

Abstract
Background and Purpose
Intracerebral hemorrhage (ICH) is a subtype of stroke with highest mortality and morbidity. Pronounced inflammation plays a significant role in the development of the secondary brain injury after ICH. Recently, Salt-inducible kinase-2 (SIK-2) was identified as an important component controlling inflammatory response. Here, we sought to investigate the role of SIK-2 in post-ICH inflammation and potential protective effects of SIK-2 inhibition after ICH.
Methods
Two hundred and ninety-three male CD-1 mice were used. ICH was induced via injection of 30 μl of autologous blood. Recombinant SIK-2 was administrated 1 hour after ICH intracerebroventricularly (ICV). SIK-2 small interfering RNA (siRNA) was injected ICV 24 hours before ICH. Bosutinib, a clinically approved tyrosine kinase inhibitor with affinity to SIK-2, was given intranasal 1 hour or 6 hours after ICH. Effects of treatments were evaluated by neurological tests and brain water content calculation. Molecular pathways were investigated by western blots and immunofluorescence studies.
Results
Endogenous SIK-2 was expressed in microglia and neurons. SIK-2 expression was reduced after ICH. Exogenous SIK-2 aggravated post-ICH inflammation leading to brain edema and the neurobehavioral deficits. SIK-2 inhibition attenuated post-ICH inflammation, reducing brain edema and ameliorating neurological dysfunctions. Bosutinib inhibited SIK-2 attenuating ICH-induced brain damage. Protective effects of Bosutinib were mediated, at least partly, by CRTC3/CREB/NF-κB pathway.
Conclusions
SIK-2 participates in inflammation induction after ICH. SIK-2 inhibition via Bosutinib or siRNA decreased inflammation, attenuating brain injury. SIK-2 effects are, at least partly, mediated by CRTC3-CREB-NF-κB signaling pathway.
Keywords: ICH, salt-inducible kinases, SIK-2 inhibition, Bosutinib
Introduction
Spontaneous intracerebral hemorrhage (ICH) accounts for 10–15% of all strokes and is associated with high mortality and morbidity. Despite intensive research, there is no effective treatment improving survival chance of ICH patients. There is mounting evidence demonstrating that microglia activation and, subsequently, releases of pro-inflammatory cytokines are one of the critical factors contributing to ICH-induced secondary brain injury. Therefore, examination of molecular pathways leading to post-ICH inflammation might be able to advance the development of new therapies for ICH.
Salt-inducible kinase (SIK) was originally identified as an enzyme induced in the adrenal glands of rats treated with a high-salt diet1. SIKs belong to a family of AMP-activated protein kinases (AMPKs) and consist of three isoforms2. It has been demonstrated recently that SIK-2, but not other SIK isoforms, contributes to brain damage development after stroke3. It has been shown that OGD (oxygen-glucose deprivation, an in-vitro model of ischemic stroke) induces degradation of SIK-2 without having the effect of the expression of another SIK isoform. Furthermore, the inhibition of SIK-2 during reperfusion was associated with increased neuronal survival. Finally, neurons from SIK-2 knock out animals were less sensitive to the OGD and SIK-2 knock out animals were more resistant to the focal cerebral ischemia induced by MCAO.
There are indications that SIK-2 is inhibited by a ‘tyrosine kinase’ inhibitor, even though it is a serine/threonine kinase. SIK-2 inhibition can decrease inflammation, result in expression of macrophages with all characteristics of regulatory macrophages, producing anti-inflammatory cytokine and, on the other hand, decreasing pro-inflammatory cytokine production4. Furthermore, in vitro screening of potential SIK-2 inhibitors revealed that an FDA approved Bosutinib can inhibit SIK-2 and that Bosutinib-induced SIK2 inhibition resulted in boost of IL-10 production in macrophages and dendritic cells4, 5. The study also revealed that SIK-2 inhibition leads to dephosphorization of CRTC-3 (CREB-regulated transcription coactivator 3), inducing the translocation of CRTC-3 to the nucleus4, which subsequently activates CREB. The CREB activation, in turn, induces the activation of anti-inflammation pathways. On the other hand, CREB activation increases the expression of NF-κB negative subunit, IκB, which inhibits the NF-κB induced inflammation. This interaction explains the strong anti-inflammatory response induced by SIK-2 inhibit6, 7.
The inflammation is a major pathophysiological factor of ICH and post-ICH neuronal death results in unresolved neurological deficits. It was therefore quite possible, that the pharmacological manipulations on SIK-2 will provide vital information able to pave the new therapeutic approaches for ICH patient.
In the present study, we sought to investigate the potential role of SIK-2 in the rise of inflammation after ICH and explore the pathway underlying it. Most importantly we intent to explore the potential therapeutic utility of FDA approved tyrosine kinase inhibitor, Bosutinib, for treatment of ICH patients.
Materials and Methods
Animals and ICH Model
Two hundred and ninety-three eight-week-old male CD1 mice (weight=30 g; Charles River, Wilmington, MA) were housed in a 12-hour light/dark cycle at a controlled temperature and humidity with unlimited access to food and water. All experiments on animals in this study were approved by the institutional animal care and use committee at Loma Linda University.
