a genome-wide signal for multiple (oligogenic) DNMs in probands, we repeated the analysis restricting to previously identified autism risk genes (SFARI845). Once again the DNM signal became stronger. We found that probands were more likely to carry two or more de novo variants within or near two or more SFARI genes compared to siblings (Figure 4C, one-sided FET p=2.6×10−4, OR=2.3, BH p=7.9×10−4). This observation is significant if we restrict to coding (p=9.3×10−4, OR=Inf, 10 probands vs. 0 siblings) and noncoding (p=0.018, OR=1.9, 35 probands, 19 siblings) and survives multiple test correction, although near the threshold of significance for multiple noncoding DNMs (BH p=1.4×10−3 and BH p=0.02, respectively). In addition to the number of individuals, we also considered these same three analyses at the level of number of DNM events. Probands with two or more mutations in autism risk genes show an overall excess of DNMs (coding and noncoding) (p=2.1×10−7, OR=2.2, 128 proband events, 58 sibling events, BH p=7.9×10−4) as well as noncoding (p=2.2×10−3, OR=1.7, 74 proband events, 42 sibling events, BH p=0.02) and coding events (p=1.0×10−6, odds ratio=Inf, 20 proband events, 0 sibling events, BH p=1.4×10−3).
Attributable fraction estimates
Finally, we calculated attributable fraction estimates for the different functional classes of DNM and simplex autism (see STAR Methods). In this analysis, we only focused on quad families from the SSC (n=1,748; Table 2) where all family members had been previously assessed by both WES and SNP microarray (excluding those in the pilot study as selection criteria was biased). We separated the families into those with “known” pathogenic mutations (n=588) and those idiopathic cases studied here, representing a random sampling of the remaining families (n=398). By sequential analysis of different classes of mutation and the subsequent removal of both probands and siblings who carried a specific class of mutation, we ensured no double-counting of samples. Among the families with “known mutations, de novo LGD mutations contributed ~6%, de novo CNVs ~4%, and individuals with multiple mutations to ~1% of cases, leading to a total contribution of ~11% of simplex autism. Among the idiopathic families, we first computed the attributable fraction estimates within study and then extrapolated what the estimates would be based on all of the simplex families (including patients with “known” mutations, see STAR Methods). This revealed a contribution of ~2% for each new DNM class, including small exonic de novo deletions, severe de novo missense mutations, and those individuals with three or more de novo VOI including noncoding regulatory mutation. This leads to a total contribution of ~6% based on this new WGS data. Overall, we estimate ~17% of simplex autism may be explained by these various functional categories. Importantly, both the known and idiopathic cases of autism highlight an enrichment of multiple DNMs in a fraction of autism cases.
Table 2.
Attributable fraction estimates in simplex autism.
| Set | Category | Proband Counts |
Sibling Counts |
Odds ratio | Attributable fraction (est) in exposed (%) |
Attributable fraction (est) in population (%) |
p-value | Extrapolated attributable fraction (est) in SSC (%) |
|---|---|---|---|---|---|---|---|---|
| Known families (n=588) | de novo LGD | 284 | 191 | 1.58 (1.30, 1.93) | 36.75 (22.67, 48.35) | 5.97 (3.46, 8.42) | < 0.001 | 5.97 |
| de novo CNV | 96 | 33 | 3.02 (2.02, 4.51) | 66.88 (50.02, 78.53) | 3.67 (2.41, 4.92) | < 0.001 | 3.67 | |
| large inherited CNV# | 36 | 32 | 1.13 (0.70, 1.82) | 11.32 (−47.64, 46.95) | 0.23 (−0.70, 1.16) | 0.624 | ||
| Multi hit (2+ of the 3 event types above) | 29 | 8 | 3.67 (1.67, 8.05) | 72.73 (38.66, 89.26) | 1.21 (0.53, 1.88) | < 0.001 | 1.21 | |
|
| ||||||||
| New families (n=398) | small de novo deletions | 16 | 6 | 2.74 (1.06, 7.07) | 63.41 (0.27, 88.40) | 2.55 (0.23, 4.81) | 0.031 | 1.69 |
| de novo missense CADD > 30 | 21 | 10 | 2.16 (1.00, 4.65) | 53.69 (−4.37, 80.80) | 2.84 (0.07, 5.52) | 0.044 | 1.89 | |
| 3+ de novo VOI | 33 | 19 | 1.80 (1.01, 3.23) | 44.51 (−2.57, 70.75) | 3.69 (0.08, 7.18) | 0.045 | 2.45 | |
Known families correspond to SSC quads where the proband carries a de novo LGD, de novo CNV, or large inherited CNV based on previous WES and SNP microarray analysis. New families represent those SSC quads that did not have any known events based on WES and SNP microarray. No pilot families were included here because of different selection criteria.
Not stratified by maternal or paternal inheritance as described previously (Krumm et al. 2015).
DISCUSSION
The genetic etiology for most children with autism is unknown. We performed WGS to gain insight into some of the most difficult cases of simplex autism—families where no large CNV or de novo gene-disruptive mutation event had been previously identified in probands. Such families are likely enriched for environmental and stochastic risk factors for autism and, therefore, represent some of the most difficult cases to investigate for underlying genetic etiology. Although the study represents a tenfold increase in sample size with respect to a previous analysis on 53 families from the SSC (Turner et al., 2016), most of the patterns we highlight suggest trends for future investigation in a larger cohort as opposed to being definitive. We developed a resource that attempts to comprehensively detect SNVs, indels, and SVs derived exclusively from deep sequencing of blood DNA. We focused specifically on DNMs, as the SSC was originally designed to enrich in DNM risk factors (Fischbach and Lord, 2010).
Other groups have recently analyzed and published WGS data from parent–child trio data from autism families (Yuen et al., 2016) or are in the process of specifically analyzing these same WGS data from quad families with a focus on assessing noncoding mutations (Werling et al., 2017) or restricting to a more detailed analysis of CNV mutations (Brandler et al., 2017). Notably, our validated mutation rate appears significantly higher than previous and unpublished reports (Werling et al., 2017; Yuen et al., 2016) possibly because of a more complete ascertainment of repetitive regions of the genome. Surprisingly, Werling and colleagues do not report the additional 33 validated LGD variants of which some clearly represent the most likely pathogenic variant in these patients (e.g., ARID1B or PHIP LGD mutations). In addition, we are not considering all possible classes of noncoding regulatory elements. Instead, we focus specifically on those that are most likely to have functional impact (promoters, UTRs and regulatory elements within the fetal brain). We caution, however, that we are comparing our results to initial draft versions of these manuscripts in BioRxiv and we have found that papers, gene lists, and conclusions often differ substantially from first drafts to published papers after peer review. Comparison and integration of these data from other labs will further enhance the quality of this resource, which will represent one of the most detailed and largest analyses of mutation data from both affected and unaffected siblings. The availability of WGS data and a comprehensive mutation analysis from both an affected individual and a genetically matched unaffected control from the same family allows for the detection of genetic signals that cannot be easily recognized from parent–child trio datasets (Yuen et al., 2016).
Our results confirm the well-established increase in de novo substitutions with paternal age (O’Roak et al, 2012; Kong et al., 2012) and extend this observation to insertion/deletion events (Figure 1). Based on our validation results, we estimate a higher mutation rate (1.5–1.7 × 10−8 substitutions per site per generation) compared to earlier estimates. This increase cannot be solely explained by differences in the average age of fathers between the studies. Instead, our data suggest that this increase is driven by a >15% higher mutation rate in repetitive DNA when compared to unique DNA. Similar increases were observed previously based on comparative sequence analysis of orthologous sequences derived from BAC inserts from human and chimpanzee (Liu et al., 2009; She et al., 2006) but have not been shown before, to our knowledge, at the level of familial DNM data. There are several possible explanations that might account for the increased substitution rate, including CpG bias, gene conversion, and/or relaxed selective constraint (Chen and Li, 2001). We hypothesize that advances in sequencing technology are providing increased access to the repetitive fraction of our genome and, as a result, early estimates provided a lower bound to the mutation rate.
