

Summary
During development, chromatin-modifying enzymes regulate both the timely establishment of cell type-specific gene programs and the coordinated repression of alternative cell fates. To dissect the role of one such enzyme - the intellectual disability-linked lysine demethylase 5C gene (Kdm5c) – in the developing and adult brain, we conducted parallel behavioral, transcriptomic and epigenomic studies in Kdm5c null and forebrain-restricted inducible knockout mice. Together, genomic analyses and functional assays demonstrate that Kdm5c plays a critical role as a repressor responsible for the developmental silencing of germ line genes during cellular differentiation, and in fine-tuning activity-regulated enhancers during neuronal maturation. Although the importance of these functions declines after birth, Kdm5c retains an important genome surveillance role preventing the incorrect activation of non-neuronal and cryptic promoters in adult neurons.
eTOC Blurb
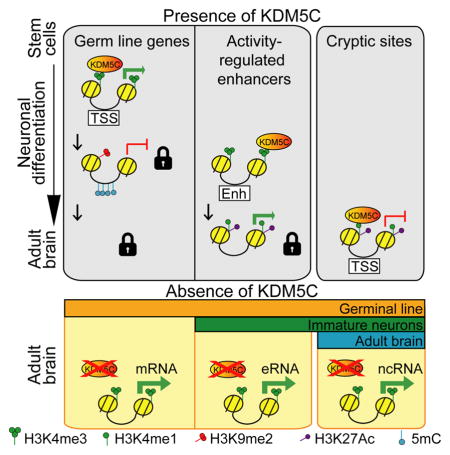
Scandaglia et al. show that Kdm5c plays critical roles constraining transcription during neuronal differentiation and maturation. Although Kdm5c contribution to neuronal transcription regulation later declines, it retains a genome surveillance role precluding spurious transcription in adult neurons. These functions likely contribute to the pathoetiology of Claes-Jensen type X-linked intellectual disability.
Introduction
The development of the nervous system is a highly organized process that requires precise spatial and temporal regulation of gene programs involved in differentiation, maturation and survival of neurons, but also the repression of alternative cell fates and restriction of cell type-specific gene expression (Lilja et al., 2013). Such dynamic expression patterns are sustained by extensive changes in the epigenome, and consequently, mutations of genes encoding chromatin-modifying enzymes can lead to severe neurodevelopmental disorders (Bjornsson, 2015; Kleefstra et al., 2014). In particular, mutations in the X-linked gene encoding for the lysine-specific demethylase 5C (KDM5C, also known as JARID1C or SMCX) can cause Claes-Jensen type X-linked intellectual disability (CJ-XLID) (Claes et al., 2000; Jensen et al., 2005). This rare syndrome accounts for approximately 1–3% of all XLID cases, and besides producing severe intellectual disability, is characterized by autistic behavior, short stature, hyperreflexia, emotional outbursts, spastic paraplegia, and epileptic seizures (Adegbola et al., 2008; Goncalves et al., 2014).
KDM5C mediates the demethylation of tri- and di-methylated lysines in position four of histone H3 (H3K4me3 and H3K4me2, respectively), functioning in non-neuronal cells as a transcriptional co-repressor of the RE1-silencing transcription factor (REST) complex (Iwase et al., 2007; Tahiliani et al., 2007) and as an enhancer modulator (Outchkourov et al., 2013; Shen et al., 2016). Notably, several other intellectual disability disorders (IDDs) also originate from mutations in enzymes that either modify or interact with the different species of K4-methylated histone H3 (Parkel et al., 2013; Vallianatos and Iwase, 2015). Furthermore, changes in H3K4 methylation are correlated with memory acquisition (Graff et al., 2011; Gupta et al., 2010; Kerimoglu et al., 2013), but whether this histone posttranslational modification (HPTM) has a functional role in the process is still unknown.
Although KDM5C is mainly expressed in brain and skeletal muscle in adult human tissues (Jensen et al., 2005), the mechanisms that link transcriptional deregulation with impaired Kdm5c function in the developing and adult brain remain largely unexplored. A recent and seminal study, upon developing the first animal model of CJ-XLID, demonstrated that the germinal loss of Kdm5c in mice causes dendritic and spine anomalies, alterations of the neural transcriptome, and behavioral deficits that resembled clinical symptoms (Iwase et al., 2016), but did not determine the specific genomic actions of Kdm5c in neurons throughout life. Shortly afterwards, Shen et al. demonstrated that loss of Kdm5c results in the activation of a set of enhancers in human breast cancer cells (Shen et al., 2016), but whether enhancer over-activation also occurs in neurons and contributes to ID remains unexplored. To address these questions, we conducted parallel behavioral and genomics screens in mice exhibiting either germinal or adult forebrain ablation of Kdm5c. Our comprehensive analyses, together with loss- and gain-of-function experiments in neuronal cultures, demonstrate the dose- and time-dependent control of cognitive development by Kdm5c and its distinct functions in developing and mature neurons.
Results
Temporal dissection of CJ-XLID endophenotypes in mice
To determine the specific role of Kdm5c in the adult brain and its contribution to cognitive processes, we generated CaMKIIα-creERT2::Kdm5Cf/f mice (from now on referred to as ifKO and their control littermates as ifWT) in which gene ablation was spatially restricted to principal neurons of the forebrain and temporarily regulated by tamoxifen administration in the adult stage (Fig. 1A–B). In order to distinguish between the adult and developmental effects of Kdm5c ablation, we conducted parallel neurological and behavioral assessments of ifKOs and Kdm5c null mice (KOs).
Figure 1. Kdm5c-KOs recapitulate CJ-XLID clinical symptoms and exhibit stronger phenotypes than ifKOs.

A. Schematics of the genetic strategy used to obtain Kdm5c-KOs and ifKOs. B. Kdm5c transcript level in hippocampus (hp), cortex (ctx), and cerebellum (cb). RT-qPCR assays were conducted using primers specific for the deleted exons (hp: KO t2=5.93, p < 0.0001; ifKO t2=10.23, p = 0.009; ctx: KO t2=82.27, p = 0.0001; ifKO t8=2.21, p = 0.06; cb KO t2=7.43, p = 0.02; ifKO t2=1.63, p = 0.2). C. Average weight at different ages (KOs: F(1,23) = 67.79, p < 0.0001, post-hoc 4 months p<0.05, post-hoc 15 months p<0.0001; ifKOs: F(1,33) = 0.13, p = 0.7). D. Fear conditioning (FC) task in KOs (left: pre/post shock comparison, WT: U = 4.00, p = 0.01; KO: U =18.50, p = 0.3; context 24h: U = 0.00, p = 0.0006; pre-cue 24h: U = 8.00, p = 0.02; post-cue: t12=5.93, p < 0.0001) and ifKOs (right: pre-shock, U = 74.00, p = 0.3; post-shock, t26= 0.99, p = 0.3; context: t27= 0.38, p = 0.7; pre-cue, U = 85.00, p = 0.5; post-cue U = 84.50, p = 0.5). E. MWM escape latency graph. Left: KOs showed deficits in the visible platform (F(1,12) = 4.913, p = 0.05), hidden platform (F(1,108) = 31.43, p < 0.0001), and reversal (F(1,66) = 5.60, p = 0.02) tests, as well as in the different probe trials (PT, see Fig. S2). Right: ifKOs showed milder impairments in the hidden (F(1,216) = 6.26, p = 0.01) and reversal (F(1,135) = 7.22, p = 0.008) tests. F. Reaction latency in the hot plate (KOs: t12= 3.05, p = 0.01; ifKOs: t21 = −1.26, p = 0.2). G. Marble burying test (KOs: t12 = 2.68, p = 0.02; ifKOs: t21 = −0.97, p = 0.3). H. Novelty suppressed feeding test (KOs: U = 49, p = 0.001; ifKOs: U = 92.00, p = 0.5). I. Mobility in the tail suspension test (KOs: t26 = 2.51, p = 0.01; ifKOs: t27 = 0.33, p = 0.7). J. Latency to fall in the accelerating rotarod task (KOs: t12 = 2.66, p = 0.02; ifKOs: t27 = 1.45, p = 0.2). K. Kainate-induced seizures measured in the Racine scale (left), and number of injections needed for exclusion (right: t6 = 6.65, p = 0.0006). L. Sensitivity to pentylenetetrazol (PTZ)-induced seizures. Data are expressed as mean ± s.e.m. ns: non significant; * = p < 0.05; ** = p < 0.005; *** = p < 0.0005 in Mann-Whitney, Student’s T-test or 2-way ANOVA.
