

Abstract
Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that mediates biological responses to endogenous and environmental chemical cues. Increasing evidence shows that the AHR plays physiological roles in regulating development, homeostasis and function of a variety of cell lineages in the immune system. However, the role of AHR in human B cell development has not been investigated. Toward this end, an in vitro feeder-free human B cell developmental model system was employed using human cord blood CD34+ hematopoietic stem/progenitor cells (HSPC). Using this model, we found that AHR activation by the high affinity ligand, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), significantly suppressed the generation of early-B cells and pro-B cells from HSPCs, indicating the impairment of B cell lineage specification and commitment. Addition of an AHR antagonist reversed TCDD-elicited suppression of early-B and pro-B cells, suggesting a role of AHR in regulating B lymphopoiesis. Gene expression analysis revealed a significant decrease in the mRNA level of EBF1 and PAX5, two critical transcription factors directing B cell lineage specification and commitment. In addition, binding of the ligand-activated AHR to the putative dioxin response elements in the EBF1 promoter was demonstrated by electrophoretic mobility shift assays and chromatin immunoprecipitation analysis, suggesting transcriptional regulation of EBF1 by AHR. Taken together, this study demonstrates a role for the AHR in regulating human B cell development, and suggests that transcriptional alterations of EBF1 by the AHR are involved in the underlying mechanism.
Keywords: human B lymphopoiesis, hematopoietic stem cell, aryl hydrocarbon receptor
Introduction
Circulating human B cells have an average half-life of 18 days (1). As a result, the homeostasis of the peripheral B cell population requires lifelong replenishment through B lymphopoiesis. The development of B cells from hematopoietic stem cells (HSC) involves stepwise cell fate choices and lineage restrictions. HSCs first generate progenitor cells with limited lineage potential, which further differentiate into cells that are irreversibly committed to the B cell lineage.
A lineage-specific transcription factor network directs B lymphopoiesis. The lymphoid priming in HSC-derived multipotent progenitors is governed by transcription factor IKZF1 (IKAROS), SPI1 (PU.1) and TCF3 (E2A) (2–4). The interplay between these transcription factors leads to the loss of myeloid lineage potential and gives rise to common lymphoid progenitors (CLP). The next step in B lymphopoiesis is B cell lineage specification, which involves the expression of genes that are functionally important for B cells. One of the critical transcription factors that governs the specification of the B cell developmental program is early B cell factor 1 (EBF1). EBF1 is present at low levels in CLPs and is under the regulation of SPI1, ETS1 and TCF3 (5). Evidence supporting a critical role of EBF1 in B cell development comes from the observation that Ebf1 knockout mice exhibit a complete block in B cell development at the CLP-like stage (6). Ectopic expression of Ebf1 can rescue B lymphopoiesis from developmentally arrested multipotent progenitors due to deletion of PU.1 or Ikaros (7, 8). In addition, the expression of EBF1 overcomes the block in B lymphopoiesis imposed by the absence of E2A (9). Moreover, enforced expression of EBF1 in HSCs skews development favoring B cell lineage commitment (10), further suggesting a critical role for EBF1 in the regulatory circuitry of B lymphopoiesis. Increased EBF1 activates expression of paired box 5 (PAX5), which reciprocally upregulates EBF1 through a positive feedback loop (5, 11). The elevated expression of EBF1 and PAX5 eliminates alternative cell fates by suppressing non-B cell genes and activates many B cell specific genes that confer B cell identity (12–14). At this stage, cells lose alternative lineage potential and are irreversibly committed to the B cell lineage.
Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that acts as a sensor of endogenous and exogenous chemicals. Upon ligand binding the AHR translocates into the nucleus and heterodimerizes with AHR nuclear translocator (ARNT) (15, 16). The ligand activated-AHR/ARNT complex functions as a transcription factor, binding dioxin responsive elements (DRE) within regulatory regions of target genes to affect gene expression (17–19).
The AHR was initially discovered in an effort to understand how 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a ubiquitous environmental contaminant, mediates biological responses (20). Thereafter, due to its high binding affinity to AHR, TCDD has been widely used as a probe to study AHR biology, including its role in the immune system (21, 22). Studies utilizing endogenous and exogenous AHR ligands, AHR antagonists, and Ahr knockout animal models have suggested physiological roles for the AHR in regulating various biological processes, including developmental, homeostatic and functional, in immunocompetent cell populations (23). Examples include a role by the AHR in the activation and proliferation of HSCs (24, 25), differentiation of Th17 cells and regulatory T cells (26, 27), maintenance of innate lymphoid cells (28), immunogenicity of dendritic cells (29), and function of mature B cells (30, 31).
Previous studies have shown that AHR activation attenuates development of B cells in mice (32, 33). Likewise, our prior studies using human CD34+ hematopoietic stem/progenitor cells (HSPC) have demonstrated an impairment of B lymphopoiesis by AHR activation (34). The underlying mechanism by which AHR regulates B lymphopoiesis remains elusive. The objective of this study was to investigate the role of AHR in B lymphopoiesis using an in vitro model of human B cell development starting from cord blood CD34+ HSPCs (35). The expression of stage specific markers confirmed that our in vitro model facilitated the B lymphopoiesis from HSPCs to pro-B cells. Activation of AHR by TCDD attenuated the generation of early-B and pro-B cells from CLPs, indicating an impairment of B lineage specification and commitment. Gene expression analysis revealed that AHR activation decreased the expression of EBF1 and PAX5, two transcription factors that are required for B lineage specification and commitment. In addition, binding of the ligand-activated AHR to putative dioxin response elements (DREs) in EBF1 promoter was demonstrated by electrophoretic mobility shift assays (EMSA) and chromatin immunoprecipitation analysis, suggesting transcriptional regulation of EBF1 by AHR.
Materials and Methods
Chemicals
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), 1-chlorodibenzo-p-dioxin (MCDD), 2,3,7-trichlorodibenzo-p-dioxin (TriCDD), and 1,2,3,4,7,8-hexachlorodibenzo-p-dioxin (HxCDD) were purchased from AccuStandard (New Haven, CT). DMSO and AHR antagonist CH223191 were purchased from Sigma Aldrich (St. Louis, MO).
CD34+ cells and in vitro cell culture
Fresh human CD34+ hematopoietic stem/progenitor cells (HSPC) isolated from cord blood from mixed donors were purchased from All Cells (Emeryville, CA). The in vitro feeder-free HSPC culture was modified based on a previous study (35). Specifically, CD34+ cells (1×104cells/well in 96-well tissue culture plates) were cultured in RPMI-1640 media (Life Technologies) supplemented with 5% human AB serum (serum from human blood type AB donors; Valley Biomedical), 100 U/ml of penicillin (Life Technologies), 100 μg/ml of streptomycin (Life Technologies), and 50 μM 2-mercaptoethanol with the addition of IL-6 (25 ng/ml; Sigma Aldrich), Flt3 ligand (25 ng/ml; Miltenyi Biotec), and stem cell factor (SCF; 25ng/ml; Miltenyi Biotec). On day 7, half of the media was replaced with fresh media containing IL7 (20 ng/ml; Miltenyi Biotec), Flt3 ligand (25 ng/ml) and SCF (25 ng/ml). After day 14, cytokine-free media was used to replace half of the media weekly.
In all cases, cells were treated with TCDD (0.01, 0.1, 1 and 10 nM) or vehicle (VH, 0.02% DMSO) only on day 0 prior to addition of cytokines. In studies using AHR antagonist CH223191, cells were treated with antagonist 30 min prior to TCDD treatment.
Human leukocyte packs and isolation of human naive B cells
Leukocyte packs were obtained from Gulf Coast Regional Laboratories (Houston, TX), diluted with HBSS (pH 7.4, Invitrogen), overlaid on Ficoll-Paque Plus density gradient (GE Healthcare, Piscataway, NJ), and centrifuged at 1300g for 25 min with low acceleration and brake rate. The peripheral blood mononuclear cells were isolated from the buffy coat post-centrifugation, washed, counted and subjected to a magnetic column-based separation that enriched CD19+CD27− naive human B cells (more than 95% purity). This negative selection was conducted using the MACS Naive human B cell isolation kits (Miltenyi Biotech, Auburn, CA) following manufacturer’s instructions.