Deep sequencing of both an autistic child and its unaffected sibling also provides a powerful genetic control for dissecting the relative impact of DNMs with respect to different genomic functional elements (Fischbach and Lord, 2010; Iossifov et al., 2014). The comparisons highlight some interesting trends. First, we observe a twofold enrichment of missense variants with high CADD scores (>30) in probands (Figure 2). This included autism risk genes (PTPN11, CACNA1G, TRIP12, and PTK7) as well as other genes of interest (SUPT16H and SCN3A) giving evidence for the importance of particularly severe de novo missense mutations (Iossifov et al., 2014) and autism. WGS also discovered an additional 2.5% of probands and 3.0% of siblings with de novo LGD mutations missed by WES. For example, we identified a frameshift variant (Table S2) in ARID1B—a known high-impact risk factor for autism and developmental disability (Hoyer et al., 2012). Other variants missed by previous WES analysis included a splice-site acceptor variant in GLIPR1L2, a frameshift variant in PHIP, a splice-site acceptor variant in the PCM1—a gene shown to bind DISC1 (candidate gene for schizophrenia) (Eastwood et al., 2010), and a stop-gain DNM in CNTNAP3—a gene differentially expressed in the blood of individuals with autism (Kong et al., 2012b).
Second, our analysis identified putative functional noncoding DNA that showed a modest but significant excess of DNM in probands. Specifically, we observe mutation enrichment in 3' UTRs and DNase I hypersensitivity sites (fetal CNS) that contain a TFBS and are either human embryonic stem cell strong enhancers or fetal brain transcriptional start sites. If the differential DNM burden between probands and their unaffected siblings is used as an indicator, we estimate that such events contribute an important role in ~5% of autism cases. These signatures suggest de novo disruption of gene regulation as an important risk factor. Several additional analyses are consistent with this model. Functional testing of a small number of these sites in mouse enhancer reporter assays reveals patterns of expression consistent with enhancer regulatory function in hindbrain, midbrain and the developing CNS (Pennacchio et al., 2006) (Figure 2, Figure S5). Ontology analysis of the genes mapping closest to these proband DNMs shows a significant enrichment of variants for genes associated with nervous system development, beta-catenin binding, and borderline enrichment for CHD8 binding. There were a greater number of DNMs intersecting CHD8 binding sites (Cotney et al., 2015) overall in probands when compared to siblings (nominal p=0.03, OR=1.1, FET). These data reinforce recent functional data regarding the importance of WNT signaling and suppression and activation of genes by CHD8 and beta-catenin in autism risk and early in neuronal development (Dong et al., 2016; Durak et al., 2016; Katayama et al., 2016; Marchetto et al., 2016).
Third and, perhaps most importantly, these data provide some of the first evidence for an oligogenic DNM model underlying simplex autism. Considering only those functional classes where we demonstrate an excess of DNMs, we compared the overall genome-wide burden between siblings and probands and observe a significant skew in the number of DNMs in probands. In fact, probands with ≥2 DNM events (at least one 3' UTR and another in a putative regulatory region) show even greater enrichment with an estimated attributable fraction in population of 6.92% [1.77%–11.80%] comparable to what has been observed for de novo LGD mutations alone (Krumm et al., 2015). This effect becomes more significant as we restrict to genes previously implicated in autism and is observed both for coding mutations as well as mutations in putative noncoding regulatory regions of multiple SFARI autism risk genes. The effect for noncoding mutations is particularly intriguing but larger numbers of genomes will be needed to confirm this observation.
It is striking that genes associated with the oligogenic burden are particularly enriched for D2+ spiny neurons from the striatum—a circuit that has been implicated in the pathophysiology of autism (Fuccillo, 2016). Dougherty and colleagues have shown that this circuit is enriched for autism candidate genes (AutDb) but not necessarily genes implicated early in development by de novo LGD events (Xu et al., 2014). Indeed, an examination of the 22 striatal genes implicated in this study with other recently published exomes/genomes (n=3,505) (De Rubeis et al., 2014; Hashimoto et al., 2016; Iossifov et al., 2014; Jiang et al., 2013; Krumm et al., 2015; Lee et al., 2014; Moreno-Ramos et al., 2015; Tavassoli et al., 2014) showed that only one of the genes (CACNA2D3) with evidence for a de novo LGD mutation in an autism proband. Our findings provide further support for this striatal circuit and suggest that multiple de novo regulatory mutations may be an important risk factor for identification of such autism risk genes. We also note that oligogenic burden appears more pronounced in autism females (Figure S6) although the relative proportion of females studied here was few. Nevertheless, our estimates of attributable fraction for both “known” and idiopathic cases of autism both point to a role for multiple DNMs in autism (Table 2).
In total, the data argue in favor of a multifactorial genetic model as has been proposed previously based on CNV and candidate gene sequencing (Girirajan et al., 2010; Jiang et al., 2004; Schaaf et al., 2011; Turner et al., 2016). It is interesting that the effect is most pronounced in females. Since females are less commonly affected than males (Fombonne, 2003) according to the multifactorial model, they require a higher burden of mutations to become affected than do males. The finding of increased CNV burden in female probands (Jacquemont et al., 2014) and a transmission disequilibrium of private LGD events from mothers to their sons (Krumm et al., 2015) are consistent with our genome-wide observations of increased mutational burden. Although many more genomes will need to be sequenced to replicate these findings, there are two important ramifications. First, genomes as opposed to exomes should be targeted for sequencing, especially for female autism probands and for effects related to differentially expressed genes in the striatum. Targeted sequencing of candidate genes (O'Roak et al., 2012a) and WES (De Rubeis et al., 2014) are currently more popular both clinically and in basic research settings. These approaches may be missing the true genetic architecture of autism in favor of an oversimplified monogenic model because once a “pathogenic” mutation is found such patients and their families are typically excluded from more detailed genetic analysis. Second, multiple DNMs may help to explain the apparent discrepancy between the female protective effect and the absence of the Carter effect (Carter, 1969; Constantino, 2014). The lack of increased familial aggregation in families with female probands could be explained, in part, by multiple DNMs in functional elements in affected females with respect to males. This high-quality genomic resource provides an important first step toward understanding the genetics of these more complex cases of simplex autism.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Evan. E. Eichler (eee@gs.washington.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Autism familial cohort
The SSC (Fischbach and Lord, 2010) contains ~2,600 families with 2,194 quads: a father, a mother, a child with autism, and a sibling without autism. Of these, 40 full quads and 13 trios (pilot study) were previously sequenced (Turner et al., 2016). For the current study (phase 1), 476 quad families (1,904 genomes) were selected, including three families previously resequenced from the pilot (11729, 13637, and 13825) but upgraded to quad status. Families were selected such that neither proband nor sibling carried the following: (1) a confirmed de novo CNV that is rare [≤0.1 population frequency based on parents and the Database of Genomic Variants (DGV)] and exonic (Krumm et al., 2015; Levy et al., 2011; Sanders et al., 2011); (2) an inherited CNV that is rare [≤0.1 population frequency based on parents and DGV] and encompasses ≥10 genes (Krumm et al., 2015; Sanders et al., 2011); and/or (3) a known, rare [≤0.1 population frequency based on EVS] exome LGD event (Iossifov et al., 2014; Iossifov et al., 2012; Krumm et al., 2015; O'Roak et al., 2011; O'Roak et al., 2012b; Sanders et al., 2012). Of the ~2,600 families, this removed 788 families of which 758 were quads (sex ratio=5.3, binomial nominal p=0.04 with female enrichment). A total of 476 quads were randomly chosen from the remaining 1,396 families for this study (phase 1). Also of note, one family (13314) was missed in the exclusion process and had a published, validated de novo exonic deletion in CHD2 (b37, chr15:93485051-93487745) (Krumm et al., 2015). In the combined pilot plus phase 1 data there were 54 affected females and 462 affected males (sex ratio of 8.6, binomial nominal p=0.05 with slight male enrichment with respect to the overall composition of the SSC). There were also 286 unaffected female siblings and 230 unaffected male siblings (sex ratio of 0.8). This study was approved for sequencing by the local institutional review board (IRB) at the New York Genome Center (Biomedical Research Alliance of New York [BRANY] IRB File # 17-08-26-385), for local SSC recontact at the University of Washington (IRB STUDY00000383 [previously IRB 48785]), and for SSC samples altogether (IRB STUDY00001619 [previously IRB 31249]) at the University of Washington.
