

This FDA approval summary provides an update on approval of pembrolizumab for treatment of patients with metastatic non‐small cell lung cancer whose tumors express PD‐L1 as determined by an FDA‐approved test. The results of KEYNOTE‐010 and KEYNOTE‐024 trials are presented.
Keywords: Non‐small cell lung cancer; Pembrolizumab; EGFR protein, Human; United States Food and Drug Administration
Abstract
On October 24, 2016, the U.S. Food and Drug Administration (FDA) approved pembrolizumab (Keytruda; Merck & Co., Inc., https://www.merck.com) for treatment of patients with metastatic non‐small cell lung cancer (mNSCLC) whose tumors express programmed death‐ligand 1 (PD‐L1) as determined by an FDA‐approved test, as follows: (a) first‐line treatment of patients with mNSCLC whose tumors have high PD‐L1 expression (tumor proportion score [TPS] ≥50%), with no epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations, and (b) treatment of patients with mNSCLC whose tumors express PD‐L1 (TPS ≥1%), with disease progression on or after platinum‐containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA‐approved therapy for these aberrations prior to receiving pembrolizumab.
Approval was based on two randomized, open‐label, active‐controlled trials demonstrating statistically significant improvements in progression‐free survival (PFS) and overall survival (OS) for patients randomized to pembrolizumab compared with chemotherapy. In KEYNOTE−024, patients with previously untreated mNSCLC who received pembrolizumab (200 mg intravenously [IV] every 3 weeks) had a statistically significant improvement in OS (hazard ratio [HR] 0.60; 95% confidence interval [CI]: 0.41–0.89; p = .005), and significant improvement in PFS (HR 0.50; 95% CI: 0.37–0.68; p < .001). In KEYNOTE‐010, patients with disease progression on or after platinum‐containing chemotherapy received pembrolizumab IV 2 mg/kg, 10 mg/kg, or docetaxel 75 mg/m2 every 3 weeks. The HR and p value for OS was 0.71 (95% CI: 0.58–0.88), p < .001 comparing pembrolizumab 2 mg/kg with chemotherapy and the HR and p value for OS was 0.61 (95% CI: 0.49–0.75), p < .001 comparing pembrolizumab 10 mg/kg with chemotherapy.
Implications for Practice.
This is the first U.S. Food and Drug Administration approval of a checkpoint inhibitor for first‐line treatment of lung cancer. This approval expands the pembrolizumab indication in second‐line treatment of lung cancer to include all patients with programmed death‐ligand 1‐expressing non‐small cell lung cancer.
Introduction
Prior to the U.S. Food and Drug Administration's (FDA) approval of pembrolizumab for first‐line treatment of non‐small cell lung cancer (NSCLC) in 2016, several immunotherapy agents, including pembrolizumab, had been approved for second‐line treatment. Identification of driver mutations in the kinase domain of the epidermal growth factor receptor (EGFR) gene and rearrangements of the anaplastic lymphoma kinase (ALK) gene in lung cancer led to development and approval of several molecularly targeted drugs that have been shown to improve objective reponse rates (ORR) and progression‐free survival (PFS) in patients with NSCLC containing these driver mutations. Epidermal growth factor receptor tyrosine kinase inhibitors, which include erlotinib [1], afatinib [2], and gefitinib [3], and the ALK inhibitor crizotinib, were approved for the first‐line treatment of patients with metastatic non‐small cell lung cancer (mNSCLC) who harbor these mutations.
In addition, two monoclonal antibodies, approved for use as first‐line treatment in combination with platinum‐doublet chemotherapy for mNSCLC have been shown to improve overall survival (OS) in randomized, controlled trials: bevacizumab (nonsquamous histology) [4] and necitumumab (squamous histology) [5].
