

Abstract
The sluggish oxidants, [FeIV(O)(TMC)(CH3CN)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) and [FeIV(O)(d12-TMCN)(OTf)]+ (3d; d12-TMCN = 1,4,7,11-tetra-d3-methyl-1,4,7,11-tetraazacyclotetradecane), are transformed into a highly reactive oxidant, [FeIV(O)(TMCO)(OTf)]+ (1; TMCO = 4,8,12-trimethyl-1-oxa-4,8,12-triazacyclotetradecane), upon replacement of a -NMe donor in the TMC and TMCN ligands by an O-atom. A rate enhancement of 5 – 6 orders of magnitude in both H-atom and O-atom transfer reactions is observed upon oxygen incorporation into the macrocyclic ligand and can be explained based upon the higher electrophilicity of the iron center and the higher availability of the more reactive S = 2 state in 1. This rationalizes nature’s preference for using O-rich ligand environments for the hydroxylation of strong C-H bonds in enzymatic reactions.
Keywords: Oxoiron(IV) Complex, Oxygen-Based Ligands, Spin State Effect, Hydrogen Atom Transfer, Oxygen Atom Transfer
Graphical Abstract
The drastically enhanced reactivity of an oxoiron(IV) center in an N3O environment relative to an N4 environment explains nature’s preference for using oxygen rich ligand environment for the hydroxylation of strong C-H bonds.
High-valent oxoiron(IV) (FeIV=O) species have been identified as key reactive intermediates in the activation of dioxygen and the oxygenation of organic substrates by mononuclear nonheme iron enzymes.[1] The FeIV=O moiety is always found in the quintet (S = 2) spin state, which is predicted by DFT calculations[2] to be more reactive in H-atom transfer (HAT) than the corresponding triplet (S = 1) spin state. The biological FeIV=O oxidants all contain at least two weak-field O-based ligands (either carboxylate or water) cis to the oxo donor (Scheme 1A),[3] which can play a significant role in the stabilization of the S = 2 state by modulating the energy gap between the Fe d(xy) and d(x2–y2) orbitals. A related species is also proposed to be the oxidant formed in the reactions of N2O with coordinately unsaturated iron(II) centers in carboxylate-based metal–organic frameworks capable of catalyzing ethane hydroxylation.[4] While no direct spectroscopic evidence is currently available, DFT studies suggest that this intermediate is an S = 2 FeIV=O species possessing an O-rich ligand environment.
Scheme 1.

Structures of biological (A) and bioinspired (B) oxoiron(IV) complexes.
The reactivity of FeIV=O complexes with weak-field O-rich ligand environments are, therefore, of particular interest. Successful synthesis and characterization of such compounds has been limited by their high reactivity and instability under ambient conditions. For example, the short half-life of 20 s at 25 °C of the S = 2 [FeIV(O)(H2O)5]2+ complex[5] has inhibited a detailed investigation of its spectroscopic and reactivity properties. On the other hand, employment of strong-field pyridine and tertiary amine donors[1a–b,6] or carbene donors[7] has led to the isolation and characterization of several well-characterized S = 1 FeIV=O model complexes with high stability and low reactivity. Efforts to stabilize the more reactive S = 2 state by replacing one or more N-donors with weak carboxylate ligands[8] have previously resulted in only a small enhancement (5 – 10 folds) in the HAT abilities of the complexes. In fact, no FeIV=O complexes supported by O-based ligands that are able to oxidize substrates containing C-H bonds with a bond dissociation energy (BDE) > 80 kcal mol−1 are presently known. This is surprising, considering nature’s preference for employing FeIV=O complexes with O-rich ligand environments to oxidize strong C-H bonds.