Kdm5c−/y males had smaller bodies and brains (Fig. 1C and S1A–B), but normal brain histology and anatomy (Fig. S1C–G), while ifKO males displayed normal weight and brain size (Fig. 1C and S1H–J). Exhaustive behavioral testing confirmed the cognitive impairments of Kdm5c KOs (Iwase et al., 2016) (Fig. 1D–E) and revealed traits related to CJ-XLID endophenotypes (Claes et al., 2000; Fujita et al., 2016; Goncalves et al., 2014) that had not been described before. These included hyperreflexia (Fig. 1F), increased impulsivity and emotional responses (Fig. 1G–I), impaired motor coordination (Fig. 1J), and epileptic seizure propensity (Fig. 1K; see also Fig. S2A–L and Table S1). In contrast, ifKOs were indistinguishable from control littermates in our battery of behavioral tasks with the exception of a significant learning delay in the Morris water maze (MWM), both in the hidden platform and reversal phases (Fig. 1D–J and 1L, S2M–O and Table S2).
Clinical investigations indicate that females carrying KDM5C mutations suffer from mild cognitive impairments (Rujirabanjerd et al., 2010; Simensen et al., 2012). To model this aspect of KDM5C-related pathology, we evaluated Kdm5c+/− females and found that they were slightly smaller than their control littermates (Fig. 2A), showed hind-paw clasping (although to a lesser extent than males, Fig. 2B and S2A), exhibited memory deficits in the fear conditioning task (Fig. 2C), and presented a learning delay in the cued phase of the MWM task (Fig. 2D–E). Other behavioral abnormalities, however, were corrected for by the presence of the wild-type allele (Table S1 and S2). We also searched for behavioral impairments in ifKO females, but did not observe any significant differences in basal conditions or after stimulating the animals through environmental enrichment (Fig. S2P–R).
Figure 2. Cognitive impairments are Kdm5c dose-dependent.

A. Kdm5c+/− females are smaller than their WT female littermates (F(1,54) = 17.86, p < 0.0001; post-hoc 12-m p < 0.0001). B. Hemizygous females show hindpaw-clasping (U = 31.50, p = 0.001), although less severe than KO males (see Fig. S2A–B). C. Fear conditioning task: Kdm5c+/− females exhibit fear memory deficits (pre-shock, t27= 0.59, p = 0.6; post-shock, t27= 0.7, p = 0.5; context 24h, U = 57.00, p = 0.04; pre-cue 24h, t27= 2.10, p = 0.05; post-cue 24h, t27= 2.49, p = 0.02). D–E. Morris water maze: The escape latency curve (D) revealed mild learning difficulties during the first day of the task (F(1,54) = 8.91, p = 0.004; post-hoc day 1 p < 0.01), but no difference in performance during the PTs (E). Data are expressed as mean ± s.e.m. * = p < 0.05; ** = p < 0.005; *** = p < 0.0005 in the Mann-Whitney test, Student’s T-test and two-way ANOVA.
Overall, these experiments validate Kdm5c–KO mice as a suitable model to investigate CJ-XLID, including the milder impairments seen in female carriers. Furthermore, the comparison of conventional and conditional KOs indicate that (i) Kdm5c plays a more prominent role during development than in the adult brain, and (ii) Kdm5c still retains some function in mature principal neurons responsible for the spatial navigation defects observed in ifKO males. We cannot, however, discard that Kdm5c ablation in other cells or brain regions could cause stronger phenotypes than those reported here for ifKOs.
Kdm5c restrains H3K4me3 content at specific promoters and enhancers in adult neurons
To identify the molecular causes of these phenotypes, we next conducted parallel genomic screens in KOs and ifKOs. We reasoned that the analysis of KOs would clarify CJ-XLID pathoetiology and Kdm5c roles during development, whereas the investigation of ifKOs could provide novel insights into the specific genomic actions of Kdm5c in mature neurons. We focused on epigenetic and transcriptional alterations in the hippocampus because this brain region is highly relevant for cognitive processes (including those affected in both KOs and ifKOs) and has a high level of Kdm5c expression (Xu et al., 2002).
The occupancy profile of Kdm5c in hippocampal chromatin confirmed the loss of Kdm5c binding in KOs (Fig 3A). No global difference was observed in bulk H3K4me3 levels for KOs and ifKOs (Fig. S3A). However, chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) revealed a prominent increase (~12%) in the number of H3K4me3-enriched regions in KOs (Fig. S3B–C), as well as local increases in H3K4me3 levels in both strains (Fig. 3B and S3D). Differential profiling analysis identified 1,423 and 540 differentially methylated H3K4me3 peaks (DHMPs) in KOs and ifKOs, respectively (Fig. 3C and Table S3). These changes largely corresponded to increases in H3K4 methylation, with each strain having a negligible number of regions showing reduced methylation. The overlap between these epigenomic changes in KOs and ifKOs was very high. For instance, 85% of the changes found in ifKOs were also observed in KOs, and about one third of the changes detected in KOs were observed in ifKOs, albeit with a smaller magnitude (Fig. 3D).
Figure 3. Lack of Kdm5c increases H3K4me3 levels throughout the neuronal epigenome.

A. Heatmap (right) and line plot (left) showing Kdm5c density at Kdm5c-enriched regions (as detected in hippocampal chromatin of wild type mice) in KO and control littermates. Color intensity in the heatmap is proportional to read density. B. Density plot showing the average H3K4me3 signal (expressed as reads per million, RPM) of DHMPs for WT, KO, ifWT and ifKO replicates. Input values are also shown. Shaded areas represent the 95% confidence interval (CI). C. Cumulative graph showing the number of DHMPs (BH p adj. < 0.05, LFC > 0.58) and their fold change with respect to the relative control (i.e., KO vs WT; ifKO vs ifWT). DHMPs were ordered by Log2FC and plotted as separate entities on the x-axis. D. Venn diagram depicting the overlap between the DHMPs of KO (vs WT) and ifKO (vs ifWT) mice. E. Distribution of DHMPs in function of gene feature, in Kdm5c-KOs and ifKOs. “Gene start” refers to peaks located between ±1 Kb of the first TSS; “Gene body” denotes internal peaks (including alternative TSSs); and “Intergenic” refers to peaks that are farther than −1 Kb from the TSS of any Refseq annotated gene. F. H3K4me3 changes at putative enhancers (intra- and intergenic H3K27ac-rich regions not overlapping with a TSS) in KOs and ifKOs. Enhancers were classified according to their H3K4me3 content (see Fig. S3G–H for additional detail).