Intracerebral hemorrhage was induced by double infusion of autologous whole blood (30 μL) as described before. Briefly, mice were randomly assigned to the experimental groups, anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) (2:1 v/v, intraperitoneal injection) and positioned prone in a stereotactic head frame (Kopf Instruments, Tujunga, CA, USA). Arterial blood was collected in a non-heparinized capillary tube, transferred into a Hamilton syringe and with a microinjection pump 5 μL of blood was infused into the right basal ganglia at bregma anterior–posterior 0.2 mm, mediolateral 1.8 mm, and dorsoventral 3.0 mm. After 5 minutes, needle was advanced and 25 μL of blood was delivered at 3.5 mm dorsoventral giving a total injection volume of 30 μL. Sham-operated animals were subjected to needle insertion only.
Following experiments were conducted.
Experiment I
to determine the time course of SIK-2 expression after ICH. The expression of SIK-2 was evaluated in ipsilateral hemisphere using Western blot analysis at 3, 6, 12, 24, 48, 72 hours after ICH and compared to sham operated animals. Spatial expression of SIK-2 was investigated by double immunostaining with Iba1 (microglia), GFAP (astrocytes) and NeuN (neuronal nuclei) using sham and ICH (24 hours after surgery) groups.
Experiment II
to evaluate the role of SIK-2 and SIK-2 inhibition in the post-ICH brain injury. Mice were randomly divided into sham, ICH + vehicle, ICH + recombinant SIK-2 (Mybiosourse, species: mouse), ICH + SIK-2-siRNA (small interfering RNA), ICH + scramble RNA (scRNA), ICH + Bosutinib group, ICH + Bosutinib + scRNA group, ICH + Bosutinib + SIK-2-siRNA group. Vehicle (sterile saline) or recombinant SIK-2 (rnSIK-2, 150 ng/5 μL of sterile saline) were administrated intracerebroventricularly (ICV.) at 1 hour after ICH as previously described8. Bosutinib (1mg/kg, 5mg/kg, and 25mg/kg) was administrated intranasally at 1 hour or 6 hours after ICH as previously described9. Scramble siRNA (500pmol/μL) or SIK-2-siRNA (500pmol/μL) was injected ICV 24 hours before ICH induction9. SIK-2 and TNF-α level were measured by Western blot 24 hours after ICH according to the vendor recommendation and as we did before10. Neurological score (Garcia test) and brain water content were measured at 24 hours, 72 hours or 14 days after ICH10.
Experiment III
to evaluate molecular pathway underlying effects observed in Experiment II. Mice were randomly divided into sham, ICH + vehicle, ICH + Bosutinib (the most effective dosage, experiment II) group, ICH + Bosutinib + scramble siRNA, ICH + Bosutinib + CRTC3-siRNA group. CRTC3 siRNA was injected ICV. 24 hours before ICH induction as described above.
Nuclear and cytoplasmic CRTC3 and NF-κB protein level was measured by Western blot and double immunostaining 24 hours after ICH. CREB/DNA binding was detected in a colorimetric assay. Neurological deficits and brain water content were measured at 24 hours after ICH.
Neurological Score
Neurological scores were assessed by two trained investigators blinded to the animal groups, using a modified Garcia test as previously described10, 11. The Garcia scoring system consists of 6 sub-tests (spontaneous activity, spontaneous movements of all limbs, forelimbs outstretching, climbing ability, body perception, and response to vibrissae stimulation). With a maximum of 21 points were scored animals without neurological deficits.
Measurement of Brain Water Content
The brain water content was measured using wet/dry method12. Briefly, mice were decapitated under deep anesthesia. Brains were immediately removed and cut into 4 mm sections around the needle track. Each section was divided into four parts: ipsilateral and contralateral basal ganglia, ipsilateral and contralateral cortex. The cerebellum was collected as an internal control. Each part was weighed on an electronic analytical balance (APX-60, Denver Instrument) and then dried at 100 °C for 24 h to determine the dry weight (DW). Brain water content (%) was calculated as [(WW−DW)/WW] × 100.
Immunofluorescence Staining
Twenty-four hours after ICH, mice were perfused under deep anesthesia with 40 ml of ice-cold PBS followed by perfusion with 40 ml formalin (10%). The brains were removed and fixed in formalin at 4 ℃ for a minimum of 3 days. Samples were then dehydrated with 30% sucrose in PBS and sectioned with cryostat (CM3050S; Leica Microsystems) in 10 μm coronal slices. Immunofluorescence was performed as previously described. Coronal brain sections (10 μm) were permeabilized with 0.3% Triton X-100 in PBS for 30 minutes. The sections then were blocked with 5% donkey serum for 1 hour before being incubated at 4°C overnight with primary antibodies: anti-Iba-1 (1:100, Abcam, Cambridge, MA), anti-GFAP (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), anti-NeuN (1:100, Abcam, Cambridge, MA), anti-SIK-2 (1:50, Santa Cruz Biotechnology, Santa Cruz, CA) and anti-CRTC3 (1:100, Abcam, Cambridge, MA). The sections then were incubated with appropriate fluorescence-conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) for 2 hours at room temperature followed by being visualized using a fluorescence microscope (Olympus OX51, Japan).