As part of our continuing efforts to uncover the structure-reactivity relationship of non-heme oxometal complexes, we report herein the synthesis and characterization of an S = 1 FeIV=O complex [FeIV(O)(TMCO)(OTf)]+ (1) (Scheme 1; TMCO = 4,8,12-trimethyl-1-oxa-4,8,12-triazacyclotetradecane),[9] bearing a weak-field N3O macrocyclic ligand in place of the popular N4-donor TMC-based ligands. 1 exhibits spectroscopic properties distinct from [FeIV(O)(TMC)(CH3CN)]2+ (TMC=1,4,8,11-tetramethyl-1,4,8,11tetraazacyclotetradecane)[10] and [FeIV(O)(d12-TMCN)(OTf)]+ (3d; d12-TMCN=1,4,7,11-tetra-d3-methyl-1,4,7,11-tetraazacyclotetradecane), and, remarkably, 5 – 6 orders of magnitude higher reactivity in HAT and O-atom transfer (OAT) reactions.
Combination of the tetradentate TMCO ligand (Figure S1) with Fe(OTf)2(CH3CN)2 yielded the expected iron(II) complex [FeII(TMCO)(CH3CN)(OTf)](OTf) (2). The X-ray structure of 2 (Figure 1 left and Table S1) displays a 6-coordinate geometry with axially bound triflate and CH3CN ligands. Notably, the macrocyclic ring exhibits a configuration, where all the methyl-groups point cis to CH3CN, with iron-ligand bond lengths (Table S2) typical of a high-spin iron(II) ion. Consistent with this spin-state assignment, the zero-field Mössbauer spectrum of 2 (Figure S2) revealed a single doublet with an isomer-shift (δ) of 1.17 mm/s and a large quadrupole splitting (ΔEQ = 3.32 mm/s). In addition, the 1H, 2D and 19F NMR spectra of 2 in CH2Cl2 display paramagnetically shifted peaks (Figure S3), indicative of coordination of both triflate anions and retention of the pseudo-Cs symmetric solid-state structure in solution.
Figure 1.
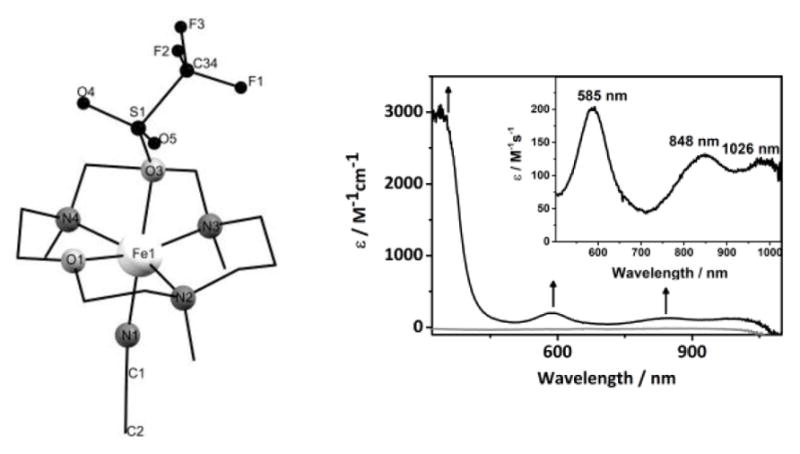
Left: X-ray crystal structure of 2. Right: UV-Absorption spectra of 1 (black) and 2 (gray) in CH2Cl2 at −90 °C. The inset shows the expansion of the 500 – 900 nm region.
The reaction of 2 with 1.5 equiv of 2-(tert-butylsulfonyl)iodosylbenzene (tBuSO2C6H4IO)[11] in anhydrous CH2Cl2 at −90 °C led to the immediate formation of a pale green intermediate 1 (t1/2 = 1.5 h; −50 °C) with absorption maxima λmax (εmax) centered at 390 nm (>3000 M−1 cm−1), 585 nm (200 M−1 cm−1), 848 nm (150 M−1 cm−1) and 1026 nm (150 M−1 cm−1) (Figure 1, right). The electrospray ionization mass spectrum (ESI-MS) of 1 (Figure S4) exhibited a signal at m/z = 464.1, which shifted by two-mass units when tBuSO2C6H4I18O was used as an oxidant, possessing a mass and isotope distribution pattern consistent with a [FeIV(O)(TMCO)(OTf)]+ formulation. The presence of the Fe=O unit in 1 was confirmed by EXAFS analysis that yields a best-fit plot (Figure S5 and Table S3) with an O/N scatterer at 1.64 Å (assigned to the Fe=O unit) and a further shell of 5 O/N scatterers at 2.10 Å (corresponding to the N/O donors of TMCO and OTf− ligands). The Fe K-edge X-ray absorption spectrum of 1 (Figure 2A) reveals an edge energy of 7124.3 eV (versus 7121.5 eV for 2) and a broad pre-edge peak assigned to 1s→3d transitions at 7114.2 eV with an area of 45 units (Table S4). Furthermore, the 1H, 2D and 19F NMR spectra of 1 (Figure S6) indicate that a single Cs-symmetric iron(IV) species is present, in which one of the OTf- anions is coordinated to the metal centre. The spectra are consistent with retention of the geometry of the starting complex 2 and led us to formulate 1 as [FeIV(O)(TMCO)(OTf)]+.