Mouse IACUC Approval
All animal work was reviewed and approved by the Lawrence Berkeley National Laboratory (LBNL) Animal Welfare and Research Committee. All mice used in this study were housed at the Animal Care Facility (ACF) of LBNL. Mice were monitored daily for food and water intake, and animals were inspected weekly by the Chair of the Animal Welfare and Research Committee and the head of the animal facility in consultation with the veterinary staff. The LBNL ACF is accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). Transgenic mouse assays were performed in healthy, wild-type Mus musculus FVB strain mice. Sample sizes were selected empirically based on our previous experience of performing transgenic mouse assays for >2,000 total putative enhancers. Mouse embryos were only excluded from further analysis if they did not express the reporter transgene or if they were not at the correct developmental stage (embryonic day 11.5). The sex of the embryos was not determined, and minor differences between sexes at the developmental stage examined are not expected to substantially influence enhancer or transgene activity. All embryo cohorts are expected to be an approximately equal mixture of males and females.
METHOD DETAILS
Sequencing and quality control
Genomes were sequenced at the New York Genome Center (NYGC) using 1 µg of DNA, an Illumina PCR-free library protocol, and sequencing on the Illumina X Ten platform. Post-sequencing, reads were aligned to the genome (BWA (Li and Durbin, 2010) mem version 0.7.8), duplicate reads marked (Picard version 1.83), base scores recalibrated (GATK (McKenna et al., 2010) version 3.4-0-g7e26428), and indels realigned (GATK version 3.4-0-g7e26428); the resulting BAM files were made available on the Amazon Cloud. Genomes were sequenced to a coverage of 34.8 ± 5.3× and with a median library insert size of 417.8 ± 111.5 bp. Quality control analysis included WGS metrics (Picard version 1.141), SAMtools (version 1.2-242-g4d56437) flagstat, and insert size (Wham-Graphening (Kronenberg et al., 2015) version v1.7.0-176-g4431). Mitochondrial haplogroups were determined by extracting mitochondrial reads from the BAM file, filtering such that only reads with <10% mismatch were retained, generating a consensus mitochondrial genome, and running through the MitoMaster (Brandon et al., 2009) API. Full-quality control statistics are available in Table S4. To assess proper relationships we utilized two methods: (1) the inheritance of mitochondrial genomes from mothers to their children and (2) kinship coefficients (ϕ) by KING (Manichaikul et al., 2010). By both of these metrics family relatedness was correct in these families.
SNV calls
SNVs and indels were called using the GATK (McKenna et al., 2010) HaplotypeCaller version 3.5-0-g36282e4 and FreeBayes version 1.0.1 (https://github.com/ekg/freebayes) on a per-family basis. de novo SNVs and indels were called using a custom pipeline using the family-level VCFs for both FreeBayes and GATK. First, BCFtools (Danecek and McCarthy, 2017) (version 1.3.1) norm was used to left-align and normalize indels. Second, candidate sites were chosen where the father’s genotype was 0/0, the mother’s genotype was 0/0, and the child’s genotype was either 0/1 or 1/1. Third, we applied allele count, read-depth and allele balance filters: the father alternate allele count=0, mother alternate allele count=0, child allele balance >0.25, father depth >9, mother depth >9, child depth >9, and either child genotype quality (GQ) >20 (GATK) or sum of quality of the alternate observations (QA) >20 (FreeBayes). Fourth, any sites in low complexity regions (https://github.com/lh3/varcmp/raw/master/scripts/LCR-hs37d5.bed.gz) were removed from further analysis. After applying the above filters, we retained sites called by both FreeBayes and GATK as the final de novo set. For X chromosome de novo variants, we applied the same procedure with the exception that we excluded variants in the pseudoautosomal regions (GRCh37: chrX:60001-2699520 and chrX:154931044-155260560) and the X/Y duplicatively transposed region (GRCh37: chrX:88456802-92375509). As part of this analysis, we also analyzed the X chromosome and identified a total of 3,276 DNM events in probands and 2,914 in siblings. While this difference is significant (nominal p=1.2 × 10−5) compared to autosomes, it is likely the result of under calling in females versus males. In support of this, if we limit the comparison to gender-matched probands and siblings and compare this to autosomes, we observe no significant difference.
SV calls
Read-depth profiles for each individual were generated by taking the BWA-mem aligned BAM files and realigning the reads to the genome using mrsFAST-ultra (Hach et al., 2010). SVs were detected using a modified implementation of dCGH (Sudmant et al., 2010), GenomeSTRiP (Handsaker et al., 2011) v. 2.00, Lumpy (Layer et al., 2014), VariationHunter (Hormozdiari et al., 2011), and two versions of Wham-Graphening—Wham (version v1.7.0-176-g4431) and Whamg (v1.7.0-296-gb406) (Kronenberg et al., 2015). We applied the dCGH pipeline with the following modifications: All samples were compared to 17 reference parental samples (9 female and 8 male) picked for high GC quality scores (percent of control windows correctly assigned copy number 2) using 500 bp tiled windows of non-repeat sequence and log2 ratios of copy number estimates. Initial consensus (across reference samples) CNV calls were generated by an edge detection algorithm as described previously (Sudmant et al., 2010). We then utilized these initial calls rather than the final genotyped polymorphic CNVs to increase our sensitivity to rare variants. Calls were filtered with estimated copy number thresholds of <1.5 and >2.5 copies and trimmed inwards to the first threshold passing 500 bp window. After filtering we corrected for hypersegmentation from the edge detection algorithm and merged adjacent (within 1 Mbp) deletions and duplications based on matching any of the following criteria: copy number for the intercall gap is consistent with a deletion or duplication; gap spans a segmental duplication (>3.5 average copies and <50 kbp); gap is <1 kbp (~2 windows); gap is <1% of the largest flanking CNV size; gap is >75% repeat and/or segmental duplication.
A GenomeSTRiP analysis was also performed on 100 genome samples at a time as well as Lumpy, VariationHunter, and Whamg on a per-family basis. Some pipelines were run in the Amazon Cloud and the code for those pipelines is available at https://github.com/eichlerlab/aws. SVs were genotyped with SVTyper (Chiang et al., 2015) (version 0.1.0) using confidence intervals of 75 bp for breakpoints in every caller except dCGH. Since dCGH is less accurate at breakpoints, we used a confidence interval of 1000 bp.