Flow cytometric analysis
Antibodies used for flow cytometry included Alexa Fluor 488 anti-human CD34 (clone: 581), BV421 anti-human CD10 (clone: HI10a), APC anti-human CD79a (clone: HM47), and PE/Cy7 anti-human CD19 (clone: HIB19) from Biolegend (San Diego, CA). At the indicated time points, cells were harvested and washed using HBSS (pH 7.4, Invitrogen). Viable cells were identified using Live/Dead Fixable Aqua Dead Cell Stain (Invitrogen). Cell surface Fc receptors were blocked by incubating cells with human AB serum (Valley Biomedical). For cell surface staining, cells were incubated with antibodies in FACS buffer (1X HBSS containing 1% BSA and 0.1% sodium azide, pH 7.4–7.6) for 30 min and then fixed using Cytofix fixation buffer (BD Biosciences) for 10 min. For intracellular staining, fixed cells were permeabilized by incubating in Perm/Wash Buffer (BD Biosciences) for 20 min and incubated with antibodies (anti-CD79a) for 30 min. In all cases, flow cytometric analyses were performed on a FACS Canto II cell analyzer (BD Biosciences) and data were analyzed using FlowJo. For data analysis, the gating strategy was to first gate on singlets and viable cells, and then gated on lymphocytes.
PrimeFlow RNA assay
PrimeFlow RNA assay, a flow cytometry based RNA detection technology, was conducted following manufacturer’s instructions (eBioscience, San Diego, CA). Specifically, 1 × 106 cells were harvested, permeabilized followed by intracellular staining using PE anti-human CD79a (clone: HM47). Cells were then incubated with EBF1 mRNA specific target probes (VA1-19733; Affymetrix, Santa Clara, CA) for 2 hours. After incubation, cells were washed and incubated with pre-Amplification and Amplification probes for 3 h, followed by incubation with fluorophore conjugated label probes for 1 h. Cells were resuspended in FACS buffer (1X HBSS containing 1% BSA and 0.1% sodium azide, pH 7.4–7.6) and analyzed by flow cytometry.
Spanning-tree progression analysis of density-normalized events (SPADE)
The SPADE (36) algorithm was used to visualize the dynamics of B lymphopoiesis and its alteration by TCDD. SPADE enables extraction of cellular hierarchy and heterogeneity from multi-parametric single-cell cytometry data in an unsupervised manner. The algorithm contains four computational modules: (i) density-dependent down-sampling of the cytometry data; (ii) agglomerative clustering of the down-sampled data into clusters of cells; (iii) construction of a minimum spanning tree connecting the clusters into a hierarchy; and (iv) mapping of each cell in the original data to the most similar cluster in the tree (36). Specifically, we used the SPADE 3.0 application (http://pengqiu.gatech.edu/software/SPADE/, accessed Jan. 17, 2017) based on MATLAB (R2014a, The MathWorks, Inc., Natick, MA). Algorithmic parameters were set to default values with number of desired clusters set as 100.
Real-time quantitative PCR
Total RNA was isolated using the RNeasy Kit (Qiagen, Valencia, CA) and was reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The expression level of target genes was assessed by TaqMan Gene Expression Assays: AHR (Hs00907314_m1), CYP1A1 (Hs01054797_g1), EBF1 (Hs03045361_m1), PAX5 (Hs00277134_m1), ETS1 (Hs00428287_m1), TCF3 (Hs00413032_m1) and SPI1 (Hs02786711_m1). Real-time qPCR was performed on ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The relative steady-state mRNA level for target genes was calculated by normalizing to the 18S ribosomal RNA and fold change was calculated by ΔΔCT method (37).
Western blot analysis
Cells were harvested and lysed using lysis buffer that consisted of 50 mM Tris-HCl (pH 7.5), 1.5 mM MgCl2, 100mM NaCl, 1mM DTT, 1 mM Na3VO4, 25 mM NaF, 0.2% (v/v) Igepal, 5% (v/v) glycerol and protease inhibitor (Roche, Indianapolis, IN). Total protein samples were prepared and the protein concentrations were determined using the BCA assay (Sigma, St Louis, MO). Proteins were separated on 4–20% Tris-HCl gels (Bio-Rad Laboratories, Hercules, CA), transferred to nitrocellulose membranes, and probed with primary antibodies: anti-EBF1rabbit mAb (Abcam EPR4183) or anti-Actin mouse mAb (Sigma A5441), and secondary antibodies: anti-rabbit IgG (Sigma A1949) or anti-mouse IgG (Sigma A3673). The blots were incubated with ECL Western blotting substrate (Pierce, Rockford, IL) and exposed to X-ray films.
Electrophoretic mobility shift assays (EMSA) and EMSA-Western analysis
a. Nuclear Protein Preparation
Nuclear protein was isolated from HEPG2 cells as previously described (38) with a few modifications. Briefly, HEPG2 cells were treated with vehicle (0.01% DMSO) or TCDD (30 nM in DMSO) for 2h at 37°C. Cells were washed twice with 10 mM HEPES (pH 7.5), incubated at 37°C for 15 min, then harvested in MDH buffer (2 mM MgCl2, 1 mM DTT, 2mM HEPES, pH 7.5, with protease inhibitor (Roche, Indianapolis, IN)) and homogenized using a Dounce homogenizer. The homogenates were centrifuged at 1000g for 5 min, washed twice with MDHK buffer (2 mM MgCl2, 1 mM DTT, 2mM HEPES, pH 7.5, and 100 mM KCl, with protease inhibitor) and centrifuged. The crude nuclear pellets were resuspended in HEDGK4 buffer (25 mM HEPES, pH 7.5, 1 mM EDTA, 1 mM DTT, 10% (v/v) glycerol, and 400 mM KCl, 200 μM phenylmethylsulfonyl fluoride, with protease inhibitor) (50 μl per plate of confluent cell) and incubated on ice for 30 min for high-salt extraction followed by centrifugation at 14,000 × g, 4°C for 15 min. The supernatants were ultracentrifuged at 99,000 × g, 4°C for 1 h. The protein concentration in supernatants was determined using the BCA assay (Sigma, St Louis, MO).
b. DRE Oligonucleotides
The consensus DRE has been previously described (39). The oligonucleotide sequence for consensus DRE is: GGCTTGCGTGCGA. The oligonucleotide sequences for three putative DREs in the promoter region of human EBF1 gene are DRE4 (−6371): CACCTTTGCGTGCTGCG, DRE6 (−5918): TGCCCTGGCGTGACCAT and DRE7 (−5789): TAGAGCTCACGCAAGCT. Complementary pairs of DRE oligomers were synthesized and HPLC purified (Integrated DNA Technologies), followed by annealing and end labeling using T4 polynucleotide kinase (New England BioLabs) and γ-32P ATP (PerkinElmer).
c. EMSA and EMSA-Western
Nuclear extracts (10 μg of protein) were incubated with double stranded poly (dI-dC) (0.5 μg) (Sigma) for 30 min at room temperature. The 32P-labeled DRE oligomer (240,000 – 480,000 cpm) (for EMSA) or unlabeled DRE oligomer (10 pmol) (for EMSA-Western) was added and incubated for another 30 min at room temperature. The final buffer condition in the binding reaction was: 25 mM Hepes (pH7.5), 1mM EDTA, 1mM DTT, 10% glycerol, 100 mM KCl. Protein:DNA complexes were resolved on a 4% non-denaturing PAGE gel in TGE buffer (25 mM Tris, 380 mM glycine, 2 mM EDTA). The radiolabeled portion of the EMSA gel was dried on 3-mm filter paper, and autoradiographed. The non-radiolabeled portion of the EMSA gel was incubated in soaking buffer (375 mM Tris-HCl, pH 7.5, 1% SDS) for 2 h at room temperature, transferred to nitrocellulose blotting membrane (GE Healthcare Life Sciences) overnight using transfer buffer (30 mM Tris, 240 mM glycine, 20% methanol). The protein:DNA complexes on the blot were blocked in TBST buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) with 5% nonfat milk for 1 h at room temperature. The anti-human AHR purified (Clone: FF3399, eBioscience) primary antibody was then added at 1:1000 dilution and incubated for 2 h at room temperature. The blot was washed using TBST buffer and incubated with the anti-mouse IgG-HRP (Sigma A3673) antibody in TBST buffer with 5% nonfat milk for 1 h. The blot was incubated with ECL Western blotting substrate (Pierce, Rockford, IL) and exposed to X-ray films.