Figure 2.
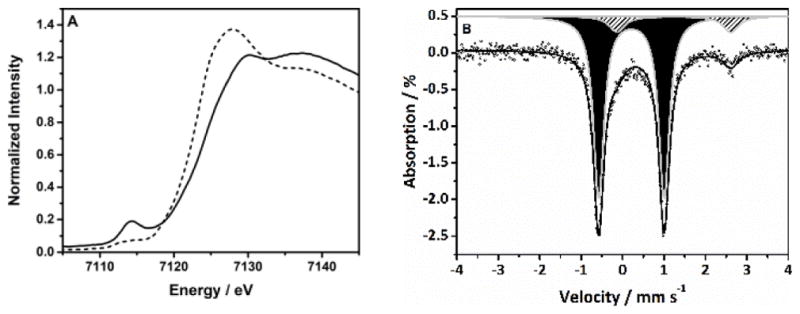
A) Normalized Fe K-edge X-ray absorption spectra of 1 (solid line) and 2 (dashed line) in a CH3CH2CN/CH2Cl2 solvent mixture (10% CH2Cl2 by volume); B) The zero field Mössbauer spectrum of 1 recorded at 4.2 K. The solid line is the simulation of the experimental spectrum (black dots) representing >95% of the absorption. The major component (90%; black shaded area) corresponds to 1; the minor component (10 %; gray shaded area) corresponds to unreacted 2.
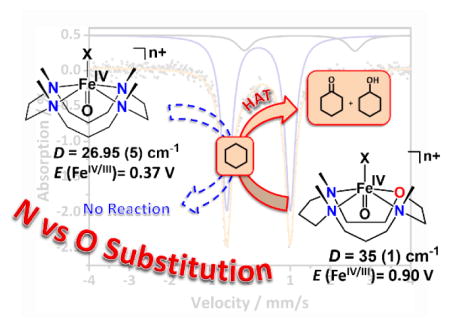
The zero field Mössbauer spectrum of 1 recorded at 4.2 K exhibits a doublet representing ca. 90% of the iron with ΔEQ = 1.58 mm s−1 and δ = 0.13 mm s−1 (Figure 2B); the remaining 10% of the signal corresponds to unreacted 2. Applied magnetic fields, B, elicit magnetic hyperfine interactions (Figure S7) with parameters typical of S = 1 FeIV=O complexes. Within resolution, the zero-field splitting (ZFS) tensor, described by the parameters D and E, was found to be axial (D = 35 (±3) cm−1; E/D = 0.0(+0.05). Notably, the D value of 1 is significantly higher than that reported for [FeIV(O)(TMC)(CH3CN)]2+ and all other oxoiron(IV) complexes (Table 1 and Table S4); this may point to a more accessible S = 2 excited state in 1.[1]
Table 1.