Western Blot
Mice were euthanized and perfused with 40 ml. of ice-cold PBS. The brains were removed, separated into ipsilateral and contralateral hemisphere, and stored at – 80 ℃ until analysis. Western Blotting was performed as previously described12. Nuclear proteins were extracted by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) following the instruction manual. Primary antibodies used are listed as follow: anti-SIK-2 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), anti-CRTC3 (1:200, Abcam, Cambridge, MA), anti-CRTC3 (1:100, Abcam, Cambridge, MA), anti-NF-κB p65 (1:200, Cell signaling, Danvers, MA), anti-TNF-α(1:200, Santa Cruz Biotechnology, Santa Cruz, CA).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad software). Data are represented as a mean ± standard deviation. One-way ANOVA followed by Tukey multiple comparisons test was used for different groups’ comparison. Chi-square test was used for mortality analyses. P values <0.05 were considered statistically significant.
Results
Mortality Rate
The total animal mortality rate of the study was 5.46% (16/293). No sham operated animal died in this study. Neither autologous blood injection-induced ICH nor administration of different agents caused significant mortality.
Endogenous SIK-2 Was Down-regulated After ICH Injury
ICH resulted in significant inhibition of SIK-2 expression as early as 3 hours after ICH. SIK-2 expression remained down-regulated 24 hours after ICH. The recovery of the SIK-2 expression was observed at 72 hours after ICH (Figure 1A).
Figure 1.
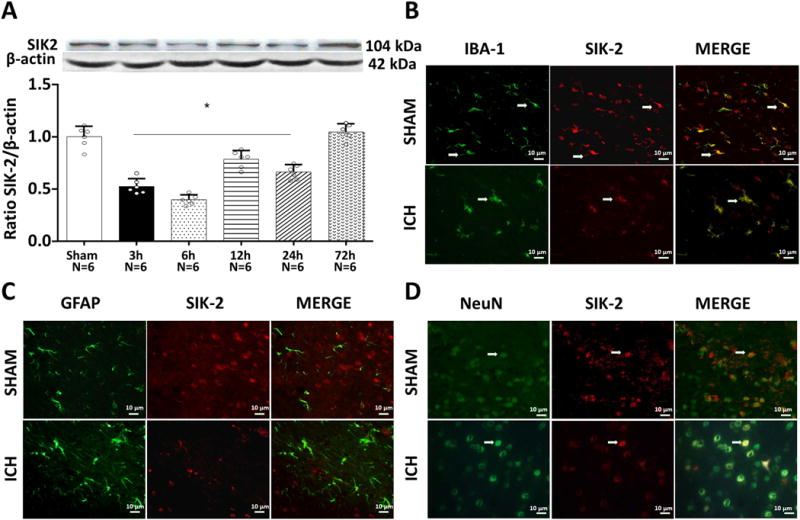
Expression time curve of SIK2 before and after intracerebral hemorrhage (ICH) injury. Western blot assay for the profiles of SIK2 expression in the ipsilateral hemisphere in sham and ICH mice 3h, 6h, 12 h, 24 h, 72 h after operation. *P<0.05 vs sham; One way ANOVA, Tukey test, n=6. (A). Representative photographs of immunofluorescence staining for SIK2 (red) expression in neurons (NeuN, green; B), microglias (Iba-1, green; C), and astrocytes (GFAP, green; D) in sham group and the peri-hematoma area 24 h after ICH.
Double immunostaining revealed that SIK-2 was expressed in neurons and microglia of the peri-hemorrhagic area 6 hours after ICH. No astrocyte SIK-2 expression was observed (Figures 1B–D).
Exogenous SIK-2 Aggravated Inflammation and Brain Injury at 24 hours after ICH
ICV administration of recombinant SIK-2 (rnSIK-2 150 ng/5 μL) resulted in increased concentration of the SIK-2 in the ipsilateral hemisphere evaluated by Western blot at 24 hours after ICH (Figure 2A). The increase of SIK-2 was accompanied with aggravation of post-ICH inflammation (TNF-α expression) (Figure 2B). Increased inflammation consequently resulted in exacerbation of brain edema and neurological dysfunctions (Figures 2C–D).
Figure 2.
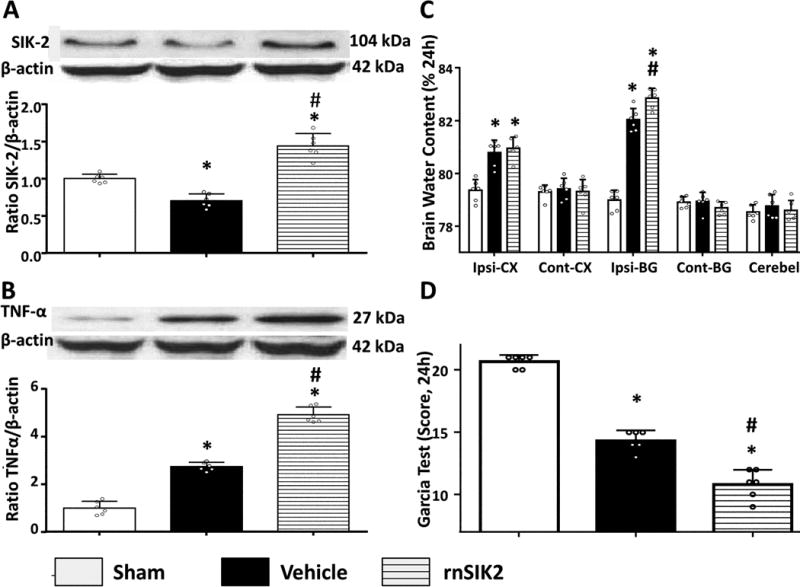
Exogenous SIK2 administration aggravated neurobehavioral deficits, brain water content and pronounced inflammatory response after ICH. Representative bands and quantification of SIK2 (A) and TNF-α (D) expression in indicated groups. Relative densities have been normalized against the sham group. n=6. The Brain water content (B) and Modified Garcia (C) test after using pretreated rnSIK2 were tested in indicated groups. *P<0.05 vs sham; #P<0.05 vs ICH+Vehicle. One way ANOVA, Tukey test.