d. Competition EMSA
Nuclear extracts (5 μg of protein) were incubated with double stranded poly (dI-dC) (1–4 μg) (Sigma) for 15 min at room temperature. The competitor DNA, which is an un-labeled consensus DRE or mutated DRE oligonucleotide, was added at 100 fold excess, relative to the labeled DRE oligonucleotides. The oligonucleotide sequence for the mutated DRE is: GGC TTG ATG TCG AAG. After incubation for 15 min, the 32P-labeled DRE oligomers (DRE4 or DRE7, 0.12 pmol, 480,000 cpm) were added incubated for an additional 15min. The final buffer condition in the binding reaction was: 25 mM HEPES (pH7.5), 1mM EDTA, 1mM DTT, 10% glycerol, 100 mM KCl. Protein:DNA complexes were resolved on a 4% non-denaturing PAGE gel in TGE buffer (25 mM Tris, 380 mM glycine, 2 mM EDTA). The competition EMSA gel was dried on 3-mm filter paper, and visualized by autoradiograph.
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR analysis
Human CD34+ HSPCs were cultured as described above for 28 days. On day 28 cells were treated with vehicle (VH, 0.02% DMSO) or TCDD (1 nM). Three hours post treatment cells were harvested for ChIP assays using ChIP-IT High Sensitivity kit following manufacturer’s instructions (Active Motif). Specifically, cells were fixed at room temperature for 15 min followed by quenching for 5 min. The cells were washed twice with ice-cold PBS and lysed on ice. The cross-linked chromatin was sheared by sonication. Chromatin immunoprecipitation was conducted by incubating sheared chromatin (18 μg) with either anti-AHR antibody (4 μg, BML-SA210, Enzo Life Sciences) or negative control antibody (4 μg, Active Motif) (mock ChIP reaction) overnight. The AHR-containing chromatin complexes bound to anti-AHR antibody were isolation by incubating with protein G agarose beads followed by filtration. The isolated chromatin complexes were incubated with proteinase K at 55°C for 30 min and at 80°C for 2 h to reverse cross-links. Thereafter, the ChIP enriched genomic DNA was purified using DNA purification column (Active Motif).
ChIP enriched DNA was analyzed using quantitative PCR (qPCR). Primers were designed to amplify regions in EBF1 promoter that contains putative AHR binding sites (DREs). The sequence of primers were: DRE4 (−6371): ACT TCC TTC GAG GGA CAA TTT (Forward), ATC ATA CAC ATC TCG CAT CCC (Reverse); DRE6 (−5918): CTT GCG GAT GTG CTT TAA TGG (Forward), CTG TAT TCT CCC GAC TCA GAA TG (Reverse); and DRE7 (−5789): CCA CAT TTA CTA TGT GAC CTC CT (Forward), ATG GGC ATC AGG AAC ATC C (Reverse). In addition, positive control and negative control PCR primer pairs were included in analysis. The positive control primer set amplifies an AHR binding region in CYP1A1 promoter. The sequence of the primer is: CTG ACC TCT GCC CCC TAG A (Forward), GGG TGG CTA GTG CTT TGA TT (Reverse). The negative control primer set amplifies a region in a gene desert on human chromosome 12 (Human Negative Control Primer Set 1, Active Motif). A standard curve was produced using Input DNA isolated from sheared chromatin. For each primer set, SYBR Green based qPCR were conducted using ChIP DNA samples along with the dilution series of Input DNA standards. The data was expressed as a percent of input.
Statistics
Statistical analyses were performed using GraphPad Prism 5.00 (Graphpad Software, San Diego, CA). Data were graphed as mean ± SEM. Statistical comparisons were performed using t-test, one-way ANOVA with Bonferroni or Dunnett’s multiple comparison posttest, or two way ANOVA with Bonferroni posttest depending on the experimental design. Data presented as fold-change were transformed using logarithmic transformation prior to statistical analysis.
Results
An in vitro feeder cell free system facilitates early-B and pro-B cell development from human CD34+ hematopoietic stem/progenitor cells (HSPC)
The in vitro human B lymphopoiesis model was established using human cord blood-derived CD34+ HSPCs based on a previous study (35). To monitor the developmental progression for B lymphopoiesis, the expression of cell markers (CD34, CD10, CD79α and CD19) demarcating discrete B cell developmental stages was quantified by flow cytometry (40) (Fig. 1). CD34+ HSPCs (94.7% in purity) were cultured in media supplemented with specific growth factors and cytokines on day 0. After two weeks (day 14), the emergence of CD10+ cells (approximately 2%) was observed, which contained common lymphoid progenitors (CLP). On day 21, a substantial population of early-B cells (CD10+ CD79α+ CD19−) (40, 41) was derived from CLPs (approximately 60% of CD10+ cells). Ultimately, pro-B cells (CD10+ CD79α+ CD19+) emerged on day 28 (approximately 23% of CD10+ cells), demonstrating the establishment of an in vitro human B lymphopoiesis model for assessing the consequence of AHR activation on B cell development.
Figure 1. The developmental process of human CD34+ hematopoietic stem/progenitor cells (HSPC) to lineage committed B cells.

A) A schematic representation of different stages in B cell development. B) Cord blood-derived human CD34+ HSPCs were cultured for up to 28 days. Cells were harvested at indicated time points and analyzed by flow cytometry for cell markers characterizing developmental stages, including CD34, CD10, cytoplasmic CD79α (cyCD79α) and CD19. Data are representative of four independent experiments.
AHR activation by TCDD suppressed the generation of early-B and pro-B cells
To assess the effects of AHR activation on human B lymphopoiesis, human HSPCs were treated with vehicle (0.2% DMSO) or TCDD (0.01, 0.1, 1 and 10 nM) on day 0. In the vehicle (VH) treated samples, the CD10+ cell population, which includes CLP, early-B and pro-B cells, increased with time (Fig. 2). The step-wise emergence of CLP, early-B and pro-B cells from HSPC cultures demonstrated the progression of B lymphopoiesis. With TCDD treatment, the CD10+ cell population was decreased in a concentration dependent manner, which is mainly attributable to the decrease in early-B cells and pro-B cells (Fig. 2). Indeed, the percentage of early-B and pro-B cells was significantly decreased by TCDD at concentrations as low as 0.1 nM, whereas no significant change was observed in CLP population (Supplement Fig. 1). In addition, the SPADE computational tool was employed for extraction of cellular hierarchy from high-dimensional cytometry data (36) to visualize the dynamics of B lymphopoiesis and its disruption by TCDD (Fig. 3A). Cells were clustered into nodes based on the expression of cell markers CD34, CD10, CD79α and CD19. Then the nodes of cell clusters were colored according to the expression intensity of CD79α, a marker for early-B and pro-B cells. In vehicle (VH) treatment, HSPCs developed along a definite trajectory of differentiation and gradually generated early-B and pro-B cells indicated by increasing expression of CD79α. With TCDD treatment, the progression of B lymphopoiesis was delayed or even arrested in a concentration-dependent manner (Fig. 3A). Quantification of the percentage of CD79α+ cells further confirmed suppression of B lymphopoiesis by TCDD (Fig. 3B).
Figure 2. TCDD treatment decreased the percentage of the CD10+ cell population during B lymphopoiesis.