A comparison of the redox potentials, zero-field splitting and second-order rate constants (k2) with C6H12 and PhSMe for 1, 3d and [FeIV(O)(TMC)(CH3CN)]2+
| Complex | D [cm−1] | E(FeIV/III) vs SCE | k2[a](C6H12) [M−1 s−1] | k2[a](PhSMe) [M−1 s−1] |
|---|---|---|---|---|
| 1 | 35±3 | 0.90 | 1 × 10−2 | 2.5 × 102 |
| 3d | - | 0.37 | no reaction | 4.0 × 10−3 |
| [FeIV(O)(TMC)(CH3CN)]2+ [1a] | 26.95 ±0.15 | 0.39 | no reaction | 1.3 × 10−3 |
@ −40 °C
The oxidative reactivity of 1 was examined in the oxidation of hydrocarbons with C–H BDEs of 77 – 99.3 kcal mol−1 (Table S5 and Figure S8). The experimentally determined (see SI for details) second-order rate constants, k2, were then adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C–H bonds of substrates; the k2′ values were correlated with the BDEs of the substrates (Figure 3 left and Table S4). This observation is consistent with the rate-determining step of the reaction being C–H bond cleavage, as is expected in HAT reactions. Furthermore, a deuterium kinetic isotope effect (KIE) of 24.8 was obtained when ethylbenzene-d10 was used as a substrate (Figure 3 right). Such a large KIE value implies a tunneling mechanism in H-atom abstraction, as is frequently proposed in C–H bond oxidation reactions of FeIV=O species.[1,6] Product analysis of the reaction mixture for cyclohexane reaction revealed the formation of cyclohexanol (35% yield), cyclohexanone (5%) and high-spin iron(III) (>90% yield; Figure S9) products. Thus, 1 performs HAT in an oxygen non-rebound mechanism,[12] as proposed earlier for other nonheme FeIV=O complexes.
Figure 3.
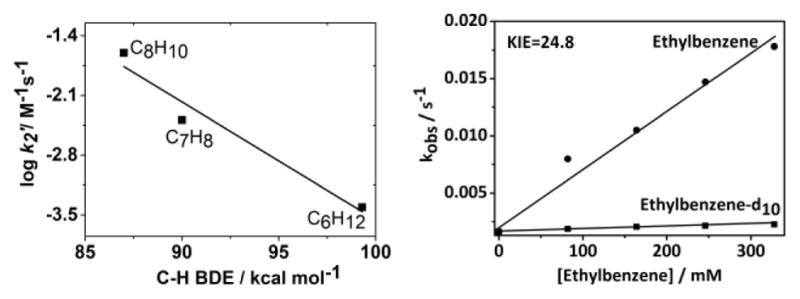
Left: Plot of log k2′ vs C-H BDE of substrates for 1. Right: Plot of the pseudo-first-order rate constants, kobs (s−1), against substrate concentrations to determine second-order rate constants, k2, and C–H kinetic isotope effect (KIE) value for the reaction of 1 with ethylbenzene.
Comparison of the HAT reactivities of 1 and other oxoiron(IV) complexes (Tables 1 and S4) shows that the rate of cyclohexane oxidation by 1 at −40 °C (0.01 M−1 s−1) is only an order of magnitude lower than that of the two most reactive complexes reported to date; namely, the S = 2 [FeIV(O)(TQA)(CH3CN)]2+ (0.37 M−1 s−1)[13] and S = 1 [FeIV(O)(Me3NTB)(CH3CN)]2+ (0.25 M−1 s−1)[14] complexes. In particular, the closely related S = 1 [FeIV(O)(TMC)(CH3CN)]2+ complex does not react at all with cyclohexane even at 25 °C. Furthermore, comparison of the OAT abilities of 1 (Table S4, Figure S10) and [FeIV(O)(TMC)(CH3CN)]2+ towards thioanisole (PhSMe) shows that the former reacts five-orders of magnitude faster than the latter at −40 °C.[15]
The TMCO ligand differs from TMC not only in being a N3O rather than a N4 macrocycle, but also in having the two propylene (and also the two ethylene) spacers adjacent, rather than alternating with respect to one another (Scheme 1). A comparison of the reactivity of 1 with that of the [FeIV(O)(TMCN)(CH3CN)]2+ (3; TMCN = 1,4,7,11-tetramethyl-1,4,7,11-tetraazacyclotetradecane) is, therefore, warranted for a proper analysis of the effect of N- vs O-donors in an otherwise identical ligand environment.