To generate a high-quality set of merged SVs, we first genotyped all SVs using SVTyper and assigned copy number estimates to calls over 1 kbp using the 500 bp windowed copy number estimates generated by the dCGH pipeline. For all read-pair-based SV callers we first excluded all large (>1 Mbp) calls without read-depth support (CN >1.5 and <2.5). Calls were then merged on a per-sample basis roughly in order of breakpoint accuracy per caller (Wham, Whamg, Lumpy, VariationHunter, GenomeSTRiP, and finally dCGH). Beginning with the first caller, we merged one caller at a time by first reducing all overlapping calls and splitting the intersecting calls from both callers into those that cover ≥50% of the merged region and recursively calling the merging procedure on those calls that cover <50% of the merged region. In the rare case where no calls covered at least 50% of the reduction, we recursively decreased the threshold by 5% until we could generate clusters of calls for merging. Once we generated a set of calls to merge, we applied the following procedure to determine which breakpoint to retain. Calls with SVTyper-supported breakpoints were prioritized followed by calls with 500 bp window copy number support. Ties were broken by retaining the calls from the previously merged caller, unless none of the calls had support, in which case we picked the largest call. Finally, we retained a set of merged calls with 2+ callers or 1 caller with SVTyper or copy number support. We also initially retained all calls with dCGH support only due to the high accuracy of these calls and increased sensitivity to duplications and segmental duplication flanked events. Our reduced set of merged calls was then re-annotated with the median per-base copy number data (from the dCGH pipeline) and filtered with more stringent symmetrical thresholds of estimated copy numbers below 1.5 or over 2.67. Each proband and sibling call was then annotated for the number of overlapping (50% reciprocal) calls in the population of parents and the estimated copy number of each CNV call in all family members, which was then used to estimate inheritance. High-quality, private de novo CNVs were considered to be those with no overlapping parental CNVs, no evidence of CNVs in the sibling or parents by copy number, SVTyper support only in the carrier, >200 bp non-repeat sequence, and <50% of the call being a segmental duplication. CRLMM was utilized for orthogonal validation of events with enough probes on SNP microarrays (method described by Krumm et al. (Krumm et al., 2015)).
Recent and ancient repeat regions
We utilized RepeatMasker 3.3.0 on the b37 human genome using the following settings: -species "Homo sapiens" -s -div 10 -dir b37_results -xsmall -no_is -e wublast -s – pa 15 to get all sequences with a divergence rate <10% from the consensus. Segmental duplications and microsatellites were downloaded from the UCSC Genome Browser. These above region categories were the recent repeats. All repeats generated by RepeatMasker for the UCSC Genome Browser were downloaded and those not in the recent repeats were considered ancient.
Validation
We selected 697 total sites for PCR amplification and used Sanger sequencing (450 sites) and PacBio sequencing (322 sites) to validate de novo variants (96.3% overall VR) using previously described methods (Turner et al., 2016). For the PacBio sequencing, we pooled PCR products from all the probands, fathers, and mothers into three pools and barcoded and prepped them for sequencing following standard protocols. The sequences were separated by barcode, mapped to the genome, and checked in IGV for the mutation event. In addition, we validated an additional 58 WES-only events by Sanger sequencing (79.3% VR).
Transgenic reporter assays
We used transgenic lacZ reporter assays as described previously (Visel et al., 2007) and tested three proband variants (Figure 2, Figure S5) for three sites from VISTA (enhancer.lbl.gov) that previously showed enhancer activity. All embryo images for the reference and variant alleles of hs737 were randomized, the labels removed, and annotation performed by at least three independent reviewers blinded to allele type. For each embryo, reviewers scored whether the enhancer activity pattern in each tissue was 1) robust and in a reproducible pattern, 2) absent, or 3) ectopic (i.e., staining was present in that tissue but not in the reproducible pattern, which can occur because of the random nature of the transgene integration). Final annotations were determined by the staining type with the most reviewer votes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Wherever possible, standard tests were applied to count data (Fisher’s exact test), differences in medians (Mann-Whitney), and burden tests (log rank test of CNV size distributions). The relationship between DNM and paternal age was calculated using both linear and exponential fits and statistics are based on Pearson’s correlation coefficient.
Oligogenic DNM burden
To model the burden of DNMs in probands and siblings, we created an oligogenic score based on the count of de novo VOI. We calculated this simple score (S) for each individual as follows:
where L is LGD variant count, M is number of missense variants with CADD score > 30, R is count of noncoding CNS DNase I hypersensitive TFBS variants, D is coding deletion count, and U is 3' UTR variant count.
Estimated attributable fraction in the population (Table 2) was calculated using the epi.2by2 function with the case.control method in epiR (http://cran.r-project.org/web/packages/epiR/index.html).
TSEA and CSEA
We used Tissue Specific Expression Analysis (Dougherty et al., 2010) (TSEA, http://genetics.wustl.edu/jdlab/tsea/) and Cell-type Specific Expression Analysis (Dougherty et al., 2010; Xu et al., 2014) (CSEA, http://genetics.wustl.edu/jdlab/csea-tool-2/) to test whether individuals with scores (S) of 3 or higher had variants in genes enriched in any tissues or cell types, respectively. We also tested individuals with scores >=3 and containing at least one pNCR TFBS and at least one 3' UTR event
Attributable fraction estimates
We assessed the contribution of the different functional classes to the cases in the SSC by utilization of the epi.2by function (method=case.control) in the epiR R package. We identified 1,786 quad families (Krumm et al. 2015) that were previously assessed by WES and SNP microarray for all four family members. We removed the pilot families (Turner et al. 2016) in this set (n=38) since the criteria used for family selection in the present study were different than in the pilot. This left 1,748 families (Table 2) with 588 families that had been removed because they had a known event (de novo LGD, de novo CNV, or very large inherited CNV). These 588 were considered the “known families”. In the phase I data, 398 families were assessed as they had undergone WES and SNP microarray analysis and represented a random sampling of the remaining families with no known cause. They are referred to as “new families” in the table. In the category column, for the known families, the different types included de novo LGD (no de novo CNV or large inherited CNV), de novo CNV (no de novo LGD or large inherited CNV), large inherited CNV (no de novo LGD, no de novo CNV), and multi-hit (contains one each of at least two of the following categories: de novo LGD, de novo CNV, large inherited CNV). In the category column for the new families, the different types included small de novo deletion (no de novo LGD, no de novo CNV, no large inherited CNV), de novo missense CADD score > 30 (no de novo LGD, no de novo CNV, no large inherited CNV, no small de novo deletion), and 3+ de novo VOI (requires at least one de novo pNCRTFBS and at least one de novo 3' UTR) (no de novo LGD, no de novo CNV, no large inherited CNV, no small de novo deletion, no de novo missense CADD score > 30). The reason for these classifications was to avoid double counting of individuals. For the known families the odds ratio, attributable prevalence, attributable prevalence in population, attributable fraction (est) in exposed (%), and attributable fraction (est) in population (%), and p-value have a denominator of 1,748 as they are representative of the full cohort, whereas the same fields for the new families have a denominator of 398. The last field (extrapolated attributable fraction (est) in population (%)) contains the percent explained by each of the significant classes wherein for the new families we treated them as a subset of unexplained cases and therefore calculated their values by the following equation: (0.664 [fraction of families that had no known variants] * attributable fraction (est) in population [in the 398 families]). The total for all extrapolated attributable fraction (est) in population (%) values was 16.88% with 6.03% as a result of the new categories from this current study.