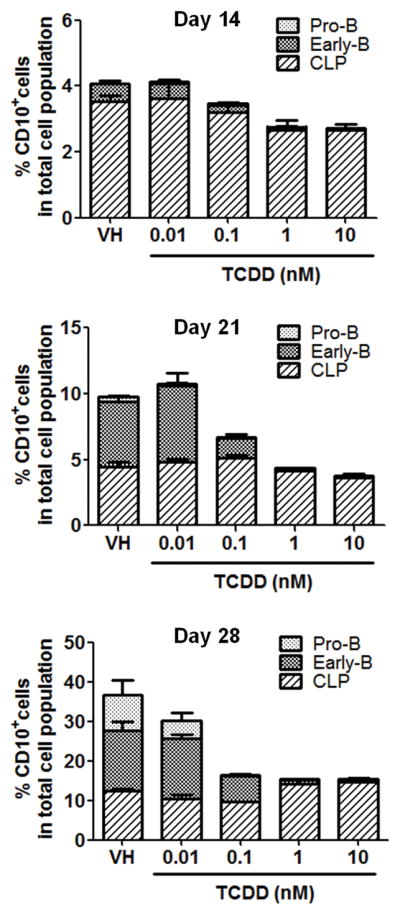
Cord blood-derived human CD34+ HSPCs were treated with vehicle (VH, 0.02% DMSO) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0 and cultured for up to 28 days. Cells were harvested on day 14, 21 and 28. The percentage of the CD10+ cell population was assessed by flow cytometry, which includes common lymphoid progenitors (CLP) (CD10+ CD79α− CD19−), early-B cells (CD10+ CD79α+ CD19−) and pro-B cells (CD10+ CD79α+ CD19+). Data are presented as mean ± SE of triplicate measurements. Data are representative of three independent experiments with similar results.
Figure 3. TCDD treatment impeded B cell development.

Human CD34+ HSPCs were treated with vehicle (VH, 0.02% DMSO) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0 and cultured for up to 28 days. A) SPADE visualization of median CD79α levels in clustered groups of cells along the HSPC to B cell developmental trajectory. Cells were clustered into nodes based on the expression of cell markers CD34, CD10, CD79α and CD19. The size of a node reflects the size of the cell population in that cluster. The nodes of cell clusters were colored according to the expression intensity of CD79α. Panels along columns represent time points, while rows represent treatments, with the first row representing vehicle treatment. Cells progressed chronologically along a lymphopoiesis trajectory with more differentiated cells (CD79α hi) towards the right of each panel. This progression is disrupted by TCDD in a concentration-dependent manner, as indicated by a depletion of the early-/pro- B cell subpopulation. B) The percentage of CD79α+ cells in the total cell population was quantified by flow cytometry. Data are presented as mean ± SE of triplicate measurements. ***p <0.001, compared to VH using two way ANOVA with Bonferroni posttest. Data are representative of three independent experiments with similar results.
AHR-mediated suppression of B lymphopoiesis by TCDD
To investigate AHR involvement in TCDD-elicited suppression of human B lymphopoiesis, the expression and functionality of AHR in hematopoietic stem and progenitor cells (HSPC) was determined. HSPCs expressed high levels of AHR mRNA, notably higher than naive peripheral blood B cells (Fig. 4A). The level of AHR mRNA in HSPCs increased modestly during the 28-day culture period, with TCDD treatment showing minimal effects (Fig. 4B). The expression of CYP1A1, a known AHR regulated gene, was markedly increased in a concentration-dependent manner in HSPCs by TCDD treatment (Fig. 4C), indicating the AHR signaling pathway to be functional in HSPCs. Next, a structure-activity-relationship experiment was conducted to confirm AHR involvement. Four polychlorinated dibenzo-p-dioxin (PCDD) congeners were used: 1-chlorodibenzo-p-dioxin (MCDD), 2,3,7-trichlorodibenzo-p-dioxin (TriCDD), 1,2,3,4,7,8-hexachlorodibenzo-p-dioxin (HxCDD) and TCDD. The rank order for AHR binding affinity is: MCDD < TriCDD < HxCDD < TCDD (20, 42). The treatment of PCDD congeners (1 nM) on HSPCs resulted in suppression of early-B and pro-B cell generation (Fig. 5). The magnitude of suppression was correlated with AHR binding affinity for respective congeners, suggesting AHR activation as a key event in TCDD-elicited suppression of B lymphopoiesis. To further confirm AHR involvement, a well-characterized AHR antagonist, CH223191, was utilized. HSPCs were treated with vehicle (0.2% DMSO), CH223191 (CH) (0.3, 1, 3, 10 μM), TCDD (1 nM) or a combination of CH and TCDD (Fig. 6). Compared to vehicle control (no CH or TCDD), 1 nM TCDD treatment significantly decreased the percentage of early-B cells that emerged from HSPCs, which was attenuated in a concentration-dependent manner by addition of the AHR antagonist (Fig. 6 and supplement Fig. 2). Therefore, the results from both the structure-activity-relationship experiments and those utilizing the AHR antagonist demonstrate AHR-mediated involvement in suppression of B lymphopoiesis by TCDD.
Figure 4. AHR expression and CYP1A1 induction in HSPCs.

A) AHR mRNA levels in human peripheral blood naive B cells and CD34+ HSPCs after 7 days of culture were determined by real-time quantitative PCR and normalized to 18s ribosomal RNA. Data are presented as mean ± SE of triplicate measurements. *p <0.05, compared to naive B cells by t-test after logarithmic transformation. B,C) Human CD34+ HSPCs were treated with vehicle (VH) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0 and cultured for up to 28 days. Cells were harvested weekly. The mRNA levels of AHR (B) and CYP1A1 (C) were determined by real-time quantitative PCR and were normalized to 18s ribosomal RNA. Data are presented as mean ± SE for triplicate measurements. * p <0.05, **p <0.01, ***p <0.001, compared to the VH using a two way ANOVA with Bonferroni posttest after logarithmic transformation. Data are representative of two independent experiments with similar results.
Figure 5. Structure-activity-relationship assay.

Human CD34+ HSPCs were treated with vehicle (VH) or with 1nM of a chlorinated dioxin congener (MCDD, TriCDD, HxCDD or TCDD) on day 0. The rank order for AHR binding affinity is: MCDD < TriCDD < HxCDD < TCDD. Cells were harvested on day 21 and analyzed by flow cytometry. Cells were first gated on CD10+. A) The gating scheme of early-B cells (CD10+ CD79α+ CD19−) and pro-B cells (CD10+ CD79α+ CD19+). B) Quantification of early-B and pro-B cell percentages in (A). Data are mean ± SE of triplicate measurements. * p <0.05, ***p <0.001, compared to VH using a one way ANOVA with Dunnett’s multiple comparison test. Data are representative of three independent experiments with similar results.
Figure 6. AHR antagonist reversed the TCDD-mediated decrease in CD10+ cell population.
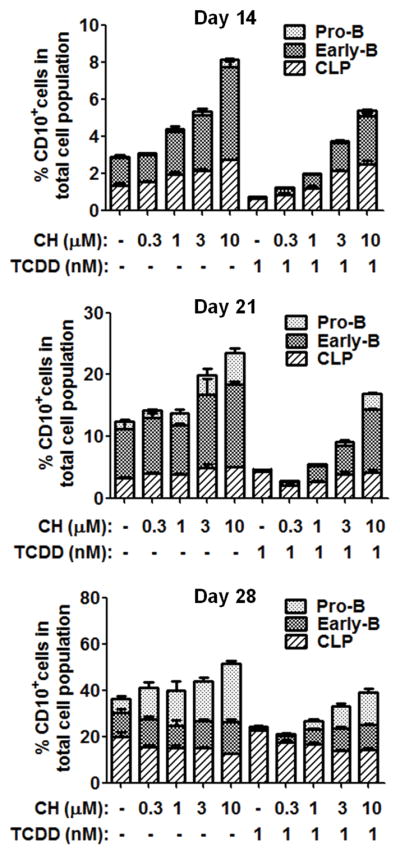
Human CD34+ HSPCs were treated on day 0 with vehicle (0.02% DMSO), AHR antagonist CH223191 (CH) (0.3, 1, 3 or 10 μM), TCDD (1 nM) or the combination of CH and TCDD. Cells were cultured for up to 28 days and harvested at the specified time points. The percentage of CD10+ cell population was assessed by flow cytometry, which includes common lymphoid progenitors (CLP) (CD10+ CD79α − CD19−), early-B cells (CD10+ CD79α+ CD19−) and pro-B cells (CD10+ CD79α+ CD19+). Data are presented as mean ± SE of triplicate measurements. Data are representative of two independent experiments with similar results.