From the 1H and 2D NMR spectra of [FeII(TMCN)(CH3CN)]2+, it was discovered that the chemically equivalent methyl groups (and ethylene and propylene units) of TMCN are magnetically inequivalent (Figure S11). This indicates that these two methyl substituents point in different directions and results in the complex being C1 symmetric. The zero-field Mössbauer spectrum of [FeII(TMCN)(CH3CN)]2+ (Figure S12) revealed a single doublet with δ = 1.20 mm s−1 and ΔEQ = 2.90 mm s−1 typical of a high-spin iron(II) ion.
The reaction of [FeII(TMCN)(CH3CN)]2+ with tBuSO2C6H4IO in CH3CN at −40 °C revealed the formation of a transient intermediate 3, whose fast decay prevented its isolation. The instability of 3 can be presumably attributed to its self-decay via intramolecular attack of a ligand methyl C–H bond (cis to the oxo group) by the Fe=O unit (see DFT calculation below). With an assumption that a large KIE would be involved for such a self-decay mechanism, the perdeuterated d12-TMCN ligand was synthesized in an effort to stabilize the corresponding complex [FeIV(O)(d12-TMCN)(OTf)]+ (3d). Indeed, the reaction of [FeII(d12-TMCN)(CH3CN)]2+ with C6H4IO in CH3CN at 0 °C led to a clean conversion to 3d (t1/2 = ~8 min at 0 °C), as confirmed by NMR (Figure S13), UV-Vis (Figure S14; λmax = 820 nm; ε = 300 M−1 cm−1) and ESI-MS (Figure S15). The oxidative reactivity of 3d, when tested in intermolecular HAT and OAT reactions, shows it to be a sluggish oxidant similar to [FeIV(O)(TMC)(CH3CN)]2+ (Tables 1 and S4).
The electrochemistry of 1, [FeIV(O)(TMC)(CH3CN)]2+, and 3d was then investigated as a means to gain insight into the observed reactivity and provide a more quantitative basis for the influence of the TMCO, TMC and d12-TMCN ligands, respectively, on the FeIV=O center (Figure S16). Cyclic voltammetry (CV) of [FeIV(O)(TMC)(CH3CN)]2+ has previously revealed an FeIV/III potential of +0.39 V vs. SCE in CH3CN.[16] A comparable FeIV/III potential of +0.37 V has been determined for 3d in CH3CN. In contrast, a large positive shift of the FeIV/III potential to +0.90 V was observed for 1 in trifluoroethanol, which justifies the much higher HAT and OAT capabilities of 1 relative to [FeIV(O)(TMC)(CH3CN)]2+ and 3d.
DFT calculations (Figure S17; Table S6) for [FeIV(O)(TMCO)(CH3CN)]2+ (1-CH3CN) and [FeIV(O)(TMC)(CH3CN)]2+ provided further insight into their differing reactivity. For both complexes, a ground-state (GS) with a triplet (S = 1) spin quantum number and a low-lying quintet (S = 2) spin state were predicted. In [FeIV(O)(TMC)(CH3CN)]2+, the S = 2 state is 5 kcal mol−1 higher in energy than the S = 1 GS, but for 1-CH3CN, this spin-state splitting is reduced to only 1 kcal mol−1. Free energies, which has been shown[17] to better rationalize the different reactivities of the oxoiron(IV) complexes than electronic energies, have also been determined for 1-CH3CN and [FeIV(O)(TMC)(CH3CN)]2+ in the temperature range 10 – 370 K (Tables S7, S8). While the S = 1 state is preferred for [FeIV(O)(TMC)(CH3CN)]2+ at all temperatures, the S = 2 state is the predicted GS at the experimental temperature of 223 K for reactivity studies. The reaction profile for HAT from cyclohexane by 1-CH3CN and [FeIV(O)(TMC)(CH3CN)]2+ in the S = 1 and S = 2 spin states is displayed in Figure S18. The obtained barriers (Table S6) are consistent with the experimental findings, with a large reduction of the barrier upon going from [FeIV(O)(TMC)(CH3CN)]2+ to 1-CH3CN in both the S = 1 and S = 2 spin states; this together with the higher accessibility of the more reactive S = 2 spin state for 1 may rationalize its enhanced oxidative reactivity relative to [FeIV(O)(TMC)(CH3CN)]2+.