In addition to H3K4me1 and H3K4me3 profiles, we also determined H3K27ac enrichment in hippocampal chromatin from wild type mice to identify putative enhancers across the genome. H3K4me3 peaks preferentially locate at transcription start sites (TSSs) and putative enhancers (i.e., at intra- and intergenic regions that show a concomitant enrichment for H3K27ac and H3K4me1; Fig. S3E). However, DHMPs were overrepresented at intra- and intergenic regions, particularly for de novo peaks (Fig. 3E and S3F), suggesting that enhancers are particularly sensitive to the absence of Kdm5c. The impact of Kdm5c loss on H3K4me3 at enhancers followed an inverted-U function, in which H3K27ac-rich enhancers with a moderate H3K4me3 content were the most affected (Fig. 3F and S3G). The same pattern was also observed in ifKOs (Fig. S3H). This indicates that regions depleted of H3K4me3 or with very high H3K4me3 content are both resilient to Kdm5c absence. Consistent with this view, the regions showing stronger Kdm5c binding in wild type mice exhibited reduced H3K4me1 signal in KOs, while the increase was not evident in the case of H3K4me3 (Fig. S3I). Therefore, Kdm5c seems to be required in regions in which it is necessary to maintain a given tri-methylation to mono-methylation balance.
Kdm5c modulates the expression of plasticity-related genes and fine-tunes activity-dependent enhancers during neuronal maturation
RNA-seq-based differential expression (DE) screens revealed that transcriptional changes were both larger and more numerous in KOs than in ifKOs (248 and 107 differentially expressed genes (DEGs), respectively) (Fig. 4A–B, S4A and Table S4). In fact, most of the DEGs in ifKOs were also altered in KOs with larger fold changes (Fig. S4B). In both strains, the changes consisted predominantly of upregulations, thereby supporting Kdm5c role in transcriptional repression. The correlation between H3K4me3 gain at TSSs and transcript levels was remarkably high for both mutants (Fig. 4C), indicating that the increment of H3K4me3 resulting from the loss of Kdm5c is highly predictive of transcriptional upregulation. In fact, in both KOs and ifKOs, 97% of the upregulated genes associated with H3K4me3 peaks showed an increase of H3K4me3 signal.
Figure 4. Transcriptional deregulation in Kdm5c-deficient mice correlates with altered H3K4me3 levels.

A. RNA-seq profile of the Kdm5c gene in the hippocampus of KOs and ifKOs confirms the absence of exons 11 and 12 (red box). Note that only neuronal cells are affected in the hippocampus of ifKOs (~70% reduction). B. Gene expression changes in KOs and ifKOs (BH p adj. < 0.05, absolute Log2FC > 0.3). Genes were ordered by Log2FC and plotted as separate entities on the x-axis. C. Scatter plots for KOs (upper panel) and ifKOs (lower panel) showing the correlation between H3K4me3 levels at peaks located in close proximity to annotated DEGs and the transcript changes. Filled dots represent DHMPs (p adj. < 0.1) and colors indicate the location of the peak with respect to the gene feature. D. Heatmap showing the DEGs common to both KOs and ifKOs (common), as well as the DEGs altered exclusively in either strain. The number of genes in each category is indicated. E. Box plot showing Log2(TPMWT+1), Log2FC of transcript level, and Log2FC of H3K4me3 at TSS for KO-exclusive DEGs. Light green dots correspond to genes with significant hippocampal expression and related to neuronal function. Pink dots: germ line-related genes associated with a Kdm5c-peak observed in embryonic stem cells (ESCs) but not in neural precursor cells (NPCs). Purple dots: germ line-related genes that are not associated with an ESC-specific Kdm5c-peak. Grey dots: DEGs not falling in previous categories. Note that the genes with the largest transcript changes (germ line-related genes) have very low TPM values. Bars represent means ± SD.
To link genomic and phenotypic alterations in Kdm5c-deficient mice, we first focused on genes exclusively altered in KOs (Fig. 4D) because we reasoned that these changes should be accountable for the severe phenotypes observed in those mice. This DEG set included a number of genes linked to neuronal terms according to Gene Ontology (GO) enrichment analysis. These genes showed appreciable hippocampal expression, and relatively modest changes in transcription and H3K4 methylation (light green dots in Fig. 4E). Downregulations affected genes important for synaptic transmission and neurodevelopment (e.g., Grik3, Nrp2, and Syt2), while upregulated genes were involved in neuronal signal transduction (e.g., Hexim1, Hexim2, Dusp5, and Dusp18). Consistent with our results, transcriptome analyses in the amygdala and frontal cortex of Kdm5c-KO mice had also revealed misexpression of neuronal genes, but whether these changes were directly linked to altered H3K4 methylation was not explored (Iwase et al., 2016). Our parallel epigenome and transcriptome screens enabled the direct comparison of both effects in vivo. The correlation between differential H3K4me3 methylation at TSSs and changes of transcript levels for DE neuronal genes was modest, suggesting that the impact of Kdm5c loss in some of these loci might rely on changes at more distant regulatory sequences.
Given the role of Kdm5c in regulating enhancer function, as well as the potential causal link between KO phenotypes (such as learning impairment and epilepsy propensity) and altered activity-driven transcription, we next investigated the contribution of Kdm5c to gene induction upon exploration of a novel environment (NE). This experience is known to trigger hippocampal transcription of numerous immediate early genes (IEGs) involved in synaptic plasticity and memory consolidation (Fig. 5A, S5A and Table S5) (Flavell and Greenberg, 2008). Strikingly, NE-induced changes in KOs represented about one third of those observed in WTs (66 versus 186 genes). This was because a significant proportion of the NE transcriptional program was moderately overexpressed in KOs in naïve situation (homecage: HC) (Fig. 5A–B), suggesting a role for Kdm5c fine-tuning activity-driven transcription. Independent RT-qPCR for the IEGs Fos and Arc confirmed this observation (Fig. 5C).
Figure 5. Kdm5c modulates neuronal activity-dependent enhancers.