EBF1 and PAX5 expression in HSPCs were reduced by AHR activation
As shown in Fig. 2 and supplement Fig. 1, AHR activation by TCDD suppressed the generation of early-B and pro-B cells but not their preceding progenitor CLPs, suggesting that AHR activation impedes B cell lineage specification and commitment. Two important transcription factors governing B cell lineage specification and commitment are EBF1 and PAX5. EBF1 is a critical regulator in driving the B cell specification program (9, 10). EBF1 activates the expression of PAX5. Together, EBF1 and PAX5 direct cell fate choice during lymphopoiesis leading to B cell lineage commitment (12–14). Gene expression analysis revealed that EBF1 mRNA levels were increased over time as cells progressed toward B cell lineage (Fig. 7A, VH groups); however, this up-regulation of EBF1 was suppressed in the presence of TCDD (Fig. 7A). By employing PrimeFlow, we quantified the EBF1 mRNA levels in individual cells by flow cytometry. First, the intracellular protein levels of CD79α and mRNA levels of EBF1 were measured simultaneously (Fig. 7B). The co-expression of EBF1 mRNA and cytoplasmic CD79α protein suggests that the expression of EBF1 is B cell lineage specific. Then, an extended concentration response using TCDD was performed. TCDD treatment decreased the percentage of EBF1-expressing cells (Fig. 7C,D); however, the average expression level of EBF1 mRNA in the EBF1-expressing cells, as represented by MFI, was not decreased (Fig. 7E), demonstrating an all-or-none (binary) decrease in EBF1 mRNA levels by TCDD treatment. The decrease in EBF1 at the protein level was also demonstrated by Western blotting (Fig. 7F,G). Given that EBF1 regulates PAX5, the effect of TCDD on PAX5 expression was also investigated. Consistent with EBF1, PAX5 mRNA increased over time, but was suppressed by TCDD treatment (Fig. 8).
Figure 7. TCDD decreased EBF1 expression.

Human CD34+ HSPCs were treated with vehicle (VH) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0. Cells were cultured for up to 28 days and harvested at the specified time points. A) The mRNA levels of EBF1 were determined by real-time quantitative PCR and were normalized to 18s ribosomal RNA. Data are presented as mean ± SE of triplicate measurements. * p <0.05, ***p <0.001, compared to the VH using a two way ANOVA with Bonferroni posttest after logarithmic transformation. B) The PrimeFlow staining of EBF1 mRNA and intracellular CD79α on day 24. C) The PrimeFlow staining of EBF1 mRNA on day 28 post treatment. D) The percentage of the EBF1 mRNA expressing cells as gated in C. E) The mean fluorescence intensity (MFI) of EBF1 mRNA in EBF1 mRNA expressing cells as gated in C. Data are presented as mean ± SE of triplicate measurements. ***p <0.001, compared to VH by one way ANOVA using a Dunnett’s multiple comparison test. F) EBF1 immunoblot on day 24 post treatment by VH or TCDD (30nM). G) Quantification of EBF1 protein levels relative to actin in (F). Data are representative of three independent experiments with similar results.
Figure 8. TCDD decreased PAX5 expression.

Human CD34+ HSPCs were treated with vehicle (VH) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0. Cells were cultured for up to 28 days and harvested at the specified time points. The mRNA levels of PAX5 were assayed by real-time quantitative PCR and were normalized to 18s ribosomal RNA. Data are presented as mean ± SE of triplicate measurements. * p <0.05, **p <0.01, ***p <0.001, compared to the VH using a two way ANOVA with Bonferroni posttest after logarithmic transformation. Data are representative of three independent experiments with similar results.
To explore the involvement of AHR in TCDD-elicited suppression of EBF1 and PAX5, an AHR antagonist was used. HSPCs were treated with vehicle (0.2% DMSO), CH223191 (CH) (0.3, 1, 3, 10 μM), TCDD (1 nM) or in combination with CH and TCDD. Compared to vehicle control (no CH or TCDD), 1 nM TCDD treatment significantly decreased EBF1 mRNA; however, the decrease was reversed in a concentration-dependent manner with the addition of increasing amounts of the AHR antagonist (Fig. 9A). Likewise, similar effects were observed for PAX5 mRNA (Fig. 9B). These findings suggest that AHR activation by TCDD leads to suppression of EBF1 and PAX5.
Figure 9. AHR antagonist reversed the TCDD-elicited suppression of EBF1 and PAX5 expression.

Human CD34+ HSPCs were treated with vehicle, AHR antagonist CH223191 (CH) (0.3, 1, 3 or 10 μM), TCDD (1 nM) or a combination of CH and TCDD on day 0. Cells were cultured for up to 28 days and harvested at the specified time points. The mRNA levels of EBF1 (A) and PAX5 (B) were determined by real-time quantitative PCR and were normalized to 18s ribosomal RNA. Data are presented as the mean ± SE of triplicate measurements. a = significantly different compared to vehicle control, b = significantly different compared to the TCDD (1 nM) treated group, by two way ANOVA with Bonferroni posttest after logarithmic transformation. Data are representative of two independent experiments with similar results.
During B lymphopoiesis, the initiation of the EBF1-PAX5 regulatory axis involves multiple transcription factors that activate EBF1 expression, including ETS1, TCF3 and SPI1 (5). To explore the possibility that the suppression of EBF1 and PAX5 by AHR activation results from the alterations of upstream regulators, we examined the expression of ETS1, TCF3 and SPI1 in the presence of TCDD (Fig. 10). The time course and concentration response study showed a modest suppression of ETS1 and TCF3 and enhancement of SPI1 by TCDD treatment, suggesting that effects on upstream regulators may also contribute, in part, to the suppression of EBF1 expression.
Figure 10. The effects of TCDD on the expression of transcription factors that regulate EBF1.

Human CD34+ HSPCs were treated with vehicle (VH) or TCDD (0.01, 0.1, 1 or 10 nM) on day 0. Cells were cultured for up to 28 days and harvested weekly. The mRNA levels of SPI1 (A), TCF3 (B) and ETS1 (C) were determined by real-time quantitative PCR and were normalized to 18s ribosomal RNA. Data are presented as the mean ± SE of triplicate measurements. * p <0.05, **p <0.01, ***p <0.001, compared to the VH using a two way ANOVA with Bonferroni posttest after logarithmic transformation. Data are representative of three independent experiments with similar results.