Calculations were also performed for the complex [FeIV(O)(TMCN)(CH3CN)]2+, for which one of the N-methyl groups was predicted to be syn to the Fe=O unit (consistent with experiment) in its most stable configuration (Figure S17C). The barrier for self-decay of [FeIV(O)(TMCN)(CH3CN)]2+ by an intramolecular HAT from the syn methyl group is 13 – 14 kcal mol−1 lower than the oxidation barrier for cyclohexane (Figure S18), explaining the inherent instability of [FeIV(O)(TMCN)(CH3CN)]2+.
In summary, we have generated a highly reactive oxoiron(IV) complex 1 by weakening the ligand field about the FeIV=O unit via introduction of an O-atom into the N4 macrocycle of the TMC or TMCN ligands. The FeIV=O center goes from being a sluggish oxidant (within an N4 macrocycle) to being an extremely reactive species (in an N3O environment) that can hydroxylate alkanes with C-H bonds as strong as 99.3 kcal mol−1. Furthermore, a dramatic enhancement of the rate of OAT to PhSMe by five-orders of magnitude was observed upon swapping to the N3O coordination sphere. Complex 1 represents the only example of a highly reactive FeIV=O complex supported by an O-containing ligand system, and explains nature’s preference for using O-rich ligand environment for the hydroxylation of strong C-H bonds.
Supplementary Material
Acknowledgments
Financial supports from the DFG (Cluster of Excellence “Unifying Concepts in Catalysis”; EXC 314-2, KR), MINECO (CTQ2014-59212-P and CTQ2015-70851-ERC, MS), GenCat (2014SGR1202, MS), FEDER (UNGI10-4E-801, MS), and COST action CM1305 “ECOSTBio” are gratefully acknowledged. K.R. also thanks the Heisenberg-Program of DFG for financial support. J.E. thanks NTU for funding. W.N. acknowledges financial support from NRF of Korea through CRI (NRF-2012R1A3A2048842) and GRL (NRF-2010-00353). XAS experiments were conducted at SSRL beamline 7-3 (SLAC National Accelerator Laboratory, USA), with support from the DOE Office of Science (DE-AC02-76SF00515) and NIH (P30-EB-009998 and P41-GM-103393).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Engelmann X, Pérez IM, Ray K. Angew Chem Int Ed. 2016;55:7632. doi: 10.1002/anie.201600507. [DOI] [PubMed] [Google Scholar]; b) Ray K, Pfaff FF, Wang B, Nam W. J Am Chem Soc. 2014;136:13942. doi: 10.1021/ja507807v. [DOI] [PubMed] [Google Scholar]; c) Fujimori DG, Walsh CT, Bollinger JM, Jr, Krebs C. Acc Chem Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 2.a) Hirao H, Kumar D, Shaik S. Acc Chem Res. 2007;40:532. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]; b) Bernasconi L, Louwerse MJ, Baerends EJ. Eur J Inorg Chem. 2007:3023. [Google Scholar]; c) Geng C, Ye S, Neese F. Angew Chem Int Ed. 2010;49:5717. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]; d) Chen H, Janardanan D, Shaik S. Nat Chem. 2011;3:19. doi: 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- 3.a) Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Schofield CJ, Hajdu J, Baldwin JE. Nature. 1997;387:827. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]; b) Burzlaff NI, Rutledge PJ, Clifton IJ, Hensgens CMH, Pickford M, Adlington RM, Roach PL, Baldwin JE. Nature. 