Administration of SIK-2 siRNA reduced brain injury at 24 hours after ICH
ICV injection of SIK-2 siRNA significantly decreased SIK-2 expression in the ipsilateral hemisphere 24 hours after ICH (Figure 3A). Furthermore, SIK-2 in vivo knockdown (siRNA) was accompanied with a decrease of TNF production (Figure 3B), consequently resulting in reduced peri-hematoma brain edema and improved neurological functions (Figures 3C–D).
Figure 3.
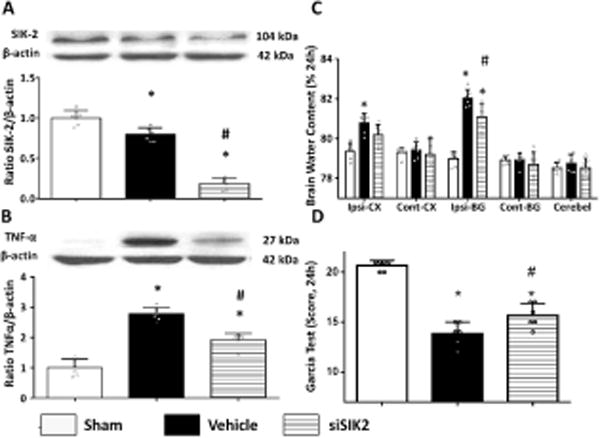
SIK2 in vivo knocked down by intraventricle siRNA inhibited inflammatory response and attenuated neurobehavioral deficits, brain water content at 24h after ICH. Representative bands and quantification of SIK2 (A) and TNF-α (B) expression were presented in indicated groups. Relative densities have been normalized against the sham group. Statistical analysis of brain water content (C) Garcia test (D) were performed. (n=6), *P<0.05 vs sham, #P<0.05 vs vehicle. One way ANOVA, Tukey test.
Bosutinib attenuated brain injury and inhibit SIK-2 expression after ICH
Compared to sham operated, all ICH animals revealed a significant increase of brain water content in ipsilateral cortex and basal ganglia as well as sever neurological deficits (Figures 4A–B). FDA approved drug, Bosutinib attenuated dose-dependently development of brain edema improving post-ICH neurological functions at 24 hours after ICH (Figures 4A–B). The effective dose of Bosutinib significantly inhibited SIK-2 expression (Figure 4C) resulting in less post-ICH inflammation (Figure 4D). In order to investigate the optimal time for SIK inhibition after ICH, we have performed additional experiment and administrated Bosutinib intranasally 6 hours after ICH. The results revealed that intranasal Bosutinib administration 1 hour or 6hours after ICU injection resulted in similar protection and attenuated brain edema and neurological deficits (Figure I in the supplement). We also performed experiment showing effects of Bosutinib given in conjunction with SIK-2 knockdown on brain edema and neurological functions. The results showed that Bosutinib given in conjunction with SIK-2 knockdown did not show any additional protection compared to Bosutinib administration only (Figure II in the supplement). This result indicated that Bosutinib had a protective effect mainly through inhibition of SIK-2 rather than multiple kinases.
Figure 4.
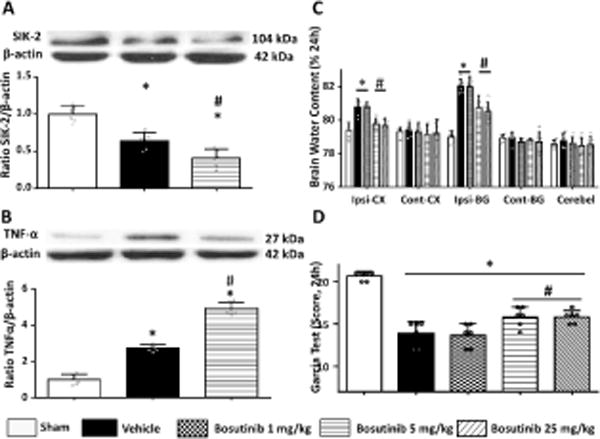
SIK2 inhibited by high and middle dose of intranasal Bosutinib reduced inflammatory response and attenuated neurobehavioral deficits, brain water content at 24h after ICH. Statistical analysis of brain water content (A) Garcia test (B) were performed. Representative bands and quantification of SIK2 (C) and TNF-α (D) expression were presented in indicated groups. Relative densities have been normalized against the sham group. (n=6), *P<0.05 vs sham, #P<0.05 vs vehicle. One way ANOVA, Tukey test.
Moreover, effective concentrations of Bosutinib attenuated development of ICH-induced brain edema evaluated 72 hours after ICH. Administration of 5 mg/kg of Bosutinib significantly improved neurological functions at this time point (Figure III in the supplement). Furthermore, we conducted the long-term study, 14 days after ICH. At this time point, we observed a tendency for increased brain water content after ICH. The treatment with Bosutinib showed the tendency to decrease the post-ICH increase of brain water content and significantly improve the neurological functions (Figure IV in the supplement). These indicate that SIK inhibition has therapeutic value on both short and long term.