Ligand-activated AHR binds to DREs in EBF1 promoter
EBF1 has been identified as a putative target for AHR in a genome-wide ChIP-on-chip and gene expression microarray study in a murine cell line (43). In addition, we have identified 13 putative AHR binding sites (DRE) in the human EBF1 promoter region (Fig. 11A). The matrix similarity scores (MSS) of these DREs were calculated based on the position weight matrix (39). All 13 DREs in the EBF1 promoter have a MSS higher than the threshold score based on experimentally confirmed DREs (39). To further explore whether AHR transcriptionally regulates EBF1 expression by direct binding within the EBF1 promoter, we selected 3 DREs (DRE 4, 6 and 7) with the highest MSS to assess AHR binding using electrophoretic mobility shift assays (EMSA) (Fig. 11B) and by EMSA-Western analysis (Fig. 11C). Briefly, nuclear protein was isolated from cells treated with vehicle (0.02% DMSO) or TCDD (30 nM), incubated with 32P-labeled (Fig. 11B) or unlabeled (Fig. 11C) DRE oligomers. The nuclear protein-DNA complex was resolved on a non-denaturing PAGE gel. TCDD-induced protein binding to DRE 4 and 7 was observed (Fig. 11B). In addition, the protein-DNA complex for DRE4 and 7 located at the same position as consensus DRE, indicating the binding of AHR to DRE4 and 7. Protein binding to DRE6, as evaluated by EMSA, appeared to be independent of TCDD treatment and located at a different position as compared to DRE4 and 7, which might be due to the binding of nuclear proteins besides AHR to the DNA oligomers. To confirm the presence of AHR, the protein-DNA complex was transferred to a nitrocellulose membrane and probed with anti-AHR antibody (Fig. 11C). Consistent with the EMSA results, AHR was detected in the TCDD-induced protein-DNA complex for all three DREs (Fig. 11C). In addition, competition EMSA were performed to demonstrate binding specificity (Fig. 11D). Nuclear extracts were incubated with 32P-labeled DRE4 or DRE7 with addition of unlabeled (cold) consensus DRE or mutated DRE oligonucleotide at 100 fold excess, relative to the labeled DREs. The TCDD-induced DNA-protein binding intensity was decreased by addition of unlabeled consensus DRE oligonucleotide; however, the same magnitude of decrease was not observed by the addition of cold mutated DRE (Fig. 11D), further demonstrating binding specificity of nuclear proteins to DRE4 and 7. Moreover, to confirm that ligand-activated AHR binding to DREs in EBF1 promoter can occur in HSPCs, chromatin immunoprecipitation (ChIP) analysis was conducted. CD34+ HSPCs were cultured for 28 days and treated with vehicle (VH, 0.02% DMSO) or TCDD (1 nM). ChIP reactions were performed using either anti-AHR antibody or negative control antibody (mock). DNA primers specific to DREs in EBF1 promoter were used to quantify the enrichment of AHR-bound chromatin using q-PCR. TCDD-induced enrichment at DRE4, 6 and 7 in EBF1 promoter indicated AHR binding to these loci in HSPCs (Fig. 11E). The consistent findings via EMSA, EMSA Western and ChIP demonstrated that TCDD-activated AHR is able to bind to DREs in the human EBF1 promoter. Together with the aforementioned findings of AHR-mediated decrease in EBF1 mRNA levels, our study suggests direct transcriptional regulation of EBF1 by AHR activation.
Figure 11. DNA binding analysis of the ligand-activated AHR to putative dioxin response elements (DRE) within the EBF1 promoter.

A) Schematic of predicted DRE sites in human EBF1 gene promoter region. B, C, D) Nuclear protein was isolated from HEPG2 cells treated with vehicle (V, 0.01% DMSO) or TCDD (T, 30nM). B) Nuclear protein was incubated with 32P-labeled DRE oligomers. Protein:DNA complexes were resolved on a 4% nondenaturing PAGE gel, dried and visualized by autoradiograph. C) Nuclear protein was incubated with unlabeled DRE oligomers. Protein:DNA complexes were resolved on a 4% nondenaturing PAGE gel, transferred to nitrocellulose, and probed with anti-AHR antibody. Arrow indicates specific binding of AHR to DRE oligomers. D) Nuclear protein was incubated with 32P-labeled DRE oligomers (DRE4 or DRE7), with addition of excess unlabeled consensus or mutated DRE oligomer relative to the labeled DREs. Protein:DNA complexes were resolved on a 4% nondenaturing PAGE gel, dried and visualized by autoradiograph. Results are representative of three independent experiments. E) Human CD34+ HSPCs were cultured as described in the Materials and Methods for 28 days. On day 28, cells were treated with vehicle (VH, 0.02% DMSO) or TCDD (1 nM). Three hours post treatment cells were harvested for chromatin immunoprecipitation (ChIP) analysis. ChIP reactions were performed using either anti-AHR antibody or negative control antibody (mock). DNA primers specific to DRE 4, 6 and 7 in EBF1 promoter were used to quantify the enrichment of AHR-bound chromatin using q-PCR. A positive control primer set that amplifies a AHR binding region in CYP1A1 promoter as well as a negative control primer set that amplifies a region in a gene desert on human chromosome 12 were also included. * p <0.05, **p <0.01, ***p <0.001, compared to VH by one-tailed t-test.
Discussion
In this study, we employed a feeder-free culture system that facilitated in vitro human B lymphopoiesis starting from cord blood-derived CD34+ cells. Activation of AHR using a high affinity agonist suppressed the generation of early-B and pro-B cells but not their preceding progenitor CLPs (Fig. 2 and supplement Fig. 1), suggesting an arrest during B cell lineage specification and commitment. This interruption of development was visualized using the SPADE algorithm (Fig. 3), showing that cells failed to progress along the development trajectory to become early-B and pro-B cells. With the addition of AHR antagonist, the arrest of B cell development was ablated. Interestingly, HSPCs treated with AHR antagonist alone showed an accelerated progression toward B cell lineage (Fig. 6 and supplement Fig. 2), suggesting a physiological role of AHR in regulating B lymphopoiesis through endogenous AHR activation. A growing list of endogenous AHR ligands has been identified which include indigoids, equilenin, tryptophan metabolites, arachidonic acid metabolites and heme metabolites (44). As a growing body of evidence suggests that endogenous AHR activation modulates immune responses (45, 46), our study provides new insights into the physiological role of AHR in human B lymphopoiesis.
Given the crucial role of EBF1 in the regulatory circuit underlying B cell lineage specification and commitment, we hypothesize that AHR-mediated suppression of EBF1 is involved in the mechanism by which AHR activation suppresses human B lymphopoiesis. The decrease in EBF1 mRNA levels by TCDD during B lymphopoiesis was demonstrated by both qPCR and PrimeFlow analyses. Interestingly, TCDD decreased EBF1 mRNA levels in an all-or-none (binary) rather than graded mode: i.e., TCDD reduces the proportion of EBF1 mRNA expressing cells in a concentration-dependent manner (Fig. 7C, D) rather than decreasing the EBF1 mRNA levels in cells that express EBF1 mRNA (Fig. 7E). This binary switch-like behavior of EBF1 expression is not unexpected, as the positive feedback loop between EBF1 and PAX5 forms a bistable memory module, which is a characteristic regulatory motif that governs cell fate decisions during cellular development (47, 48).
Within the B lymphopoiesis regulatory circuit, the expression of PAX5 is activated by EBF1 (11), which is consistent with our observation that the up-regulation of EBF1 mRNA precedes that of PAX5 during B cell development (Fig. 7A, Fig. 8 and Fig 9, VH groups). In our study, AHR activation suppressed both EBF1 and PAX5. Given that EBF1 and PAX5 act in a hierarchical manner, we speculate the suppression of PAX5 is a consequence of AHR-mediated suppression of EBF1. To explore whether the down-regulation of EBF1 results from AHR-mediated alterations of upstream transcription factors, we examined the expression of ETS1, TCF3 and SPI1, which are known to initiate EBF1 expression during B lymphopoiesis (5). Unlike the responses of EBF1 to AHR activation (Fig. 7A), all three transcription factors exhibited modest changes that occurred only in the late stage of the 28 day culture period (Fig. 10). Hence, we hypothesize that AHR can exert direct transcriptional regulation of EBF1.
It is established that AHR is a ligand-activated transcription factor, which regulates gene expression by binding to DREs in regulatory regions of target genes. Here we demonstrate the binding of the ligand-activated AHR to three putative DREs within the EBF1 promoter using EMSA and EMSA-Western analysis. EMSA has high sensitivity but falls short in specificity. Bioinformatic predictions suggest that there are multiple putative binding sites for transcription factors besides the AHR, even within the short 17-bp oligonucleotide probe we used in this assay. Indeed, the failure to detect TCDD-induced protein binding to DRE6 is likely the result of other nuclear proteins binding to DRE6 in addition to AHR, which masked visualization of AHR-DRE binding by EMSA. To increase the specificity of detecting AHR-DRE binding, we conducted EMSA-Western analysis. The advantage of the EMSA-Western analysis is the ability to identify specific DNA binding proteins using antibodies, which in this case were used to identify the AHR. Due to the specificity of the EMSA-Western assay, AHR-DRE6 binding was detected, which was otherwise masked in EMSA analysis by the binding of other proteins to the DNA probe. Consistent with EMSA and EMSA-Western assays, the ChIP analysis in HSPCs also demonstrated that the ligand-activated AHR is capable of binding to DREs in EBF1 promoter, suggesting that AHR transcriptionally impairs EBF1 expression.