A. Heatmap of DEGs upon novelty exposure (NE) in WT mice together with their values for naïve (HC) and NE KOs. B. Scatter plot comparing NE effect with genotype effect in KOs for the set of NE-induced genes. C. RT-qPCR assays for Arc (F(1,20)condition = 8.73, p = 0.008; F(1,20)genotype = 2.22, p = 0.2; post-hoc WT p < 0.05) and Fos (F(1,20) condition = 12.67, p = 0.002; F(1,20) genotype = 0.72, p = 0.4; post hoc WT p < 0.01) using hippocampal RNA. D. Number of H3K4me3 peaks detected in WT-HC, WT-NE, KO-HC and KO-NE replicates. E. Venn diagram depicting the overlap between the DHMPs exclusive of WT-NE (vs. WT-HC) and KO-HC (vs. WT-HC). F. Left: Genomic profiles for Npas4. Right: Bar graphs representing the read density for H3K4me3 at the TSS (F(1,4)condition = 11.56, p = 0.03; F(1,4)genotype = 0.0073, p = 0.9; post-hoc ns) and upstream enhancer (F(1,4)condition = 1.31, p = 0.3; F(1,4)genotype = 100.9, p = 0.0006; post-hoc HC and NE p<0.01) of Npas4; and for the transcripts corresponding to Npas4 mRNA (F(1,8)condition = 19.33, p = 0.002; F(1,8)genotype = 0.05, p = 0.8; post-hoc WT p < 0.01, post-hoc KO p > 0.05) and its putative eRNA (F(1,8)condition = 0.70, p = 0.4; F(1,8)genotype = 20.03, p = 0.002, post-hoc ns). G. RT-qPCR assays of IEG-associated eRNAs. Npas4-enhancer (t4 = 1.93, p = 0.13); Fos-enhancer 5 (t4 = 4,74, p = 0.009). H. Up: Scheme of PNC experiment; Down: Immunostaining of Kdm5cf/f PNC co-infected with LVs expressing Cre and hKDM5C. I. RT-qPCR assays confirmed the loss of the endogenous Kdm5c, the expression of hKDM5C and their modulation of IEG expression (Kdm5c: Cre− vs Cre+, t4 = 4.92, p = 0.008; Cre− vs Cre++hKdm5c, t4 = 6.54, p = 0.003; Cre+ vs Cre++hKdm5c, t4 = 13.74, p = 0.0002; Arc: Cre− vs Cre+, t4 = 3.62, p = 0.02; Cre+ vs Cre++hKdm5c, t4 = 5.43, p = 0.006. Fos: Cre− vs Cre+, t4 = 4.28, p = 0.01; Cre+ vs Cre++hKdm5c, t4 = 4.17, p = 0.01. Npas4: Cre− vs Cre+, t4 = 3.58, p = 0.02; Cre+ vs Cre++hKdm5c, t4 = 3.07, p = 0.04). Data are expressed as means + s.e.m. * = p < 0.05, ** = p < 0.005; *** = p < 0.0005 (genotype effect); # = p < 0.05, ## = p < 0.005; ### = p < 0.0005 (condition effect) in 2-way ANOVA (C and F) and Student t-test (G and I).
We next investigated the impact of NE on H3K4me3 profiles and its relationship with Kdm5c loss. Our ChIP-seq experiment revealed that NE caused a 5% increase in the number of detected peaks in hippocampal chromatin (Fig. 5D), demonstrating that the levels of H3K4me3, like the transcriptome, are susceptible to modulation by this experience. Notably, NE-induced changes in H3K4me3 profiles were partially occluded in KOs (Fig. 5D–E), which is consistent with Kdm5c playing a role in the regulation of activity-dependent enhancers. H3K4me3 levels at proximal enhancers of important IEGs involved in neuroplasticity were significantly elevated in naïve Kdm5c-KO mice, even when the TSS was not affected. A clear example of this is the IEG Npas4, which encodes a transcription factor that regulates cognition-related transcription and GABAergic synapse formation (Ramamoorthi et al., 2011). H3K4me3 levels at the enhancer region 10 Kb upstream of the Npas4 TSS were higher in KOs than in WTs, and this increase was associated with augmented transcription of the enhancer RNA (eRNA) at the basal state (Fig. 5F–G). A similar scenario was also observed at enhancers 2 and 5 of Fos (Fig. S5B–C) and at the Arc enhancer (Fig. S5D–E), although for the latter, H3K4me3 levels at the promoter were also higher in KOs’ chromatin.
In contrast to KOs, basal expression and induction of IEGs and H3K4me3 profiles at these activity-regulated enhancers were normal in ifKOs (Fig. S5F–H). This suggests that Kdm5c plays a specific role in establishing the ground activity of IEGs during neuronal maturation. To tackle this model, we investigated the fine-tuning of activity-regulated enhancers using primary neuronal cultures (PNCs), and found that Kdm5c ablation in PNCs from Kdm5cf/f embryos using a Cre recombinase-expressing lentivirus (LV) enhanced basal expression of IEGs such as Fos, Arc and Npas4 (Fig. 5H–I). Moreover, normal basal IEG expression was observed upon co-infection with a second LV driving the expression of wild type human KDM5C (hKDM5C) (Fig. 5H–I). These experiments show that these loci are still susceptible to epigenetic tuning at this stage, and demonstrate that Kdm5c is both necessary and sufficient for proper modulation of activity-regulated enhancers during neuronal maturation.
Germinal loss of Kdm5c precludes the silencing of germ line genes
The exploration of DEG exclusive of KOs also revealed a subset comprised of genes related to reproduction and gametogenesis, such as D1Pas1, Naa11, Ddx4 and Ccnb1iP1, that had not been reported in previous screens. These genes had very low or undetectable expression in the hippocampus of control mice, and presented large, highly correlated changes in transcript and H3K4me3 levels in the hippocampus of Kdm5c-KO mice (pink and purple dots in Fig. 4E). In fact, most of the genes presenting the largest differences between KO and wild type littermates belonged to this category (Fig. 4D). Interestingly, some of these genes (pink dots) showed robust Kdm5c binding at their promoters in the chromatin of embryonic stem cells (ESCs), although this occupancy decreased over time, from neuroprogenitor cells (NPCs) to mature neurons (MNs) (Fig. 6A–B). Kdm5c disappearance coincides with the strong DNA methylation of these loci in wild-type mature neurons, according to available MeDIP data (Halder et al., 2016) (Fig. 6B and S6A, pink track). To confirm this model, we next examined DNA methylation levels in the hippocampi of KO mice and control littermates. Reduced CpG methylation at the promoter of the germ line genes D1Pas1 and Naa11 (Fig. 6C) was concomitant with their enhanced transcription (Fig. 6D). Further supporting our hypothesis, PNCs from Kdm5c−/y embryos also showed de-repression of D1Pas1 and Naa11 (Fig. 6E–F), whereas PNCs from Kdm5cf/f embryos transduced with Cre recombinase showed normal germ line silencing despite of a rapid and strong reduction in the levels of Kdm5c (Fig. S6B). These results indicate that Kdm5c is necessary for germ line gene silencing during early development but becomes dispensable at later developmental stages once a repressed status has been established through other mechanisms such as DNA methylation (Fig. S6C).
Figure 6. Germinal loss of Kdm5c prevents the silencing of germ line genes.

A. Developmental loss of Kdm5c occupancy at the TSS of germ line genes (pink dots in Fig 4E). RPM: reads per million. B. Genomic profiles for representative germ line genes. From top to bottom, we show (a) Kdm5c-ChIP-seq profiles in ESC, NPC (Outchkourov et al., 2013), PNC (Iwase et al., 2016), and adult brain (MN, this study); (b) MeDIP profiles in adult brain of wild type mice (Halder et al., 2016), and (c) RNAseq and H3K4me3-ChIPseq experiments in adult hippocampus of WT and KO mice (this study). C. DNA methylation at specific CpGs (red boxes in Fig. 6B) at the promoter of D1Pas1 and Naa11 in hippocampal chromatin of WT and KO littermates (star symbols label significant DNA methylation reduction in KOs). Testis DNA was used as control for un-methylated DNA. D. RT-qPCR assays confirm the expression of germ line genes in the hippocampi of KOs (D1Pas1: t10 = 8.53, p < 0.0001; Naa11: t10 = 3.90, p = 0.003). E. Scheme of the experiment (top). Functionality of the hKDM5C construct was confirmed by transient transfection and examination of H3K4me3 levels in EYFP-expressing neurons (bottom). F. RT-qPCR assay confirmed the overexpression of germ line genes in PNCs from Kdm5c−/y embryos, infected or not with a hKDM5C-expressing lentivirus (Kdm5c, WT vs KO: t8 = 4.64, p = 0.002, KO vs KO+hKDM5C: t7 = 15.57, p < 0.0001, WT vs KO+hKDM5C: t9 = 18.54, p < 0.0001; D1Pas1, WT vs KO: t8 = 3.49, p = 0.008; KO vs KO+hKDM5C: t8 = 1.27, p = 0.2; Naa11, WT vs KO: t9 = 5.05, p = 0.0007; KO vs KO+hKDM5C: t8 = 0.0007, p = 1.0). Data are expressed as mean + s.e.m. ns: non-significant; * = p < 0.05; ** = p < 0.005; *** = p < 0.0005 in Student’s t test.