Bosutinib induced protection was mediated by CRTC3/CREB pathway
While ICH had no effect on the total CRTC3 expression (data not shown), ICH induced translocation of CRTC3 into the nucleus, resulting in the increased ratio of nuclear/cytoplasmic CRTC3 (Figure 5A). Bosutinib escalated the nuclear translocation of CRTC3, resulting in the further increase of the nuclear/cytoplasmic ratio in Bosutinib- compared to vehicle-treated animals evaluated by western blot (24 hours after ICH, Figure 5A). Immunostaining study confirmed results of western blot and clearly demonstrated that Bosutinib treatment induced nucleus translocation of CRTC3 in microglia (Iba-1 positive cells, Figure 5B).
Figure 5.
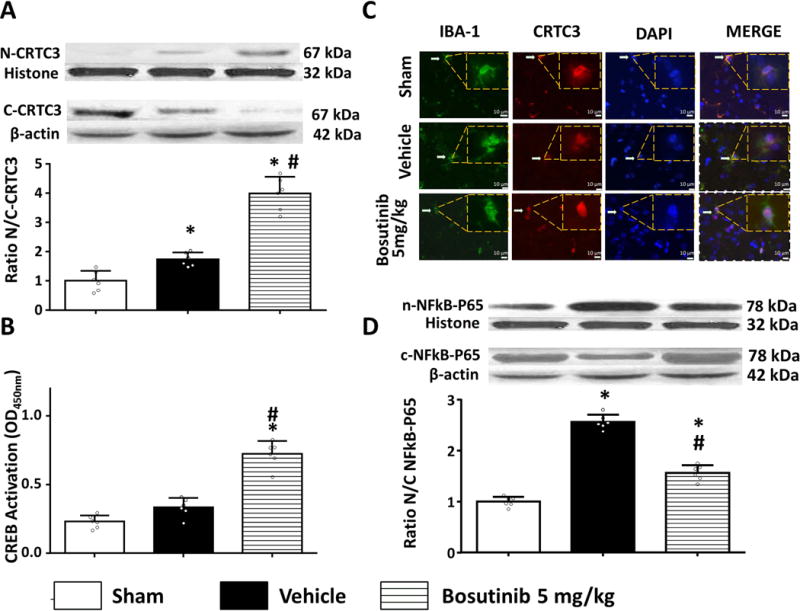
Intranasal administration of bosutinib (5mg/kg) induced translocation of CRTC3 from cytoplasm to nucleus and activated CREB and inhibited NFkB 24h after ICH. (A) Western Blot bands and quantification of the ratio of nuclear against cytoplasmic CRTC3 were presented in indicated groups. Relative densities have been normalized against the sham group. (B) Representative photographs of immunofluorescence staining (Magnification: (800*)) shows location of CRTC3 (red) in microglia (IBA-1, green) of indicated groups. White arrows indicate positive cells. The inset of each image (boxed) represent CRTC3 and IBA1 colocalization at a higher magnification (2000*), Bar=10 μm. (C) Quantitative detection of CREB that binds to a DNA consensus sequence in a colorimetric assay. Levels of CREB expressed as average absorbance +/− SD of six wells. (D) Western Blot bands and quantification of the ratio of nuclear against cytoplasmic NFkB-p65 were presented in indicated groups. Relative densities have been normalized against the sham group. n=6, *P<0.05 vs sham; #P<0.05 vs ICH+Vehicle. One way ANOVA, Tukey test.
The nuclear translocation of CRTC3 resulted in CREB activation monitored by CREB ability to bind to the DNA via colorimetric assay. While ICH alone did not activate CREB, Bosutinib significantly activated CREB 24 hours after ICH (Figure 5C).
Pro-inflammatory effects were, furthermore, evaluated by monitoring of NF-κB p65 spatial expression. Compared to sham, ICH induced significant increase of nuclear portion of NF-κB p65 24 hours after ICH. Bosutinib treatment attenuated this effect (Figure 5D).
CRTC3 in vivo knockdown attenuated protective effects of Bosutinib
SiRNA-induced knockdown of CRTC3 resulted in a significant decrease of CRTC3 expression (Figure V in the supplement). In vivo knockdown of CRTC3 attenuated the protective effects of Bosutinib. Compared with scramble RNA, in vivo knockdown resulted in less CREB activation in Bosutinib treated animals (Figure 6A), increasing nuclear accumulation of NF-κB-p65 (Figure 6B). Additionally, animals treated with a combination of Bosutinib/(CRTC3 siRNA) had higher brain water content of the ipsilateral hemisphere, consequently resulting in aggravation of neurological dysfunction (Figures 6C–D).
Figure 6.