Despite these advances, the standard of care in the first line for patients with advanced, inoperable NSCLC whose tumors do not harbor EGFR mutations or ALK rearrangements remained chemotherapy with cisplatin or carboplatin‐based doublets. Clinical trials of various first‐line platinum‐based chemotherapy regimens demonstrated ORRs from 12% to 37%, median PFS from 4 to 7 months, median OS from 8 to 13 months, and a 1‐year survival rate of approximately 33% [1], [6] in these patients. The toxicity of platinum‐based therapy can be significant, with myelosuppression, infection, neuropathy, and gastrointestinal symptoms as the most common grade 3–5 toxicities. Treatment‐related deaths have been reported in 1%–3% of patients [7]. Treatment options for patients with mNSCLC who progress after platinum‐based chemotherapy (second line and beyond) included nivolumab, docetaxel (with or without ramucirumab), pemetrexed, atezolizumab, and pembrolizumab.
Pembrolizumab (Keytruda Merck & Co., Inc., https://www.merck.com) is a monoclonal antibody that binds to the programmed cell death protein 1 (PD‐1) receptor and blocks its interaction with programmed death‐ligands 1 (PD‐L1) and 2 (PD‐L2), releasing PD‐1 pathway‐mediated inhibition of antitumor immune response. Up‐regulation of PD‐1 ligands occurs in some tumors and signaling through this pathway can contribute to inhibition of active T‐cell immune surveillance of tumors. On October 2, 2015, the FDA granted accelerated approval to pembrolizumab for treatment of patients with mNSCLC whose tumors express PD‐L1 as determined by an FDA‐approved test with disease progression on or after platinum‐containing chemotherapy. The approval specified that patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA‐approved therapy for these aberrations prior to receiving pembrolizumab. Approval was based on durable ORR in a subgroup of patients whose tumors expressed PD‐L1 with a tumor proportion score (TPS) ≥50%, defined as ≥50% of tumor cells expressing PD‐L1, observed in KEYNOTE‐001, an open‐label, single‐arm, multicenter trial [8].
Subsequently, Merck submitted two supplemental Biologic License Applications (sBLAs) for approval of pembrolizumab for (a) first‐line treatment of mNSCLC patients whose tumors have high PD‐L1 expression with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for mNSCLC; and (b) treatment of mNSCLC patients whose tumors express PD‐L1 and who have disease progression on or after platinum‐containing chemotherapy. The applications were supported with data from two randomized controlled trials—KEYNOTE‐024 [9] and KEYNOTE‐010 [10]—demonstrating statistically significant improvements in PFS and OS for patients randomized to pembrolizumab compared with chemotherapy. KEYNOTE‐010 fulfilled Merck's postmarketing requirement to verify the clinical benefit of pembrolizumab in a randomized clinical trial, establishing the superiority of pembrolizumab over available therapy in patients with PDL1‐positive mNSCLC previously treated with platinum‐containing chemotherapy.
Clinical Trial Designs
KEYNOTE‐024 was a randomized, multicenter, open‐label, actively‐controlled trial in patients with mNSCLC whose tumors had high PD‐L1 expression (TPS ≥50%) as determined by immunohistochemistry (IHC) at a central laboratory and who had not received prior systemic treatment for mNSCLC. Patients with EGFR or ALK genomic tumor aberrations were ineligible. Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (PS; 0 vs. 1), histology (squamous vs. nonsquamous), and geographic region (East Asia [Japan] vs. non‐East Asia). Eligible patients were randomized in a 1:1 ratio to receive pembrolizumab 200 mg intravenously (IV) every 3 weeks (Q3W) or investigator's choice of any of the following standard‐of‐care (SOC) platinum‐containing chemotherapy regimens for 4–6 cycles: (a) pemetrexed 500 mg/m2 Q3W and carboplatin area under the curve (AUC) 5–6 mg/mL per minute Q3W on day 1 for 4–6 cycles followed by optional pemetrexed 500 mg/m2 Q3W for patients with nonsquamous histologies; (b) pemetrexed 500 mg/m2 Q3W and cisplatin 75 mg/m2 Q3W on day 1 followed by optional pemetrexed 500 mg/m2 Q3W (for nonsquamous histologies only); (c) gemcitabine 1,250 mg/m2 on days 1 and 8 and cisplatin 75 mg/m2 Q3W on day 1; (d) gemcitabine 1,250 mg/m2 on days 1 and 8 and carboplatin AUC 5 to 6 mg/mL per minute Q3W on day 1; or (e) paclitaxel 200 mg/m2 Q3W and carboplatin AUC 5 to 6 mg/mL per minute Q3W on day 1 followed by optional pemetrexed maintenance (for nonsquamous histologies only).