To examine whether the spurious expression of germ line genes in neurons can be rescued by restoring Kdm5c activity, PNCs from Kdm5c−/y embryos were transduced with a hKDM5C-expressing lentivirus (Fig. 6D). Interestingly, hKDM5C expression did not restore the silent status of germ line genes (Fig. 6E), which suggests that the mechanism of Kdm5c repression requires of cofactors that are absent in postmitotic neurons. Together these experiments show that Kdm5c is necessary to assure the silencing of germ line genes during early development. Consistent with this view and underscoring the clinical relevance of our finding, blood cells of CJ-XLID patients show multilocus loss of DNA methylation (Grafodatskaya et al., 2013).
Kdm5c retains a role preventing transcription from cryptic and non-neuronal promoters and enhancers in adult neurons
We next explored the transcriptional changes occurring in both strains (referred to as common DEGs in Fig. 4D). These genes are likely to be direct and permanent targets of Kdm5c and could thus provide insight into the somatic actions of this enzyme. Moreover, although these genes may not play a primary role in the most severe behavioral deficits observed in KOs, their deregulation was associated with impaired spatial navigation in ifKOs.
The integration of DHM and DE data revealed that common DEGs behaved remarkably similar in both strains. Prominently upregulated transcripts of this set (FC > 2) showed significant H3K4me3 increases at their TSSs, and characteristically low hippocampal expression in control mice (< 10 transcripts per million, TPM) (Fig. 7A). Their transcriptional and H3K4me3 profiles were thereby similar to those observed at de-repressed germ line genes. According to GO enrichment analyses, the common DEGs were associated with biological processes that are not related to neuron function, such as Muscle contraction and Extracellular matrix assembly (Fig. 7B). The set was comprised of both protein-coding genes and a number of non-coding RNAs (ncRNAs) (25%, including both miRNAs and lincRNAs). Although some of these strongly upregulated transcripts corresponded to full-length messenger RNAs (mRNAs) (e.g., Tnxb, Fig. 7C), intriguingly, others were truncated mRNAs or partially overlapping ncRNAs. For example, upregulation of the muscle genes Dnah1 (Fig. 7C) and Acta1 (Fig. S7A) was restricted to their terminal exons and started at an internal DHMP. These truncated transcripts did not coincide with shorter isoforms of assigned biological function, but instead resembled a recently described class of eRNAs referred to as multiexonic eRNAs (meRNAs) that act as alternate promoters and utilize splicing and poly-adenylation signals from their protein-coding hosts (Kowalczyk et al., 2012).
Figure 7. Kdm5c loss causes the activation of cryptic and non-neuronal regulatory sequences in adult neurons.

A. Representation of Log2FC for common DEGs respect to the transcript levels in WT counterparts, for both KO (left) and ifKO (right) strains. TPM: transcripts per million; DHMP: differentially methylated H3K4me3 peak. B. GO enrichment for biological processes (GOBP) for common DEGs. Terms in grey correspond to an overlap of less than 5 genes. C. Genomic profiles for Tnxb and Dnah1, two representative examples of non-neuronal genes exhibiting spurious transcription and H3K4 methylation in Kdm5c-deficient strains. In the case of Dnah1, upregulation was restricted to the terminal exons and might correspond to the production of a meRNA. D. Scatter plots showing H3K4me3 level in KOs at peaks overlapping the TSS of non-annotated DE transcripts. E. Genomic profiles for one of the non-annotated ncRNAs identified in our de novo transcriptome assembly screen. F. Transcript upregulation in intergenic DHMPs in KOs. Reads for the plus and minus strand were independently scored 2 Kb either side of the peak center. G. Graphs show the presence of enhancer marks (H3K27ac and H3K4me1; right) and Kdm5c occupancy (left) at the same regions. ESC: embryonic stem cells; NPC: neuroprogenitor cells; PNC: primary neuronal cultures; MN: mature neurons. H. RT-qPCR assays confirmed spurious transcription of non-neuronal genes in Kdm5c−/y PNC (left, Tnxb: t9 = 4.16, p = 0.003; Dnah1: t3 = 6.00, p = 0.009) and in Kdm5cf/f PNC infected with Cre-expressing LV (right, Tnxb: t4 = 13.62, p = 0.0002; Dnah1: t3 = 8.75, p = 0.003). I. Zfhx2 downregulation in both strains coincides with upregulation of an antisense lncRNA that overlaps with Zfhx2os. Data are expressed as mean + s.e.m. ns: non-significant; * = p < 0.05; ** = p < 0.005; *** = p < 0.0005 in Student’s t test.
To further explore this phenomenon in an unbiased manner, we extended our DE screen to non-annotated transcripts using de novo transcriptome assembly. This approach allowed us to identify several dozen additional Kdm5c-dependent transcripts, including both novel isoforms transcribed from internal TSSs, and non-annotated intergenic transcripts (Table S4 and Fig. S7B). Like for annotated genes, there was a large overlap between the expression changes in KOs and ifKOs (100% of ifKO changes were observed in KOs) and a high correlation with H3K4me3 levels (Fig. 7D, see examples in Fig. 7E and S7C). Indeed, examination of RNA-seq data around DHMPs located at intergenic regions revealed a robust and consistent association between elevated H3K4me3 and the production of transcripts (Fig. 7F and S7D). These DHMPs showed H3K27ac and Kdm5c occupancy in the chromatin of control mice, suggesting that these regions may have a regulatory or structural role in wild type mice (Fig. 7G and S7E). Re-analysis of publicly available transcriptomic data (Iwase et al., 2016) confirmed that the spurious transcription described here also occurs in other brain regions of Kdm5c-KOs (Fig. S7F). Notably, the spurious activation of these loci was also observed in PNCs from Kdm5cf/f embryos few days after triggering gene ablation (Fig. 7H), which demonstrate that, contrary to the situation described above for germ line genes, Kdm5c is constantly required to prevent the transcription from these cryptic promoters/regulatory elements.
The overexpression of some of these ncRNAs can directly alter the expression of protein-coding genes. For example, the downregulation of Zfhx2, which encodes a homeobox transcription factor highly expressed in the developing brain and whose germinal loss causes behavioral abnormalities (Komine et al., 2012), was not associated with H3K4me3 changes at its promoter, but rather with the upregulation of an anti-sense ncRNA (partially overlapping with the previously described Zfhx2os (Komine et al., 2006)) transcribed from a downstream DHMP (Fig. 7I). A similar type of interference, in which the absence of Kdm5c results in the production of an anti-sense ncRNA, may also occur at other gene loci (e.g., Ssh1 and Depdc5). It is also possible that some of these ncRNAs are trans-acting, which could explain the downregulation of neuronal genes with no apparent H3K4me3 changes at their promoters and proximal regulatory regions.