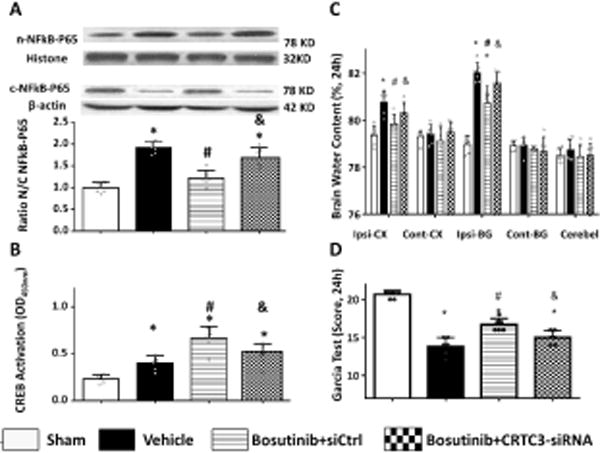
CRTC3 in vivo knock-down with intraventricle siRNA abolished the attenuation intranasal Bosutinib on brain edema, neurological deficiency and neuroinflammation 24h after ICH. Statistical analysis of brain water content (A) Garcia test (B) were performed. (C) Quantitative detection of CREB that binds to a DNA consensus sequence in a colorimetric assay. Levels of CREB expressed as average absorbance +/− SD of six wells. (D) Representative Western Blot bands and quantification of NFkB-p65 in indicated groups. Relative densities have been normalized against the sham group. n=6, *P<0.05 vs sham; #P<0.05 vs ICH+Vehicle; and &P<0.05 vs ICH+scramble siRNA.
Discussion
Inflammation is a major factor contributing to both high mortality and morbidity after ICH. In the current study, we investigated for the first time the role of SIK-2 in the rise of inflammation after ICH. We were able to demonstrate SIK-2 was expressed in microglia and neurons and that the expression was transient inhibited in ICH animals. SIK-2 inhibition via Bosutinib or in vivo knockdown of SIK-2 suppressed ICH-induced inflammation, resulting in fewer brain injuries and consequently, in better neurological functions. Finally, we established that anti-inflammatory effects of SIK-2 inhibition were, at least partly, mediated via CRTC3-CREB-NF-κB pathway.
SIK was originally identified as a kinase induced in the adrenal glands of rats fed with a high-salt diet and shortly after it as a kinase induced by membrane depolarization of neurons1, 13. SIK belongs to an AMP-activated protein kinase family and plays a crucial role in the regulation of stress response14. SIK consists of three isoforms and potential participation of different isoforms in stress response is controversially discussed in the literature. There are indications that over expression of SIK-3 and SIK-1 but not SIK-2 inhibits NF-κB activation in response to TLR4 stimulation and decreases production of pro-inflammatory cytokines15. SIK-3, but not SIK-1 and SIK-2, deficiency is responsible for the profound inflammatory response upon LPS16. While SIK-1 and SIK-3 inhibition seems to be pro-inflammatory, the inhibition of SIK-2, on the contrary, is beneficial and increase neuronal survival both in cell culture and in whole animals after ischemic stroke3. Ischemic stroke induced SIK-2 degradation while no effects of ischemia on another SIK isoforms were established in in-vivo or in-vitro part of the study3. Furthermore, it has been demonstrated that inhibition of SIK-2 induced a switch of macrophages towards a regulatory phenotype, resulting in the decrease of pro-inflammatory cytokines release and increase of anti-inflammatory cytokines4. Interesting enough the anti-inflammatory effect of SIK-2 inhibition was lost on macrophages expressing mutated, drug-resistant form of SIK-2 isoform, indicating that the SIK-2 is a key molecular switch between pro- and anti-inflammatory macrophage phenotype4. SIK-2 appeared to be a promising target, able to decrease inflammation and increase neuronal survival after ICH.
In our study, we demonstrated that ICH induced transient reduction of SIK-2 production as early as 3 hours after ICH. The production remained down-regulated 24 hour and returned to the normal level at 72 hours after ICH. It was in agreement with previous reports, demonstrating in in vitro ischemic stroke model that SIK-2 was down regulated in early stage of reoxygenation without recovery during 24 hours of the reoxygenation3. We have hypothesized the spontaneously down-regulation of SIK-2 as a negative feedback to inflammatory factors increase in the early stage of ICH. However, the endogenous inhibition of SIK-2 is not sufficient to attenuate the neuroinflammation. Further inhibition of SIK-2 with exogenous inhibitors is therefore beneficial. Furthermore, we demonstrated that SIK-2 was expressed on neurons and microglia of both ICH and sham operated animals. Neuronal SIK-2 expression was demonstrated before3. We showed for the first time that SIK-2 is expressed on microglia as well. However, giving the numerous demonstrations of SIK-2 expression on macrophage, and the fact that microglia is resident macrophages of CNS; this finding does not contradict previous publications4, 6. Although previous publication showed expression of SIK-2 on astrocytes, no SIK-2 expression on astrocytes was observed in our study. It is, however, worth to mention that the authors of previous study used cortical cell culture and western blot as a detection method. Under these conditions, they could see a signal of SIK-2 on astrocytes, which was only a marginal, compared to neuronal signal of SIK-2. We believe that lack of SIK-2 positive astrocytes in the basal ganglia is not in direct conflict with previous study and is to explain with different experimental conditions.
In order to investigate the role of SIK-2 in the rise of inflammation after ICH, we administrated recombinant SIK-2 using ICV delivery. As expected, administration of recombinant SIK-2 significant increased the concentration of the kinase in the brain. Giving that dramatic increase of TNF-α expression in post-ICH brain is well established event, the change of TNF-α expression was used to investigate the effect of SIK-2 on post-ICH inflammation8. We demonstrated that the increase of SIK-2 was associated with significant up-regulation of TNF-α production, clearly demonstrating the involvement of SIK-2 in an inflammatory response. TNF-α is able to directly disrupt blood-brain barrier, contributing to development of brain edema17. Anti-TNF-α therapy improved recovery in murine model of ICH, indicating that TNF-α is a key factor for the development of brain injury after ICH18, 19. It is therefore not surprisingly that SIK-2-induced increase of TNF-α expression had an opposite effect and led to advanced brain edema in ICH animals treated with SIK-2 compared to the ICH alone and to more severe neurological deficits. We confirmed the finding by conducting of in-vitro knocking down of SIK-2 via siRNA20. Well in agreement with previous experiment, knockdown of SIK-2 significantly decrease post-ICH production of TNF- α resulting in attenuation of brain edema and improving neurological functions.