DATA AND SOFTWARE AVAILABILITY
Data availability
Approved researchers can obtain the SSC population dataset described in this study by applying at https://base.sfari.org/. BAM and VCF files are provided in SFARI Base with accession number SFARI Base Accession Numbers: SFARI_SSC_WGS_1 and SFARI_SSC_WGS_1_Turner.
Code availability
Code for pipelines that ran in the Amazon Cloud is available at https://github.com/eichlerlab/aws.
Supplementary Material
Figure S1. Breakdown of SSC families (related to STAR Methods). Those marked as PILOT + PHASE 1 are the focus of this current study. The KNOWN (nominal p=0.04), PHASE 1 (nominal p=0.03), and PILOT + PHASE 1 (nominal p=0.05) families are all significantly enriched for their sex ratio by a two-sided binomial test in comparison to the full dataset. The PILOT (nominal p=8.2×10−4) and KNOWN (2.7×10−3) families are significantly enriched for having significantly different full-scale IQs than the full cohort by two-sided Mann-Whitney test and upon visualization they have a lower IQ. There is no statistically significant difference for the social responsiveness scale, ADOS calibrated severity score, or normalized head circumference.
Table S3. Gene ontology enrichment analysis for proband variants in CNS DNase I hypersensitive, conserved transcription factor binding sites (related to Figure 2). Analysis was performed on http://geneontology.org/ using the PANTHER overrepresentation test (release 20170413) and the GO ontology database release 20170424. Bonferroni correction was applied to calculate p-values. Biological process (BP), molecular function (MF), and cellular component (CC) were all tested by this analysis. Only significant results are shown. Proband genes include ABAT, ADGRB1, ADNP-AS1, AEBP2, AGMO, AIF1L, ANKRD44, ANTXR2, ANXA10, AQP5, ARRDC3-AS1, ASTN1, ATG4C, AUTS2, BBX, BCL9, C9orf92, CACNA2D3, CASZ1, CBWD6, CBX8, CDH8, CDKAL1, CEP112, CEP128, CFAP20, CFAP61, CHD6, CHRM2, CHSY3, CLMP, CNIH3, CNTNAP4, COBL, CORO1C, CPS1, CSGALNACT1, DISC1, DLX2-AS1, DMRT1, ECT2, EMX1, EPHB1, ERAP1, EVA1C, FH, GACAT3, GALNT16, GLI3, GPR180, GRIP1, GRM3, GXYLT1, HCN1, HIVEP3, IDH3A, IMMP2L, KC6, KCNH7, KCNJ4, KCNN3, LINC00525, LINC00536, LINC00861, LINC00923, LINC01065, LINC01085, LINC01122, LINC01163, LINC01239, LINC01435, LINC01696, LINC01822, LINC01923, LMF2, LOC100506136, LOC101927237, LOC101928591, LOC101928622, LOC101929497, LOC102724623, LOC103344931, LOC105374428, LOC105374972, LOC729159, LRRIQ3, MAP7, MGMT, MIR4532, MIR4790, MIR9-3HG, MIR99AHG, MSI2, MTERF1, NEB, NEIL3, NEUROG1, NKAIN2, NLGN1, NRXN3, OLFML2B, PARK2, PDZRN3-AS1, PHC2, PITPNB, PLAUR, PLEKHA6, PLXNA1, POU2F1, PPP4R3A, PTMS, PTPRD, PVRL3-AS1, RASEF, RHOG, RND3, RNF220, ROR1, RUNX1T1, SCAP, SEPHS1, SHOX2, SKIL, SLC15A5, SLIT2, SLIT3, SNTA1, SNX1, SOCS2, SOX9, STARD13, STK32B, STMN1, TCF7L1, THRB, TLX1NB, TMEM132C, TSHZ2, UBE2Q1, UNC80, USP3, UST, UVSSA, VPS13C, WWOX, ZEB2, ZFPM2, and ZNF827. Sibling genes include ACTR2, ADHFE1, AMBN, ANPEP, ASAP2, ATF7, ATXN1, C11orf74, C7orf66, CAMKK2, CAMTA1, CASC18, CD83, CHD6, CLNK, CPXM2, CSMD1, CTTNBP2, CXXC5, CYP7B1, DAOA-AS1, DHRS4L1, DMRTB1, DOK5, EBF3, EIF1, EPHB1, ERBB4, EVPL, FAM160A1, FAM181A, FAM19A5, FAM208A, FANCC, FBXO15, FGF18, FGFR2, FOS, FOXP1, FSCB, FSHR, FSTL5, GATA3-AS1, GBE1, GCG, HAND1, HS6ST3, IFT43, IGDCC3, INPP5D, KANK1, KY, LAMC1, LCORL, LINC00334, LINC00474, LINC00844, LINC01101, LINC01122, LINC01166, LINC01333, LINC01828, LINC01995, LOC101928767, LOC101929019, LOC101929153, LOC101929710, LOC105378068, LOC283038, LOC339529, LOC339975, LOC90768, MARK2, MCM9, MEGF10, MGMT, MIR4445, MIR4475, MIR5093, MMP15, NAB1, NELL1, NFATC3, NFIB, NOG, NOL4L, NRAP, NRP1, NTRK3, OLIG3, PALLD, PDCD6IP, PKN2-AS1, PNOC, POLR3E, PPP1R9A, PQLC2L, PRRX1, PRTFDC1, PRTG, PTPN3, RAD51B, RBFOX1, ROBO1, RORB, RPUSD1, SEMA5A, SERTAD3, SETBP1, SHQ1, SIL1, SLC6A11, SPNS2, SRCIN1, STEAP2-AS1, TACR1, TCF7L2, TIAM1, TPRG1-AS1, TRAF3IP2-AS1, TRIML1, TRIP4, UNC13C, XAB2, ZNF423, ZNF516, ZNF536, and ZXDC.
Table S4. Genome sequencing quality control metrics (related to STAR Methods). Included are the family identifier, individual identifier, blood cell identifier, sex, phase of study (pilot refers to Turner et al. 2016), mean sequence coverage, median library insert size, standard deviation of insert size, percent properly paired reads, mitochondrial haplogroup, and genotype concordance with SNP microarray.
Figure S2. Noncoding variants and paternal age (related to Figure 1). (A) Noncoding de novo SNVs per individual based on father’s age at child’s birth and (B) noncoding de novo indels per individual based on father’s age at child’s birth both showing significant correlation of number of variants with father’s age at birth (noncoding SNV p=1.0×10−94, r=0.59) (noncoding indel p=1.6×10−8, r=0.19).
Figure S3. Exome versus genome comparisons (related to Figure 1). (A) Venn diagram of events found within the exome by WES and/or by WGS. (B,C,D) Low sequence depth coverage exons, in WES but with good coverage in WGS, containing novel LGD events. Shown is the average coverage across the exome capture regions in (B) ARID1B, (C) CNTNAP3, and (D) DNM1 in WES and WGS data. The WES data was from 9,014 individuals analyzed in the Krumm et al. (Krumm et al., 2015) paper and the WGS data was from 2,064 individuals in the present study. The capture regions with a black circle around them are those with novel LGD variants only detected by WGS. (E,F,G) Genes of interest with multiple low-coverage exons based on WES data but covered well in WGS data. Shown is the average coverage across the exome capture regions in (E,F) HYDIN and (G) SHANK3 in WES and WGS data (E) HYDIN is a gene of interest because it is a human-specific duplicated gene (Sudmant et al., 2010). In (E) the capture regions are plotted in genomic space but because there are many exons that are close together and visualization is difficult, we also plotted them with the capture regions equal distance from each other (F). (G) SHANK3 is a gene of interest because of its known role in autism (Durand et al., 2007).