Discussion
Dose- and time-dependent control of cognitive development by Kdm5c
Our comprehensive characterization and exhaustive analyses of conventional and inducible forebrain-restricted knockout mice demonstrate that this chromatin-modifying enzyme exerts a tight dose- and time-dependent control of cognitive development. The correlation in the magnitude of epigenetic, transcriptional and cognitive dysfunctions across the two mouse strains, and the result of our loss- and gain-of-function experiments in neuronal cultures strongly supports a role for epigenetic dysregulation in the etiology of the syndrome. More specifically, our results suggest that the inability of Kdm5c-deficient neurons to silence germ line genes, to fine-tune the neuronal epigenome, and to preclude spurious transcription are probable causes of neurological symptoms. This insight opens up new avenues for therapy and pinpoints specific genes as potential biomarkers for this disorder. Moreover, since Kdm5c directly interacts with other ID-linked epigenetic regulators, our findings go beyond CJ-XLID and can also benefit other chromatin-related IDDs that target the same processes and gene networks.
Kdm5c is essential for the silencing of germ line genes in early development
During the development of multicellular organisms gene silencing is achieved through the implementation of precise DNA methylation patterns by DNA methyltransferases (DNMTs). The recruitment of these DNMTs requires the previous action of lysine demethylases (KDM) that remove methyl group from specific lysine residues associated with active transcription (Ooi et al., 2007), including (as we showed here) Kdm5c. Particularly interesting is our discovery that Kdm5c plays an essential role in the repression of germ line genes. Intriguingly, some of these genes are also upregulated in mouse embryos deficient for enzymes that introduce repressive marks in the chromatin, such as the methyltransferase of H3K9 Ehmt2 and the DNA methyltransferase Dnmt3b (Auclair et al., 2016), underscoring the interplay between DNA methylation, and H3K9 and H3K4 methylation (Du et al., 2015). Dnmt3b silences this class of genes by recruiting the transcriptional repressor E2F6 (Velasco et al., 2010). The ablation of other transcriptional repressors, such as Myc-associated Factor X (Max) and polycomb group ring finger 6 (Pcgf6), also leads to robust de-repression of germ cell-related genes affecting cell growth and viability (Endoh et al., 2017; Maeda et al., 2013). Most of these proteins (MAX, PCGF6, E2F6, EHMT2) seem to interact with KDM5C (Endoh et al., 2017; Outchkourov et al., 2013; Tahiliani et al., 2007), indicating that a large multimeric repressive complex is responsible for the silencing of germ line in somatic cells. This, together with our results, suggests that two mechanisms are necessary for promoting the silencing of these germ line genes: the removal of H3K4me3 by Kdm5c and the addition of H3K9me2 by Ehmt2. If either one of these processes is disrupted, these loci will not be inactivated by CpG methylation and will consequently remain transcriptionally active. Although the specific impact that the spurious expression of germ line genes has on neuronal differentiation and maturation remains unknown, it is striking that several chromatin-modifying enzymes linked to neurodevelopmental defects target the same or similar loci (Auclair et al., 2016; Hansen et al., 1999; Katz et al., 2009; Schaefer et al., 2009), which suggests that their defective silencing may play a role in the etiology of intellectual disability.
Kdm5c fine-tunes activity-regulated enhancers during neuronal maturation
Our study also clarifies conflicting in vitro results regarding the function of Kdm5c at enhancers (Outchkourov et al., 2013; Shen et al., 2016). Recent studies have blurred the functional boundary between enhancers and promoters by demonstrating that enhancers are often major sites of extragenic noncoding transcription (Andersson, 2015; Li et al., 2016; Paralkar et al., 2016). The lack of Kdm5c would tip the balance between H3K4me3 (a HPTM strongly associated with active promoters) and H3K4me1 (which is enriched at enhancers), thereby favoring eRNA transcription from Kdm5c-regulated enhancers. Among the enhancers fine-tuned by Kdm5c, we identified regulatory regions that influence the transcription of genes of singular importance in the context of neuronal plasticity, such as Fos, Arc and Npas4. Kdm5c modulation of the ground activity of activity-regulated enhancers seems to take place during neuronal maturation because the phenomenon was reproduced in floxed PNCs but not in ifKO brains, which indicates that these regions become resilient to the H3K4 methylation imbalance caused by Kdm5c loss in the adult brain. Tuning down activity-driven genes is essential for the maturation of dendritic arbors and the control of neural circuit responses to upcoming signals (Yang et al., 2016). It is thereby tempting to speculate that some of the phenotypes observed in KOs that recapitulate clinical symptoms, such as epilepsy propensity and learning disability, could be caused by the mistuning of activity-regulated enhancers. This opens the possibility that compounds that tune down neuronal activity could have therapeutic value in the treatment of CJ-XLID.
A role for Kdm5c in neuronal genome surveillance
The comparison of genomic effects after gene ablation in the germinal line, developing neurons and mature neurons, presented here indicates that the biological relevance of Kdm5c in neurons declines over time, probably in parallel with the progressive locking of the cell type-specific epigenetic status (Smith and Meissner, 2013) that narrows the possibility of spurious transcription. Nonetheless, Kdm5c seems to retain a life-long surveillance role in mature neurons preventing extemporaneous transcription from numerous intragenic and intergenic regulatory sites with transcriptional potential. Even though spurious transcription likely occurs in all tissues, the clinical picture of CJ-XLID indicates that this form of deregulation particularly affects the central nervous system. The function of most of these Kdm5c-sensitive loci and associated lncRNAs is unknown. However, their weak transcription in control animals and apparent tolerance to transcriptional deregulation in vivo would suggest that many of these transcripts do not have a critical function, and that the evolutionary pressure for establishing a locked state is lower than in protein coding genes. However, some of these Kdm5c-regulated ncRNAs may play key regulatory roles in specific cell types or developmental stages (e.g., the consequences of altering the Zfhx2/Zfhx2os expression balance are likely more severe during development than in mature neurons). Furthermore, the consequences of the spurious activation of intergenic sites may be more severe in humans than mice since a recent study linked the expression of thousands of unannotated, non-exonic differentially expressed regions (DERs) in the human prefrontal cortex to a higher risk of developing brain disorders and variations in neuronal phenotype (Jaffe et al., 2015). It is also possible that more prominent phenotypes could emerge in Kdm5c-ifKOs upon exposure to specific situations or challenges. For instance, Kdm5c ablation in breast cancer cells was not deleterious by itself, but did favor the emergence of a tumorigenic phenotype (Shen et al., 2016).
In the context of cancer research, KDM5C-inactivating mutations have been associated with genome instability and poor prognosis (Rondinelli et al., 2015). In agreement with our results in neurons, these cancer cells overexpressed lincRNAs that are normally silent. Recent studies have underscored the relevance of genome instability in brain pathology, particularly in the context of aging and age-related neurodegenerative disorders (Madabhushi et al., 2014; McKinnon, 2013). Neurons are very long-living cells that have to withstand challenge from numerous stressors over their lifetime, which makes critical to safeguard their DNA integrity (Pan et al., 2014). The lifelong role of Kdm5c in genome surveillance described here can not only explain its retained expression in adult neurons and the deficits observed in ifKOs, but also pinpoints new possible mechanisms of pathoetiology.
Experimental Procedures
For details, refer to Supplemental Experimental Procedures.