Targeting of specific protein by siRNA represents unquestionably promising treatment strategy, which, unfortunately, has only limited clinical relevance up to now21. In order to increase translatability of our results into clinical practice, we used Bosutinib (an FDA-approved small molecular tyrosine kinase inhibitor) to inhibit SIK-2. Bosutinib was originally approved for treatment of chronic myelogenous leukemia22. Recent publication demonstrated that Bosutinib can inhibit SIK by depriving tyrosine kinases of access to the Cdc37-Hsp90 molecular chaperone system on which they depend for their cellular stability, leading to their ubiquitylation and degradation.5. Furthermore, Bosutinib had beneficial effects in the model of neurodegenerative diseases, such as AD23. In our study, we first investigated the dose dependent response of Bosutinib. Authors of previous publication demonstrated effectiveness of intraperitoneal delivery of the drug. However, they had applied Bosutinib for 6 weeks and used a model of chronic inflammation (AD)23. Since Bosutinib has a high molecular weight, for treatment of acute post-ICH inflammation, we choose an intranasal delivery of the drug. Intranasal delivery is a reliable way, allowing a quick delivery of the high molecular weight drug into brain9, 24. We demonstrated that Bosutinib dose-dependently decreased brain edema and improved neurological functions. According to our long-term study, Bosutinib can improve neurological functions by the time of 14 days after ICH, when brain edema and neuroinflammation secondary to ICH already become less predominant. It indicated that besides neuroinflammation, Bosutinib may also affect neuronal survival directly. The direct investigation into the treatment effects on the neuronal survival is a goal of our future study.
It has been revealed previously that SIK-2 inhibition leads to dephosphorization of CREB-regulated transcription coactivators (CRTCs) including CRTC1, CRTC2 and CRTC3. While CRTC1 and CRTC2 are mainly expressed on neurons, CRTC3 is primarily expressed on microglia. In our current study we focused on relationship between SIK-2 and inflammatory response, such as TNF-α production. Giving the factor that the inflammatory cytokines are mostly produced by microglia, we focused on SIK-2 induced CRTC3 regulation in the mechanism study. SIK-2 inhibition leads to dephosphorization of CRTC-3 (CREB-regulated transcription coactivator 3), inducing the translocation of CRTC-3 to the nucleus4. The translocation activates CREB, promoting CREB-dependent gene transcription and inhibiting NF-κB4, 7, 25, 26. Well in agreement with these findings, we demonstrated that Bosutinib significantly increases the nuclear fraction of CRTC3, resulting in CREB activation. The activated CREB inhibited NF-κB and consequently decreased inflammation in brain of mice after ICH.
Taken together, our results indicated that SIK-2 contributed to inflammation after ICH and this effect was mediated, at least partly, through CRTC3-CREB pathway. Most importantly, we demonstrated that Bosutinib inhibited SIK-2, decreasing inflammation and attenuating brain injury after ICH. Considering Bosutinib is an FDA approved well tolerated drug, our results might provide a clue to a new strategy for treatment of ICH patients.
Supplementary Material
Acknowledgments
Funding Sources:
Natural Science Foundation of China: 81601029 to LM, and NIH NS078755, NS082124 and NS091042 to JHZ.
Footnotes
Disclosures: None. Authors have declared no conflict of interests.