Figure S4. Breakdown of event types within the different de novo score categories (related to Figure 3). Shown are LGD=likely gene-disrupting, MIS30=missense with CADD score >30, DEL=exonic deletion; UTR=untranslated region, TFBS=putative noncoding regulatory with a transcription factor binding site events.
Figure S5. Two of the three de novo sequence variants identified in autism probands that were tested for in vivo enhancer activity in the CNS (reference allele from enhancer.lbl.gov) (Visel et al., 2007) (related to Figure 2). We extended the previous assessment for the reference allele in our current study by also testing the variant allele. For each locus, we show, from top to bottom, the human genome reference allele, the patient variant (red text), the location of conserved TFBS near the variant, the VISTA enhancer with hs number (blue bar), and representative transgenic embryonic day 11.5 mouse embryos for the reference and variant alleles, respectively displaying the enhancer activity pattern (blue staining). Testing of the variants in (A) hs311 and (B) hs1386 shows no difference between reference and variant alleles. (C) Replication of hs737 data.
Figure S6. Barplot showing the difference by gender for autism probands and their unaffected siblings (related to Figure 3). This plot excludes 3' UTR mutations. VOI=variants of interest. Because of the potential for a female protective effect in autism (Jacquemont et al., 2014; Krumm et al., 2015; Turner et al., 2015), we hypothesized that females with autism may generally have the higher scores. No significant difference was found considering all variant types. However, if we excluded the 3' UTR, we found that female probands have higher scores compared to unaffected female siblings (one-sided Mann-Whitney nominal p=6.3×10−3). It is striking that while 41% of female probands contain a VOI (score ≥ 1); only 24% of unaffected female siblings carry a VOI. No difference in oligogenic DNM burden was observed comparing male probands and unaffected males (one-sided Mann-Whitney nominal p=0.35) suggesting that this oligogenic effect DNM is driven primarily by females.
Table S1. DNM counts by individual (related to Figure 2 and Figure 4). This table shows for each mutation category the number of individuals with a specific number of variants. For example, 0: 502 means that there are 502 people with no (0) variants in that category. * 476 families assessed for CNVs as opposed to 516 for all SNV/indel categories.
Table S2. Summary of de novo LGD and missense CADD >30 (MIS30) events detected (related to Figure 2). * indicates the event maps to a segmental duplication or a repeat. Dataset (G=genome, GNED=genome no exome data available, E=exome). Effect (SSA=splice site acceptor, SSD=splice site donor, FS=frame shift, SG=stop gained and for missense it is the reference amino acid, amino acid position, and alternate amino acid). Validation is the Sanger sequencing of PCR amplicons (Y=yes and AF=attempt failed).
Comprehensive CNV/SNV dataset from whole-genome sequencing of 516 autism families
Estimated human germline mutation rate of 1.5 × 10−8 substitutions/site/generation
Autism probands enriched for de novo missense, promoter and enhancer mutations
Oligogenic de novo mutation signal for genes enriched in striatal neuron expression
Acknowledgments
We thank Tonia Brown for assistance in editing this manuscript. We are grateful to the NYGC sequencing production team for providing high-quality WGS data and Tom Maniatis, the scientific director of the NYGC, for his support and encouragement; Phil Green, Kelley Harris, and David Reich for helpful insights into the mutation rate analyses; Marta Benadetti and Stephan Sanders for working with us on initial family selection as well as Natalia Volfovsky for assistance on general collection questions. We are thankful for discussions with Brian McNally and Skylar Thompson on IT optimizations during data production. Thanks also to Jason Underwood and Katy Munson for useful advice on experimental design of the validation experiments. This work was supported by grants from the Simons Foundation Autism Research Initiative (SFARI 303241 and 385035, E.E.E.) and the National Institutes of Health (NIH grants R01MH101221 to E.E.E., UM1 HG008901 to R.B.D.). The Centers for Common Disease Genomics are funded by the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute and the GSP Coordinating Center (U24HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. Research conducted at the E.O. Lawrence Berkeley National Laboratory was additionally supported by NIH grants R01HG003988 and U54HG006997 (L.A.P.) and performed under Department of Energy Contract DE-AC02-05CH11231, University of California. This work was also supported by a postdoctoral fellowship grant from the Autism Science Foundation (#16-008, T.N.T.). We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren and E. Wijsman). We appreciate obtaining access to phenotypic data on SFARI Base. E.E.E. and R.B.D. are investigators of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, T.N.T., D.E.D., L.A.P., and E.E.E.; Methodology, T.N.T., B.P.C., D.E.D., B.J.N., M.C.Z., L.A.P., R.B.D., E.E.E.; Software, T.N.T., B.P.C., B.J.N., M.C.Z., Z.N.K., F.H., A.R.; Validation, T.N.T., D.E.D, K.H., L.A.P.; Formal Analysis, T.N.T., B.P.C., D.E.D., L.E.P., E.E.E.; Investigation, T.N.T., B.P.C., D.E.D., K.H., B.J.N., M.C.Z., A.R., L.A.P., E.E.E.; Resources, M.C.Z., L.A.P., R.B.D, E.E.E.; Data Curation, T.N.T., B.P.C., B.J.N.; Writing – Original Draft, T.N.T., B.P.C., D.E.D., L.A.P., E.E.E.; Writing – Review & Editing, T.N.T., B.P.C., D.E.D., K.H., B.J.N., M.C.Z., Z.N.K., F.H., A.R., L.A.P., R.B.D., E.E.E.; Visualization, T.N.T., D.E.D., L.A.P., E.E.E.; Supervision, M.C.Z., L.A.P., R.B.D, E.E.E.; and Funding Acquisition, T.N.T., L.A.P., R.B.D, E.E.E.