Animals and neuronal cultures
The generation of Kdm5c−/y, Kdm5cf/f (Iwase et al., 2016) and CaMKIIα-creERT2 (Erdmann et al., 2007) mice have been previously described. Experiments were conducted in 2–6 months old animals, except when it is otherwise indicated. Primary cortical and hippocampal neurons were obtained from E17.5–E18.5 embryos, and were infected with lentiviral pseudovirions at the times indicated in the text. Mice were maintained and bred under standard conditions, consistent with Spanish and European regulations and approved by the Institutional Animal Care and Use Committee.
Behavioral testing
Independent cohorts of adult Kdm5c-KO males, Kdm5c−/+ females and Kdm5c-ifKOs from both sexes were tested in series of behavioral tasks. The different tasks are described in detail in Supplemental Experimental Procedures.
Histology
Nissl staining and immunostainings were performed as described (Ito et al., 2014). The primary antibodies used in this study are α-NeuN (Chemicon, MAB377), α-MAP2 (Sigma, M9942), α-GFAP (Sigma, G9269), α-DCX (Abcam, ab18723), α-Parvalbumin (Swant, PV235), α-Cre (gift from Schütz’s lab), α-H3K4me3 (Millipore 07-473), and α-GFP (Aves Labs, GFP-1020, valid also for recognizing EYFP). Nuclei were counterstained with a 1 nM DAPI solution (Invitrogen) before mounting. For magnetic resonance imaging (MRI), 4-month old Kdm5c-KO mice and wild-type littermates were examined.
Western blots, DNA isolation and pyrosequencing
Western blot analyses were carried out using Western Lightning ECL kit (Perkin-Elmer, Boston, MA), and α-H3K4me3 (Millipore 07-473) and α-actin (Sigma F5441) antibodies. DNA was extracted from adult hippocampi with QIAamp DNA Mini Kit (Qiagen). Bisulfite conversion of genomic DNA was performed with the EpiTect Fast bisulfite kit (Qiagen). Primer sequences for PCR amplification and pyrosequencing are provided in Supplemental Experimental Procedures.
RT-qPCR and RNA-seq
Total RNA from hippocampal cultures and tissue was extracted with TRI reagent (Sigma-Aldrich) and reverse transcribed to cDNA. Each independent sample was assayed in duplicate and normalized using GAPDH levels. RNA preparation for sequencing was performed as described in (Fiorenza et al., 2016). Information on library preparation and size, mapping to reference genome and differential expression and functional genomics analyses can be found in Supplemental Experimental Procedures.
ChIP assays and ChIP-seq
Chromatin immunoprecipitation (ChIP) experiments were conducted as described (Lopez-Atalaya et al., 2013) using anti-H3K4me3 (Millipore 07-473) anti-H3K4me1 (Abcam ab8895) and anti-Kdm5c (Iwase et al., 2016). See Supplemental Experimental Procedures for additional details.
Statistical analyses
All statistical analyses were two-tailed. P-values were considered to be significant when α < 0.05. Mean ± s.e.m. and percentages are represented in bar graphs. For additional details see Supplemental Experimental Procedures.
Accession numbers
RNA-seq (GSE85874) and ChIP-seq (GSE85873) data can be accessed at the GEO repository.
Supplementary Material
Table S3. Related to Figure 3. DHMPs in Kdm5c-KO and ifKO mice (Excel file with two spreadsheets).
Table S4. Related to Figure 4. DEGs in Kdm5c-KO and ifKO mice (Excel file with three spreadsheets).
Table S5. Related to Figure 5. DEGs upon NE (Excel file with two spreadsheets).
Highlights.
Dose- and time-dependent control of cognitive development by Kdm5c
Kdm5c is required for germ line gene silencing during early development
Kdm5c functions as a fine-tuner of enhancers in maturing neurons
Kdm5c retains a genome surveillance role in adult neurons
Acknowledgments
We thank Juan Medrano, Jesús Pacheco and Romana Tomasoni for their assistance and advice, and Eloisa Herrera and Luis M. Valor for useful discussions. M.S. and A.M.F. are recipients of “Formación de Personal Investigador” fellowships, and B.B. is the recipient of a “Juan de la Cierva – Incorporación” contract, all from the Spanish Ministry of Economy and Competitivity (MINECO). J.P.L.A. research is supported by a “Ramón y Cajal” contract and by the grant SAF2014-60233-JIN from MINECO co-financed by the European Regional Development Fund (ERDF). A.B. research is supported by grants SAF2014-56197-R, PCIN-2015-192-C02-01 and SEV-2013-0317 from MINECO co-financed by ERDF, grant PROMETEO/2016/006 from the Generalitat Valenciana, NARSAD Independent Investigator grant from the Brain & Behavior Research Foundation, and a grant from the Alicia Koplowitz Foundation. S.I. research is supported by the NIH grant NS089896 and the Farrehi Family Foundation. KDM5C research in Y.S. lab is supported by the NIH grant MH096066. Y.S. is an American Cancer Society Research Professor. The Instituto de Neurociencias is a “Centre of Excellence Severo Ochoa”.
Footnotes
Authors’ contributions: M.S. performed and analyzed cellular and molecular biology experiments with the collaboration of A.M.F., B.B., M.L. and R.O. in specific assays. A.M. and M.S. collected and analyzed behavioral data. J.P.L.A. conducted bioinformatics analyses. M.T.L.C., M.S. and A.B collaborated in bioinformatics analyses. S.I. and Y.S. generated the Kdm5c null and floxed strains. E.B. conducted seminal experiments related to KDM5C. A.B. conceived the study and secured funding. A.B. and M.S. designed the experiments and wrote the manuscript. All authors read and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD) Am J Med Genet A. 2008;146A:505–511. doi: 10.1002/ajmg.a.32142. [DOI] [PubMed] [Google Scholar]
- Andersson R. Promoter or enhancer, what’s the difference? Deconstruction of established distinctions and presentation of a unifying model. Bioessays. 2015;37:314–323. doi: 10.1002/bies.201400162. [DOI] [PubMed] [Google Scholar]
- Auclair G, Borgel J, Sanz LA, Vallet J, Guibert S, Dumas M, Cavelier P, Girardot M, Forne T, Feil R, Weber M. EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res. 2016;26:192–202. doi: 10.1101/gr.198291.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015;25:1473–1481. doi: 10.1101/gr.190629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes S, Devriendt K, Van Goethem G, Roelen L, Meireleire J, Raeymaekers P, Cassiman JJ, Fryns JP. Novel syndromic form of X-linked complicated spastic paraplegia. Am J Med Genet. 2000;94:1–4. doi: 10.1002/1096-8628(20000904)94:1<1::aid-ajmg1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16:519–532. doi: 10.1038/nrm4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M, Endo TA, Shinga J, Hayashi K, Farcas A, Ma KW, Ito S, Sharif J, Endoh T, Onaga N, et al. PCGF6-PRC1 suppresses premature differentiation of mouse embryonic stem cells by regulating germ cell-related genes. eLife. 2017:6. doi: 10.7554/eLife.21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann G, Schutz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC neuroscience. 2007;8:63. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorenza A, Lopez-Atalaya JP, Rovira V, Scandaglia M, Geijo-Barrientos E, Barco A. Blocking miRNA Biogenesis in Adult Forebrain Neurons Enhances Seizure Susceptibility, Fear Memory, and Food Intake by Increasing Neuronal Responsiveness. Cereb Cortex. 2016;26:1619–1633. doi: 10.1093/cercor/bhu332. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita A, Waga C, Hachiya Y, Kurihara E, Kumada S, Takeshita E, Nakagawa E, Inoue K, Miyatake S, Tsurusaki Y, et al. Different X-linked KDM5C Mutations in Affected Male Siblings: Is Maternal Reversion Error Involved? Clin Genet. 2016 doi: 10.1111/cge.12767. [DOI] [PubMed] [Google Scholar]