AHR exhibits diverse mechanisms of action in regulating gene expression. In addition to the classic pathway of dimerizing with ARNT to influence gene expression, AHR has also been reported to interact with a variety of coactivators and corepressors (49). The potential transcriptional impairment of EBF1 by AHR might result from interactions with corepressors. Alternatively, AHR binding to promoter regions of EBF1 might directly interfere with the ability of other transcription factors to bind to their cognate DNA sequences, leading to the disruption of EBF1 transcription (50).
In this study, we employed a feeder-free culture system to demonstrate that the activation of AHR by TCDD impairs human B lymphopoiesis. In a prior study (34), we also observed decreased B lymphopoiesis with TCDD treatment using culture systems containing various stromal cell components; however, the magnitude of suppression was not as profound as in the current study. Differences in sensitivity to TCDD using the different culture systems are not surprising and can likely be attributed to several factors. Because TCDD is highly lipophilic, one likely contributing factor is that in addition to binding CD34+ cells, a portion of the TCDD is also bound to stromal cells therefore reducing the overall TCDD exposure of HSPCs. A second likely contributing factor is that stromal cells or soluble cytokines produced by stromal cells promote B lymphopoiesis and therefore attenuated the suppressive effect of TCDD. Since the influence of stromal cells and there secretory factors are poorly defined, here we employed a feeder-free culture system to explore the direct effects of AHR activation on human B lymphopoiesis and the underlying mechanism.
To study the role of AHR, we used a high affinity AHR agonist, TCDD. TCDD is a ubiquitous environment contaminant, which exhibits a wide spectrum of toxicity including immunotoxicity (51, 52). TCDD has been shown to suppress humoral immunity by disrupting the function of human mature B cells (31, 53, 54). Here the reported impairment of B cell development may represent an additional mechanism of immune suppression by TCDD. Due to the relatively short life span of circulating B cells, the homeostasis of the peripheral B cell population requires lifelong replenishment through B lymphopoiesis. As a result, suppression of B lymphopoiesis by TCDD could impair the competency of humoral immunity. In addition, TCDD exposure has been epidemiologically associated with increased incidence of non-Hodgkin’s lymphoma and multiple myeloma (55–59). As the disruption of EBF1 and PAX5 has been frequently implicated in human leukemia (60, 61), the TCDD-elicited decrease of EBF1 and PAX5, as reported in this study, might contribute to the carcinogenic effect of TCDD.
In summary, we show that AHR activation suppresses B cell lineage specification and commitment, which is attenuated by AHR antagonist, suggesting a physiological role of AHR in regulating human B lymphopoiesis. The AHR-mediated suppression of EBF1, together with the binding of ligand-activated AHR to DREs in EBF1 promoter, suggests that transcriptional regulation of EBF1 is involved in the mechanism by which AHR regulates B lymphopoiesis.
Supplementary Material
Acknowledgments
We would like to acknowledge helpful advice provided by Peng Qiu for SPADE analysis of flow cytometry data. We also thank Ms. Kimberly Hambleton for assistance with submission of this manuscript.
This work was supported by NIH ES002520 and NIH P42004911.
Abbreviations used in this paper
- AHR
Aryl hydrocarbon receptor
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem/progenitor cell
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- DRE
dioxin response element
- EMSA
electrophoretic mobility shift assays
- CLP
common lymphoid progenitors
- EBF1
early B cell factor 1
- PAX5
paired box 5
- ARNT
AHR nuclear translocator
- PCDD
polychlorinated dibenzo-p-dioxin
- MCDD
1-chlorodibenzo-p-dioxin
- TriCDD
2,3,7-trichlorodibenzo-p-dioxin
- HxCDD
1,2,3,4,7,8-hexachlorodibenzo-p-dioxin
- SPADE
spanning-tree progression analysis of density-normalized events
- MMS
matrix similarity scores
- ChIP
chromatin immunoprecipitation
References
- 1.Macallan DC, Wallace DL, Zhang Y, Ghattas H, Asquith B, de Lara C, Worth A, Panayiotakopoulos G, Griffin GE, Tough DF, Beverley PC. B-cell kinetics in humans: rapid turnover of peripheral blood memory cells. Blood. 2005;105:3633–3640. doi: 10.1182/blood-2004-09-3740. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida T, Ng SYM, Zuniga-Pflucker JC, Georgopoulos K. Early hematopoietic lineage restrictions directed by Ikaros. Nature immunology. 2006;7:382–391. doi: 10.1038/ni1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU. 1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid–myeloid progenitors. Immunity. 1997;6:437–447. doi: 10.1016/s1074-7613(00)80287-3. [DOI] [PubMed] [Google Scholar]
- 4.Dias S, Månsson R, Gurbuxani S, Sigvardsson M, Kee BL. E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity. 2008;29:217–227. doi: 10.1016/j.immuni.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roessler S, Gyory I, Imhof S, Spivakov M, Williams RR, Busslinger M, Fisher AG, Grosschedl R. Distinct promoters mediate the regulation of Ebf1 gene expression by interleukin-7 and Pax5. Mol Cell Biol. 2007;27:579–594. doi: 10.1128/MCB.01192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin H, Grosschedl R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature. 1995;376:263–267. doi: 10.1038/376263a0. [DOI] [PubMed] [Google Scholar]
- 7.Medina KL, Pongubala JM, Reddy KL, Lancki DW, Dekoter R, Kieslinger M, Grosschedl R, Singh H. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–617. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Reynaud D, I, Demarco A, Reddy KL, Schjerven H, Bertolino E, Chen Z, Smale ST, Winandy S, Singh H. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nature immunology. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seet CS, Brumbaugh RL, Kee BL. Early B cell factor promotes B lymphopoiesis with reduced interleukin 7 responsiveness in the absence of E2A. J Exp Med. 2004;199:1689–1700. doi: 10.1084/jem.20032202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Cotta CV, Stephan RP, deGuzman CG, Klug CA. Enforced expression of EBF in hematopoietic stem cells restricts lymphopoiesis to the B cell lineage. Embo J. 2003;22:4759–4769. doi: 10.1093/emboj/cdg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zandi S, Mansson R, Tsapogas P, Zetterblad J, Bryder D, Sigvardsson M. EBF1 is essential for B-lineage priming and establishment of a transcription factor network in common lymphoid progenitors. J Immunol. 2008;181:3364–3372. doi: 10.4049/jimmunol.181.5.3364. [DOI] [PubMed] [Google Scholar]
- 12.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 13.Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, Diefenbach A, Grosschedl R. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nature immunology. 2013;14:867–875. doi: 10.1038/ni.2641. [DOI] [PubMed] [Google Scholar]
- 14.Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27:49–63. doi: 10.1016/j.immuni.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Probst MP, Reisz-Porszasz S, Agbunag RV, Ong MS, Hankinson O. Role of the Aryl Hydrocarbon Receptor Nuclear Translocator Protein in Aryl Hydrocarbon (Dioxin) Receptor Action. Mol Pharmacol. 1993;44:511–518. [PubMed] [Google Scholar]
- 16.Pollenz RS, Sattler CA, Poland A. The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence microscopy. Molecular pharmacology. 1994;45:428–438. [PubMed] [Google Scholar]
- 17.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicological sciences : an official journal of the Society of Toxicology. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah Receptor Nuclear Translocator Protein (ARNT) as a Component of the DNA Binding Form of the Ah Receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 19.Watson AJ, Hankinson O. Dioxin and Ah receptor-dependent protein binding to xenobiotic responsive elements and G-rich DNA studied by in vivo footprinting. J Biol Chem. 1992;266:6874–6878. [PubMed] [Google Scholar]
- 20.Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. The Journal of biological chemistry. 1976;251:4936–4946. [PubMed] [Google Scholar]