COMPETING FINANCIAL INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
References
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychological medicine. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic acids research. 2009;37:D832–836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Kleiber ML, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, Pang T, et al. Paternally inherited noncoding structural variants contribute to autism. bioRxiv. 2017 doi: 10.1126/science.aan2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon MC, Ruiz-Pesini E, Mishmar D, Procaccio V, Lott MT, Nguyen KC, Spolim S, Patil U, Baldi P, Wallace DC. MITOMASTER: a bioinformatics tool for the analysis of mitochondrial DNA sequences. Human mutation. 2009;30:1–6. doi: 10.1002/humu.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CO. Genetics of common disorders. British medical bulletin. 1969;25:52–57. doi: 10.1093/oxfordjournals.bmb.a070671. [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–456. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, Hall IM. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nature methods. 2015;12:966–968. doi: 10.1038/nmeth.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN. Recurrence rates in autism spectrum disorders. Jama. 2014;312:1154–1155. doi: 10.1001/jama.2014.9841. [DOI] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, McCarthy SA. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics (Oxford, England) 2017 doi: 10.1093/bioinformatics/btx100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS computational biology. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Jiang J, McSweeney C, Zou D, Liu L, Mao Y. Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Human molecular genetics. 2016;25:2738–2751. doi: 10.1093/hmg/ddw131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, Heintz N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic acids research. 2010;38:4218–4230. doi: 10.1093/nar/gkq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai LH. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nature neuroscience. 2016;19:1477–1488. doi: 10.1038/nn.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature genetics. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Walker M, Hyde TM, Kleinman JE, Harrison PJ. The DISC1 Ser704Cys substitution affects centrosomal localization of its binding partner PCM1 in glia in human brain. Human molecular genetics. 2010;19:2487–2496. doi: 10.1093/hmg/ddq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. Journal of autism and developmental disorders. 2003;33:365–382. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- Fuccillo MV. Striatal Circuits as a Common Node for Autism Pathophysiology. Frontiers in neuroscience. 2016;10:27. doi: 10.3389/fnins.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratten J, Wray NR, Peyrot WJ, McGrath JJ, Visscher PM, Goddard ME. Risk of psychiatric illness from advanced paternal age is not predominantly from de novo mutations. Nature genetics. 2016;48:718–724. doi: 10.1038/ng.3577. [DOI] [PubMed] [Google Scholar]
- Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, Sahinalp SC. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nature methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Archives of general psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Nakazawa T, Tsurusaki Y, Yasuda Y, Nagayasu K, Matsumura K, Kawashima H, Yamamori H, Fujimoto M, Ohi K, et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. Journal of human genetics. 2016;61:199–206. doi: 10.1038/jhg.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Hajirasouliha I, McPherson A, Eichler EE, Sahinalp SC. Simultaneous structural variation discovery among multiple paired-end sequenced genomes. Genome research. 2011;21:2203–2212. doi: 10.1101/gr.120501.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Penn O, Borenstein E, Eichler EE. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015;25:142–154. doi: 10.1101/gr.178855.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. American journal of human genetics. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014 doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, Beckmann JS, Rosenfeld JA, Eichler EE. A higher mutational burden in females supports a "female protective model" in neurodevelopmental disorders. Am J Hum Genet. 2014;94:415–425. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. American journal of medical genetics Part A. 2004;131:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. American journal of human genetics. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537:675–679. doi: 10.1038/nature19357. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012a;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee IH, Brewster SJ, Hanson E, Harris HK, Lowe KR, et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PloS one. 2012b;7:e49475. doi: 10.1371/journal.pone.0049475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G, Wexler E, Rosen E, Mukamel Z, Osborn GE, Chen L, Lu D, Gao F, Gao K, Lowe JK, et al. Modeling the functional genomics of autism using human neurons. Molecular psychiatry. 2012;17:202–214. doi: 10.1038/mp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, Elde NC, Yandell M. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput Biol. 2015;11:e1004572. doi: 10.1371/journal.pcbi.1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–588. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lin MC, Kornblum HI, Papazian DM, Nelson SF. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Human molecular genetics. 2014;23:3481–3489. doi: 10.1093/hmg/ddu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GE, Alkan C, Jiang L, Zhao S, Eichler EE. Comparative analysis of Alu repeats in primate genomes. Genome research. 2009;19:876–885. doi: 10.1101/gr.083972.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England) 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria KC, Beltrao-Braga PC, Trujillo CA, Mendes AP, Padmanabhan K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Molecular psychiatry. 2016 doi: 10.1038/mp.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Ramos OA, Olivares AM, Haider NB, de Autismo LC, Lattig MC. Whole-Exome Sequencing in a South American Cohort Links ALDH1A3, FOXN1 and Retinoic Acid Regulation Pathways to Autism Spectrum Disorders. PloS one. 2015;10:e0135927. doi: 10.1371/journal.pone.0135927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012a;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. American journal of human genetics. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Human molecular genetics. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She X, Liu G, Ventura M, Zhao S, Misceo D, Roberto R, Cardone MF, Rocchi M, Green ED, Archidiacano N, et al. A preliminary comparative analysis of primate segmental duplications shows elevated substitution rates and a great-ape expansion of intrachromosomal duplications. Genome Res. 2006;16:576–583. doi: 10.1101/gr.4949406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. Journal of child psychology and psychiatry, and allied disciplines. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli T, Kolevzon A, Wang AT, Curchack-Lichtin J, Halpern D, Schwartz L, Soffes S, Bush L, Grodberg D, Cai G, et al. De novo SCN2A splice site mutation in a boy with Autism spectrum disorder. BMC medical genetics. 2014;15:35. doi: 10.1186/1471-2350-15-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner T, Pihur V, Chakravarti A. Quantifying and modeling birth order effects in autism. PloS one. 2011;6:e26418. doi: 10.1371/journal.pone.0026418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman HA, et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet. 2016;98:58–74. doi: 10.1016/j.ajhg.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Sharma K, Oh EC, Liu YP, Collins RL, Sosa MX, Auer DR, Brand H, Sanders SJ, Moreno-De-Luca D, et al. Loss of delta-catenin function in severe autism. Nature. 2015;520:51–56. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic acids research. 2007;35:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, Brand H, An J-Y, Stone MR, Glessner JT, Zhu L, Collins RL, Dong S, Layer RM, Markenscoff-Papadimitriou E-C. Limited contribution of rare, noncoding variation to autism spectrum disorder from sequencing of 2,076 genomes in quartet families. bioRxiv 2017 [Google Scholar]
- Xu X, Wells AB, O'Brien DR, Nehorai A, Dougherty JD. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:1420–1431. doi: 10.1523/JNEUROSCI.4488-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature neuroscience. 2017;20:602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T, et al. Genome-wide characteristics of de novo mutations in autism. NPJ genomic medicine. 2016;1:160271–1602710. doi: 10.1038/npjgenmed.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Breakdown of SSC families (related to STAR Methods). Those marked as PILOT + PHASE 1 are the focus of this current study. The KNOWN (nominal p=0.04), PHASE 1 (nominal p=0.03), and PILOT + PHASE 1 (nominal p=0.05) families are all significantly enriched for their sex ratio by a two-sided binomial test in comparison to the full dataset. The PILOT (nominal p=8.2×10−4) and KNOWN (2.7×10−3) families are significantly enriched for having significantly different full-scale IQs than the full cohort by two-sided Mann-Whitney test and upon visualization they have a lower IQ. There is no statistically significant difference for the social responsiveness scale, ADOS calibrated severity score, or normalized head circumference.