1999;401:721. doi: 10.1038/44400. [DOI] [PubMed] [Google Scholar]; c) Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. J Am Chem Soc. 2007;129:6168. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 4.Xiao DJ, Bloch ED, Mason JA, Queen WL, Hudson MR, Planas N, Borycz J, Dzubak AL, Verma P, Lee K, Bonino F, Crocellà V, Yano J, Bordiga S, Truhlar DG, Gagliardi L, Brown CM, Long JR. Nat Chem. 2014;6:590. doi: 10.1038/nchem.1956. [DOI] [PubMed] [Google Scholar]
- 5.Pestovsky O, Stoian S, Bominaar EL, Shan X, Münck E, Que L, Jr, Bakac A. Angew Chem Int Ed. 2005;44:6871. doi: 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- 6.a) Puri M, Que L., Jr Acc Chem Res. 2015;48:2443. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nam W. Acc Chem Res. 2015;48:2415. [Google Scholar]
- 7.a) Meyer S, Klawitter I, Demeshko S, Bill E, Meyer F. Angew Chem Int Ed. 2013;52:901. doi: 10.1002/anie.201208044. [DOI] [PubMed] [Google Scholar]; b) Kupper C, Meyer S, Andris E, Navrátil R, Krahe O, Mondal B, Atanasov M, Bill E, Roithová J, Ye S, Meyer F, Neese F. J Am Chem Soc. 2016;138:4312. doi: 10.1021/jacs.6b07708. [DOI] [PubMed] [Google Scholar]; c) Kupper C, Mondal B, Serrano-Plana J, Klawitter I, Neese F, Costas M, Ye S, Meyer F. J Am Chem Soc. 2017;139:8939. doi: 10.1021/jacs.7b03255. [DOI] [PubMed] [Google Scholar]
- 8.a) Sastri CV, Lee J, Oh K, Lee YJ, Lee J, Jackson TA, Ray K, Hirao H, Shin W, Halfen JA, Kim J, Que L, Jr, Shaik S, Nam W. Proc Natl Acad Sc. 2007;104:19181. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McDonald AR, Guo Y, Vu VV, Bominaar EL, Münck E, Que L., Jr Chem Sci. 2012;1:1680. doi: 10.1039/C2SC01044E. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rohde JU, Stubna A, Bominaar EL, Münck E, Nam W, Que L., Jr Inorg Chem. 2006;45:6435. doi: 10.1021/ic060740u. [DOI] [PubMed] [Google Scholar]
- 9.Amorim MTS, Chaves S, Delgado R, Fraústo da Silva JJR. Dalton Trans. 1991:3065. [Google Scholar]
- 10.Rohde J-U, J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 11.Macikenas D, Skrzypczak-Jankun E, Protasiewicz JD. J Am Chem Soc. 1999;121:7164. doi: 10.1002/1521-3773(20000602)39:11<2007::aid-anie2007>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 12.Cho KB, Wu X, Lee YM, Kwon YH, Shaik S, Nam W. J Am Chem Soc. 2012;134:20222. doi: 10.1021/ja308290r. [DOI] [PubMed] [Google Scholar]
- 13.Biswas AN, Puri M, Meier KK, Oloo WN, Rohde GT, Bominaar EL, Münck E, Que L., Jr J Am Chem Soc. 2015;137:2428. doi: 10.1021/ja511757j. [DOI] [PubMed] [Google Scholar]
- 14.Seo MS, Kim NH, Cho KB, So JE, Park SK, Clémancey M, Garcia-Serres R, Latour JM, Shaik S, Nam W. Chem Sci. 2011;2:1039. [Google Scholar]
- 15.Hong S, So H, Yoon H, Cho KB, Lee YM, Fukuzumi S, Nam W. Dalton Trans. 2013;42:7842. doi: 10.1039/c3dt50750e. [DOI] [PubMed] [Google Scholar]
- 16.Lee YM, Kotani H, Suenobu T, Nam W, Fukuzumi S. J Am Chem Soc. 2008;130:434. doi: 10.1021/ja077994e. [DOI] [PubMed] [Google Scholar]
- 17.Lee NY, Mandal D, Bae SH, Seo MS, Lee Y-M, Shaik S, Cho KB, Nam W. Chem Sci. 2017;8:5460. doi: 10.1039/c7sc01738c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.