- 21.Sherr DH, Monti S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Seminars in immunopathology. 2013;35:705–716. doi: 10.1007/s00281-013-0390-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacological reviews. 2013;65:1148–1161. doi: 10.1124/pr.113.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanieh H. Toward understanding the role of aryl hydrocarbon receptor in the immune system: current progress and future trends. BioMed research international. 2014;2014:520763. doi: 10.1155/2014/520763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh KP, Garrett RW, Casado FL, Gasiewicz TA. Aryl hydrocarbon receptor-null allele mice have hematopoietic stem/progenitor cells with abnormal characteristics and functions. Stem cells and development. 2011;20:769–784. doi: 10.1089/scd.2010.0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, Schultz PG, Cooke MP. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 27.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 28.Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19961–19966. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sulentic CE, Kaminski NE. The long winding road toward understanding the molecular mechanisms for B-cell suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicological sciences : an official journal of the Society of Toxicology. 2011;120(Suppl 1):S171–191. doi: 10.1093/toxsci/kfq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu H, Crawford RB, Suarez-Martinez JE, Kaplan BL, Kaminski NE. Induction of the aryl hydrocarbon receptor-responsive genes and modulation of the immunoglobulin M response by 2,3,7,8-tetrachlorodibenzo-p-dioxin in primary human B cells. Toxicological sciences : an official journal of the Society of Toxicology. 2010;118:86–97. doi: 10.1093/toxsci/kfq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh KP, Wyman A, Casado FL, Garrett RW, Gasiewicz TA. Treatment of mice with the Ah receptor agonist and human carcinogen dioxin results in altered numbers and function of hematopoietic stem cells. Carcinogenesis. 2009;30:11–19. doi: 10.1093/carcin/bgn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahrenhoerster LS, Tate ER, Lakatos PA, Wang X, Laiosa MD. Developmental exposure to 2,3,7,8 tetrachlorodibenzo-p-dioxin attenuates capacity of hematopoietic stem cells to undergo lymphocyte differentiation. Toxicology and applied pharmacology. 2014;277:172–182. doi: 10.1016/j.taap.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Phadnis-Moghe AS, Crawford RB, Kaminski NE. Aryl hydrocarbon receptor activation by 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs human B lymphopoiesis. Toxicology. 2017;378:17–24. doi: 10.1016/j.tox.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraus H, Kaiser S, Aumann K, Bonelt P, Salzer U, Vestweber D, Erlacher M, Kunze M, Burger M, Pieper K, Sic H, Rolink A, Eibel H, Rizzi M. A feeder-free differentiation system identifies autonomously proliferating B cell precursors in human bone marrow. J Immunol. 2014;192:1044–1054. doi: 10.4049/jimmunol.1301815. [DOI] [PubMed] [Google Scholar]
- 36.Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Denison MS, Rogers JM, Rushing SR, Jones CL, Tetangco SC, Heath-Pagliuso S. Analysis of the aryl hydrocarbon receptor (AhR) signal transduction pathway. Current protocols in toxicology Chapter. 2002;4(Unit4 8) doi: 10.1002/0471140856.tx0408s11. [DOI] [PubMed] [Google Scholar]
- 39.Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic acids research. 2004;32:4512–4523. doi: 10.1093/nar/gkh782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LeBien TW. Fates of human B-cell precursors. Blood. 2000;96:9–23. [PubMed] [Google Scholar]
- 41.Rose RC, Nahrwold DL. Electrolyte transport by gallbladders of rabbit and guinea pig: effect of amphotericin B and evidence of rheogenic Na transport. The Journal of membrane biology. 1976;29:1–22. doi: 10.1007/BF01868949. [DOI] [PubMed] [Google Scholar]
- 42.Sulentic CE, Holsapple MP, Kaminski NE. Putative link between transcriptional regulation of IgM expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin and the aryl hydrocarbon receptor/dioxin-responsive enhancer signaling pathway. The Journal of pharmacology and experimental therapeutics. 2000;295:705–716. [PubMed] [Google Scholar]
- 43.De Abrew KN, Kaminski NE, Thomas RS. An Integrated Genomic Analysis of Aryl Hydrocarbon Receptor-Mediated Inhibition of B-Cell Differentiation. Toxicological sciences : an official journal of the Society of Toxicology. 2010;118:454–469. doi: 10.1093/toxsci/kfq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen LP, Bradfield CA. The Search for Endogenous Activators of the Aryl Hydrocarbon Receptor. Chemical research in toxicology. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, Iannitti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, Lanz TV, Platten M, Della Fazia MA, Piobbico D, Zelante T, Funakoshi H, Nakamura T, Gilot D, Denison MS, Guillemin GJ, DuHadaway JB, Prendergast GC, Metz R, Geffard M, Boon L, Pirro M, Iorio A, Veyret B, Romani L, Grohmann U, Fallarino F, Puccetti P. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511:184–190. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiNatale BC, I, Murray A, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ, Perdew GH. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicological sciences : an official journal of the Society of Toxicology. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Kline DE, Bhattacharya S, Crawford RB, Conolly RB, Thomas RS, Andersen ME, Kaminski NE. All-or-none suppression of B cell terminal differentiation by environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and applied pharmacology. 2013;268:17–26. doi: 10.1016/j.taap.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong W, Ferrell JE., Jr A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen TA, Hoivik D, Lee JE, Safe S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Archives of biochemistry and biophysics. 1999;367:250–257. doi: 10.1006/abbi.1999.1282. [DOI] [PubMed] [Google Scholar]
- 50.Masten SA, Shiverick KT. The Ah receptor recognizes DNA binding sites for the B cell transcription factor, BSAP: a possible mechanism for dioxin-mediated alteration of CD19 gene expression in human B lymphocytes. Biochem Biophys Res Commun. 1995;212:27–34. doi: 10.1006/bbrc.1995.1931. [DOI] [PubMed] [Google Scholar]
- 51.Peterson RE, Theobald HM, Kimmel GL. Developmental and reproductive toxicity of dioxins and related compounds: cross-species comparisons. Critical reviews in toxicology. 1993;23:283–335. doi: 10.3109/10408449309105013. [DOI] [PubMed] [Google Scholar]
- 52.Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annual review of pharmacology and toxicology. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 53.Wood SC, Holsapple MP. Direct suppression of superantigen-induced IgM secretion in human lymphocytes by 2,3,7,8-TCDD. Toxicology and applied pharmacology. 1993;122:308–313. doi: 10.1006/taap.1993.1200. [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Crawford RB, Kaplan BL, Kaminski NE. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated disruption of the CD40 ligand-induced activation of primary human B cells. Toxicology and applied pharmacology. 2011;255:251–260. doi: 10.1016/j.taap.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kramarova E, Kogevinas M, Anh CT, Cau HD, Dai LC, Stellman SD, Parkin DM. Exposure to Agent Orange and occurrence of soft-tissue sarcomas or non-Hodgkin lymphomas: an ongoing study in Vietnam. Environmental health perspectives. 1998;106(Suppl 2):671–678. doi: 10.1289/ehp.106-1533419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Becher H, Flesch-Janys D, Kauppinen T, Kogevinas M, Steindorf K, Manz A, Wahrendorf J. Cancer mortality in German male workers exposed to phenoxy herbicides and dioxins. Cancer causes & control : CCC. 1996;7:312–321. doi: 10.1007/BF00052936. [DOI] [PubMed] [Google Scholar]
- 57.Kogevinas M, Becher H, Benn T, Bertazzi PA, Boffetta P, Bueno-de-Mesquita HB, Coggon D, Colin D, Flesch-Janys D, Fingerhut M, Green L, Kauppinen T, Littorin M, Lynge E, Mathews JD, Neuberger M, Pearce N, Saracci R. Cancer mortality in workers exposed to phenoxy herbicides, chlorophenols, and dioxins. An expanded and updated international cohort study. American journal of epidemiology. 1997;145:1061–1075. doi: 10.1093/oxfordjournals.aje.a009069. [DOI] [PubMed] [Google Scholar]
- 58.Floret N, Mauny F, Challier B, Arveux P, Cahn JY, Viel JF. Dioxin emissions from a solid waste incinerator and risk of non-Hodgkin lymphoma. Epidemiology. 2003;14:392–398. doi: 10.1097/01.ede.0000072107.90304.01. [DOI] [PubMed] [Google Scholar]
- 59.Viel JF, Daniau C, Goria S, Fabre P, de Crouy-Chanel P, Sauleau EA, Empereur-Bissonnet P. Risk for non Hodgkin’s lymphoma in the vicinity of French municipal solid waste incinerators. Environmental health : a global access science source. 2008;7:51. doi: 10.1186/1476-069X-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 61.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–1266. doi: 10.1038/sj.leu.2404691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.