References
- 1.Wang Z, Takemori H, Halder SK, Nonaka Y, Okamoto M. Cloning of a novel kinase (sik) of the snf1/ampk family from high salt diet-treated rat adrenal. FEBS letters. 1999;453:135–139. doi: 10.1016/s0014-5793(99)00708-5. [DOI] [PubMed] [Google Scholar]
- 2.Katoh Y, Takemori H, Horike N, Doi J, Muraoka M, Min L, et al. Salt-inducible kinase (sik) isoforms: Their involvement in steroidogenesis and adipogenesis. Molecular and cellular endocrinology. 2004;217:109–112. doi: 10.1016/j.mce.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Sasaki T, Takemori H, Yagita Y, Terasaki Y, Uebi T, Horike N, et al. Sik2 is a key regulator for neuronal survival after ischemia via torc1-creb. Neuron. 2011;69:106–119. doi: 10.1016/j.neuron.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, et al. Phosphorylation of crtc3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundberg TB, Choi HG, Song JH, Russell CN, Hussain MM, Graham DB, et al. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:12468–12473. doi: 10.1073/pnas.1412308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, et al. Pge(2) induces macrophage il-10 production and a regulatory-like phenotype via a protein kinase a-sik-crtc3 pathway. J Immunol. 2013;190:565–577. doi: 10.4049/jimmunol.1202462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henriksson E, Sall J, Gormand A, Wasserstrom S, Morrice NA, Fritzen AM, et al. Sik2 regulates crtcs, hdac4 and glucose uptake in adipocytes. J Cell Sci. 2015;128:472–486. doi: 10.1242/jcs.153932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang P, Manaenko A, Xu F, Miao L, Wang G, Hu X, et al. Role of pdgf-d and pdgfr-beta in neuroinflammation in experimental ich mice model. Experimental neurology. 2016;283:157–164. doi: 10.1016/j.expneurol.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Chen Y, Wu J, Manaenko A, Yang P, Tang J, et al. Activation of dopamine d2 receptor suppresses neuroinflammation through alphab-crystalline by inhibition of nf-kappab nuclear translocation in experimental ich mice model. Stroke. 2015;46:2637–2646. doi: 10.1161/STROKEAHA.115.009792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manaenko A, Lekic T, Ma Q, Zhang JH, Tang J. Hydrogen inhalation ameliorated mast cell-mediated brain injury after intracerebral hemorrhage in mice. Critical care medicine. 2013;41:1266–1275. doi: 10.1097/CCM.0b013e31827711c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manaenko A, Fathali N, Chen H, Suzuki H, Williams S, Zhang JH, et al. Heat shock protein 70 upregulation by geldanamycin reduces brain injury in a mouse model of intracerebral hemorrhage. Neurochemistry international. 2010;57:844–850. doi: 10.1016/j.neuint.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manaenko A, Fathali N, Khatibi NH, Lekic T, Hasegawa Y, Martin R, et al. Arginine-vasopressin v1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochemistry international. 2011;58:542–548. doi: 10.1016/j.neuint.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldman JD, Vician L, Crispino M, Hoe W, Baudry M, Herschman HR. The salt-inducible kinase, sik, is induced by depolarization in brain. Journal of neurochemistry. 2000;74:2227–2238. doi: 10.1046/j.1471-4159.2000.0742227.x. [DOI] [PubMed] [Google Scholar]
- 14.Mihaylova MM, Shaw RJ. The ampk signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yong Kim S, Jeong S, Chah KH, Jung E, Baek KH, Kim ST, et al. Salt-inducible kinases 1 and 3 negatively regulate toll-like receptor 4-mediated signal. Molecular endocrinology. 2013;27:1958–1968. doi: 10.1210/me.2013-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanosaka M, Fujimoto M, Ohkawara T, Nagatake T, Itoh Y, Kagawa M, et al. Salt-inducible kinase 3 deficiency exacerbates lipopolysaccharide-induced endotoxin shock accompanied by increased levels of pro-inflammatory molecules in mice. Immunology. 2015;145:268–278. doi: 10.1111/imm.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K, Takata F, et al. Tumor necrosis factor-alpha mediates the blood-brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. Journal of pharmacological sciences. 2010;112:251–254. doi: 10.1254/jphs.09292sc. [DOI] [PubMed] [Google Scholar]
- 18.Zhou QH, Boado RJ, Hui EK, Lu JZ, Pardridge WM. Brain-penetrating tumor necrosis factor decoy receptor in the mouse. Drug metabolism and disposition: the biological fate of chemicals. 2011;39:71–76. doi: 10.1124/dmd.110.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei B, Dawson HN, Roulhac-Wilson B, Wang H, Laskowitz DT, James ML. Tumor necrosis factor alpha antagonism improves neurological recovery in murine intracerebral hemorrhage. Journal of neuroinflammation. 2013;10:103. doi: 10.1186/1742-2094-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y, Hu Q, Manaenko A, Zhang Y, Peng Y, Xu L, et al. 17beta-estradiol attenuates hematoma expansion through estrogen receptor alpha/silent information regulator 1/nuclear factor-kappa b pathway in hyperglycemic intracerebral hemorrhage mice. Stroke; a journal of cerebral circulation. 2015;46:485–491. doi: 10.1161/STROKEAHA.114.006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukuda AM, Badaut J. Sirna treatment: “A sword-in-the-stone” for acute brain injuries. Genes. 2013;4:435–456. doi: 10.3390/genes4030435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintas-Cardama A, Cortes J. Kinase inhibitors in chronic myelogenous leukemia. Clinical advances in hematology & oncology: H&O. 2006;4:365–374. [PubMed] [Google Scholar]
- 23.Lonskaya I, Hebron ML, Selby ST, Turner RS, Moussa CE. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in alzheimer’s disease models. Neuroscience. 2015;304:316–327. doi: 10.1016/j.neuroscience.2015.07.070. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Zhang Y, Yang P, Enkhjargal B, Manaenko A, Tang J, et al. Recombinant osteopontin stabilizes smooth muscle cell phenotype via integrin receptor/integrin-linked kinase/rac-1 pathway after subarachnoid hemorrhage in rats. Stroke; a journal of cerebral circulation. 2016;47:1319–1327. doi: 10.1161/STROKEAHA.115.011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datusalia AK, Sharma SS. Nf-kappab inhibition resolves cognitive deficits in experimental type 2 diabetes mellitus through creb and glutamate/gaba neurotransmitters pathway. Current neurovascular research. 2016;13:22–32. doi: 10.2174/1567202612666151030104810. [DOI] [PubMed] [Google Scholar]
- 26.Ge L, Liu L, Liu H, Liu S, Xue H, Wang X, et al. Resveratrol abrogates lipopolysaccharide-induced depressive-like behavior, neuroinflammatory response, and creb/bdnf signaling in mice. European journal of pharmacology. 2015;768:49–57. doi: 10.1016/j.ejphar.2015.10.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.