Table S3. Gene ontology enrichment analysis for proband variants in CNS DNase I hypersensitive, conserved transcription factor binding sites (related to Figure 2). Analysis was performed on http://geneontology.org/ using the PANTHER overrepresentation test (release 20170413) and the GO ontology database release 20170424. Bonferroni correction was applied to calculate p-values. Biological process (BP), molecular function (MF), and cellular component (CC) were all tested by this analysis. Only significant results are shown. Proband genes include ABAT, ADGRB1, ADNP-AS1, AEBP2, AGMO, AIF1L, ANKRD44, ANTXR2, ANXA10, AQP5, ARRDC3-AS1, ASTN1, ATG4C, AUTS2, BBX, BCL9, C9orf92, CACNA2D3, CASZ1, CBWD6, CBX8, CDH8, CDKAL1, CEP112, CEP128, CFAP20, CFAP61, CHD6, CHRM2, CHSY3, CLMP, CNIH3, CNTNAP4, COBL, CORO1C, CPS1, CSGALNACT1, DISC1, DLX2-AS1, DMRT1, ECT2, EMX1, EPHB1, ERAP1, EVA1C, FH, GACAT3, GALNT16, GLI3, GPR180, GRIP1, GRM3, GXYLT1, HCN1, HIVEP3, IDH3A, IMMP2L, KC6, KCNH7, KCNJ4, KCNN3, LINC00525, LINC00536, LINC00861, LINC00923, LINC01065, LINC01085, LINC01122, LINC01163, LINC01239, LINC01435, LINC01696, LINC01822, LINC01923, LMF2, LOC100506136, LOC101927237, LOC101928591, LOC101928622, LOC101929497, LOC102724623, LOC103344931, LOC105374428, LOC105374972, LOC729159, LRRIQ3, MAP7, MGMT, MIR4532, MIR4790, MIR9-3HG, MIR99AHG, MSI2, MTERF1, NEB, NEIL3, NEUROG1, NKAIN2, NLGN1, NRXN3, OLFML2B, PARK2, PDZRN3-AS1, PHC2, PITPNB, PLAUR, PLEKHA6, PLXNA1, POU2F1, PPP4R3A, PTMS, PTPRD, PVRL3-AS1, RASEF, RHOG, RND3, RNF220, ROR1, RUNX1T1, SCAP, SEPHS1, SHOX2, SKIL, SLC15A5, SLIT2, SLIT3, SNTA1, SNX1, SOCS2, SOX9, STARD13, STK32B, STMN1, TCF7L1, THRB, TLX1NB, TMEM132C, TSHZ2, UBE2Q1, UNC80, USP3, UST, UVSSA, VPS13C, WWOX, ZEB2, ZFPM2, and ZNF827. Sibling genes include ACTR2, ADHFE1, AMBN, ANPEP, ASAP2, ATF7, ATXN1, C11orf74, C7orf66, CAMKK2, CAMTA1, CASC18, CD83, CHD6, CLNK, CPXM2, CSMD1, CTTNBP2, CXXC5, CYP7B1, DAOA-AS1, DHRS4L1, DMRTB1, DOK5, EBF3, EIF1, EPHB1, ERBB4, EVPL, FAM160A1, FAM181A, FAM19A5, FAM208A, FANCC, FBXO15, FGF18, FGFR2, FOS, FOXP1, FSCB, FSHR, FSTL5, GATA3-AS1, GBE1, GCG, HAND1, HS6ST3, IFT43, IGDCC3, INPP5D, KANK1, KY, LAMC1, LCORL, LINC00334, LINC00474, LINC00844, LINC01101, LINC01122, LINC01166, LINC01333, LINC01828, LINC01995, LOC101928767, LOC101929019, LOC101929153, LOC101929710, LOC105378068, LOC283038, LOC339529, LOC339975, LOC90768, MARK2, MCM9, MEGF10, MGMT, MIR4445, MIR4475, MIR5093, MMP15, NAB1, NELL1, NFATC3, NFIB, NOG, NOL4L, NRAP, NRP1, NTRK3, OLIG3, PALLD, PDCD6IP, PKN2-AS1, PNOC, POLR3E, PPP1R9A, PQLC2L, PRRX1, PRTFDC1, PRTG, PTPN3, RAD51B, RBFOX1, ROBO1, RORB, RPUSD1, SEMA5A, SERTAD3, SETBP1, SHQ1, SIL1, SLC6A11, SPNS2, SRCIN1, STEAP2-AS1, TACR1, TCF7L2, TIAM1, TPRG1-AS1, TRAF3IP2-AS1, TRIML1, TRIP4, UNC13C, XAB2, ZNF423, ZNF516, ZNF536, and ZXDC.
Table S4. Genome sequencing quality control metrics (related to STAR Methods). Included are the family identifier, individual identifier, blood cell identifier, sex, phase of study (pilot refers to Turner et al. 2016), mean sequence coverage, median library insert size, standard deviation of insert size, percent properly paired reads, mitochondrial haplogroup, and genotype concordance with SNP microarray.
Figure S2. Noncoding variants and paternal age (related to Figure 1). (A) Noncoding de novo SNVs per individual based on father’s age at child’s birth and (B) noncoding de novo indels per individual based on father’s age at child’s birth both showing significant correlation of number of variants with father’s age at birth (noncoding SNV p=1.0×10−94, r=0.59) (noncoding indel p=1.6×10−8, r=0.19).
Figure S3. Exome versus genome comparisons (related to Figure 1). (A) Venn diagram of events found within the exome by WES and/or by WGS. (B,C,D) Low sequence depth coverage exons, in WES but with good coverage in WGS, containing novel LGD events. Shown is the average coverage across the exome capture regions in (B) ARID1B, (C) CNTNAP3, and (D) DNM1 in WES and WGS data. The WES data was from 9,014 individuals analyzed in the Krumm et al. (Krumm et al., 2015) paper and the WGS data was from 2,064 individuals in the present study. The capture regions with a black circle around them are those with novel LGD variants only detected by WGS. (E,F,G) Genes of interest with multiple low-coverage exons based on WES data but covered well in WGS data. Shown is the average coverage across the exome capture regions in (E,F) HYDIN and (G) SHANK3 in WES and WGS data (E) HYDIN is a gene of interest because it is a human-specific duplicated gene (Sudmant et al., 2010). In (E) the capture regions are plotted in genomic space but because there are many exons that are close together and visualization is difficult, we also plotted them with the capture regions equal distance from each other (F). (G) SHANK3 is a gene of interest because of its known role in autism (Durand et al., 2007).
Figure S4. Breakdown of event types within the different de novo score categories (related to Figure 3). Shown are LGD=likely gene-disrupting, MIS30=missense with CADD score >30, DEL=exonic deletion; UTR=untranslated region, TFBS=putative noncoding regulatory with a transcription factor binding site events.
Figure S5. Two of the three de novo sequence variants identified in autism probands that were tested for in vivo enhancer activity in the CNS (reference allele from enhancer.lbl.gov) (Visel et al., 2007) (related to Figure 2). We extended the previous assessment for the reference allele in our current study by also testing the variant allele. For each locus, we show, from top to bottom, the human genome reference allele, the patient variant (red text), the location of conserved TFBS near the variant, the VISTA enhancer with hs number (blue bar), and representative transgenic embryonic day 11.5 mouse embryos for the reference and variant alleles, respectively displaying the enhancer activity pattern (blue staining). Testing of the variants in (A) hs311 and (B) hs1386 shows no difference between reference and variant alleles. (C) Replication of hs737 data.
Figure S6. Barplot showing the difference by gender for autism probands and their unaffected siblings (related to Figure 3). This plot excludes 3' UTR mutations. VOI=variants of interest. Because of the potential for a female protective effect in autism (Jacquemont et al., 2014; Krumm et al., 2015; Turner et al., 2015), we hypothesized that females with autism may generally have the higher scores. No significant difference was found considering all variant types. However, if we excluded the 3' UTR, we found that female probands have higher scores compared to unaffected female siblings (one-sided Mann-Whitney nominal p=6.3×10−3). It is striking that while 41% of female probands contain a VOI (score ≥ 1); only 24% of unaffected female siblings carry a VOI. No difference in oligogenic DNM burden was observed comparing male probands and unaffected males (one-sided Mann-Whitney nominal p=0.35) suggesting that this oligogenic effect DNM is driven primarily by females.
Table S1. DNM counts by individual (related to Figure 2 and Figure 4). This table shows for each mutation category the number of individuals with a specific number of variants. For example, 0: 502 means that there are 502 people with no (0) variants in that category. * 476 families assessed for CNVs as opposed to 516 for all SNV/indel categories.
Table S2. Summary of de novo LGD and missense CADD >30 (MIS30) events detected (related to Figure 2). * indicates the event maps to a segmental duplication or a repeat. Dataset (G=genome, GNED=genome no exome data available, E=exome). Effect (SSA=splice site acceptor, SSD=splice site donor, FS=frame shift, SG=stop gained and for missense it is the reference amino acid, amino acid position, and alternate amino acid). Validation is the Sanger sequencing of PCR amplicons (Y=yes and AF=attempt failed).
Data Availability Statement
Approved researchers can obtain the SSC population dataset described in this study by applying at https://base.sfari.org/. BAM and VCF files are provided in SFARI Base with accession number SFARI Base Accession Numbers: SFARI_SSC_WGS_1 and SFARI_SSC_WGS_1_Turner.