- Goncalves TF, Goncalves AP, Fintelman Rodrigues N, dos Santos JM, Pimentel MM, Santos-Reboucas CB. KDM5C mutational screening among males with intellectual disability suggestive of X-Linked inheritance and review of the literature. European journal of medical genetics. 2014;57:138–144. doi: 10.1016/j.ejmg.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiological reviews. 2011;91:603–649. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- Grafodatskaya D, Chung BH, Butcher DT, Turinsky AL, Goodman SJ, Choufani S, Chen YA, Lou Y, Zhao C, Rajendram R, et al. Multilocus loss of DNA methylation in individuals with mutations in the histone H3 lysine 4 demethylase KDM5C. BMC Med Genomics. 2013;6:1. doi: 10.1186/1755-8794-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Vizcaino JC, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102–110. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Magalska A, Alcaraz-Iborra M, Lopez-Atalaya JP, Rovira V, Contreras-Moreira B, Lipinski M, Olivares R, Martinez-Hernandez J, Ruszczycki B, et al. Loss of neuronal 3D chromatin organization causes transcriptional and behavioural deficits related to serotonergic dysfunction. Nature communications. 2014;5:4450. doi: 10.1038/ncomms5450. [DOI] [PubMed] [Google Scholar]
- Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, Tomassy GS, Kasza T, Lin G, Thompson A, et al. A Mouse Model of X-linked Intellectual Disability Associated with Impaired Removal of Histone Methylation. Cell reports. 2016;14:1000–1009. doi: 10.1016/j.celrep.2015.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jaffe AE, Shin J, Collado-Torres L, Leek JT, Tao R, Li C, Gao Y, Jia Y, Maher BJ, Hyde TM, et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci. 2015;18:154–161. doi: 10.1038/nn.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz DJ, Edwards TM, Reinke V, Kelly WG. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell. 2009;137:308–320. doi: 10.1016/j.cell.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerimoglu C, Agis-Balboa RC, Kranz A, Stilling R, Bahari-Javan S, Benito-Garagorri E, Halder R, Burkhardt S, Stewart AF, Fischer A. Histone-Methyltransferase MLL2 (KMT2B) Is Required for Memory Formation in Mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. The genetics of cognitive epigenetics. Neuropharmacology. 2014;80:83–94. doi: 10.1016/j.neuropharm.2013.12.025. [DOI] [PubMed] [Google Scholar]
- Komine Y, Nakamura K, Katsuki M, Yamamori T. Novel transcription factor zfh-5 is negatively regulated by its own antisense RNA in mouse brain. Mol Cell Neurosci. 2006;31:273–283. doi: 10.1016/j.mcn.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Komine Y, Takao K, Miyakawa T, Yamamori T. Behavioral abnormalities observed in Zfhx2-deficient mice. PLoS ONE. 2012;7:e53114. doi: 10.1371/journal.pone.0053114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, et al. Intragenic enhancers act as alternative promoters. Mol Cell. 2012;45:447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- Lilja T, Heldring N, Hermanson O. Like a rolling histone: epigenetic regulation of neural stem cells and brain development by factors controlling histone acetylation and methylation. Biochim Biophys Acta. 2013;1830:2354–2360. doi: 10.1016/j.bbagen.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Lopez-Atalaya JP, Ito S, Valor LM, Benito E, Barco A. Genomic targets, and histone acetylation and gene expression profiling of neural HDAC inhibition. Nucleic Acids Res. 2013;41:8072–8084. doi: 10.1093/nar/gkt590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda I, Okamura D, Tokitake Y, Ikeda M, Kawaguchi H, Mise N, Abe K, Noce T, Okuda A, Matsui Y. Max is a repressor of germ cell-related gene expression in mouse embryonic stem cells. Nature communications. 2013;4:1754. doi: 10.1038/ncomms2780. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16:1523–1529. doi: 10.1038/nn.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outchkourov NS, Muino JM, Kaufmann K, van Ijcken WF, Groot Koerkamp MJ, van Leenen D, de Graaf P, Holstege FC, Grosveld FG, Timmers HT. Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell reports. 2013;3:1071–1079. doi: 10.1016/j.celrep.2013.02.030. [DOI] [PubMed] [Google Scholar]
- Pan L, Penney J, Tsai LH. Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J Mol Biol. 2014;426:3376–3388. doi: 10.1016/j.jmb.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paralkar VR, Taborda CC, Huang P, Yao Y, Kossenkov AV, Prasad R, Luan J, Davies JO, Hughes JR, Hardison RC, et al. Unlinking an lncRNA from Its Associated cis Element. Mol Cell. 2016;62:104–110. doi: 10.1016/j.molcel.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkel S, Lopez-Atalaya JP, Barco A. Histone H3 lysine methylation in cognition and intellectual disability disorders. Learn Mem. 2013;20:570–579. doi: 10.1101/lm.029363.112. [DOI] [PubMed] [Google Scholar]
- Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, Lin Y. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science. 2011;334:1669–1675. doi: 10.1126/science.1208049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli B, Rosano D, Antonini E, Frenquelli M, Montanini L, Huang D, Segalla S, Yoshihara K, Amin SB, Lazarevic D, et al. Histone demethylase JARID1C inactivation triggers genomic instability in sporadic renal cancer. J Clin Invest. 2015;125:4625–4637. doi: 10.1172/JCI81040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujirabanjerd S, Nelson J, Tarpey PS, Hackett A, Edkins S, Raymond FL, Schwartz CE, Turner G, Iwase S, Shi Y, et al. Identification and characterization of two novel JARID1C mutations: suggestion of an emerging genotype-phenotype correlation. Eur J Hum Genet. 2010;18:330–335. doi: 10.1038/ejhg.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron. 2009;64:678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X, Hu Z, et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell. 2016;165:331–342. doi: 10.1016/j.cell.2016.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simensen RJ, Rogers RC, Collins JS, Abidi F, Schwartz CE, Stevenson RE. Short-term memory deficits in carrier females with KDM5C mutations. Genet Couns. 2012;23:31–40. [PubMed] [Google Scholar]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. 2015;7:503–519. doi: 10.2217/epi.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco G, Hube F, Rollin J, Neuillet D, Philippe C, Bouzinba-Segard H, Galvani A, Viegas-Pequignot E, Francastel C. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc Natl Acad Sci U S A. 2010;107:9281–9286. doi: 10.1073/pnas.1000473107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Burgoyne PS, Arnold AP. Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet. 2002;11:1409–1419. doi: 10.1093/hmg/11.12.1409. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 2016;353:300–305. doi: 10.1126/science.aad4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Related to Figure 3. DHMPs in Kdm5c-KO and ifKO mice (Excel file with two spreadsheets).
Table S4. Related to Figure 4. DEGs in Kdm5c-KO and ifKO mice (Excel file with three spreadsheets).
Table S5. Related to Figure 5. DEGs upon NE (Excel file with two spreadsheets).