Treatment with pembrolizumab continued until Response Evaluation Criteria in Solid Tumors (RECIST) 1.1‐defined progression of disease as determined by a blinded independent radiology committee (BIRC), unacceptable toxicity, or for up to 24 months. The primary objective was to compare PFS per RECIST 1.1 as assessed by BIRC review with SOC chemotherapies. Secondary objectives were to evaluate safety and tolerability, OS, and ORR. Approximately 305 patients were needed to observe 175 PFS events and 110 deaths in the 2 randomized arms, providing an approximate 97% power to detect a hazard ratio (HR) of 0.55 with a 1‐sided alpha of 2%. The final OS analysis was planned to occur at 170 deaths, and would provide 75% power to observe an HR <1 assuming approximately 70% crossover from the chemotherapy arm to the pembrolizumab arm.
KEYNOTE‐010 was a multicenter, worldwide, randomized, adaptively designed trial of IV pembrolizumab at two dosing schedules versus docetaxel in patients with mNSCLC with PD‐L1 positive tumors (TPS ≥1%) who experienced disease progression after at least two cycles of platinum‐containing systemic therapy. Patients were randomized in a 1:1:1 ratio to receive pembrolizumab at 2 mg/kg administered IV over 30 minutes Q3W, pembrolizumab at 10 mg/kg IV over 30 minutes Q3W, or docetaxel at 75 mg/m2 IV over 1 hour Q3W.
Patients were stratified by PD‐L1 status (weak [TPS = 1%–49%] vs. strong [TPS ≥50%]), ECOG PS (0 vs. 1), and geographic region of the enrolling site (East Asia [Japan, Korea, and Taiwan] vs. non‐East Asia). Programmed death‐ligand 1 expression levels were measured in fresh NSCLC tumor tissues by IHC, analyzed by the FDA‐approved Dako Clinical Trial Assay by using the 22C3 clone against PD‐L1. Enrollment was limited to patients with mNSCLC having TPS ≥1%, that is, ≥1% of tumor cells staining positive for PD‐L1 expression. Treatment with pembrolizumab continued until unacceptable toxicity, documented disease progression, 2 years of uninterrupted pembrolizumab, or 35 administrations of pembrolizumab. Clinically stable patients with unconfirmed disease progression were eligible to continue treatment with pembrolizumab at the discretion of the treating physician until confirmed progression at the next tumor staging evaluation.
Primary objectives were to compare the OS and PFS in the strongly positive PD‐L1 stratum compared with docetaxel; to compare OS and PFS in the weakly positive PD‐L1 stratum compared with docetaxel; and to evaluate the safety and tolerability profile of pembrolizumab in the strong and weak groups. The study was considered to have met its primary objective in the primary population of subjects with overall positive or strongly positive PD‐L1 (TPS >50%) expression if at least one pembrolizumab arm was superior to docetaxel either in PFS or in OS at an interim analysis or the final analysis.
Clinical Pharmacology
Updated population pharmacokinetic analyses, pooling data from KEYNOTE‐024 and KEYNOTE‐010 trials with data from Merck trials KEYNOTE‐001 (solid tumors, including melanoma and NSCLC), KEYNOTE‐002 (melanoma), and KEYNOTE‐006 (melanoma), suggested that the observed pembrolizumab exposure in mNSCLC patients receiving the 200 mg Q3W regimen was within the therapeutic exposure range previously observed from 2 mg/kg Q3W to 10 mg/kg Q2W in melanoma and mNSCLC patients. Previous analyses indicated that the observed pembrolizumab exposures in patients with squamous cell carcinoma of the head and neck (HNSCC) treated with pembrolizumab 200 mg Q3W were similar to the pembrolizumab exposure data observed in patients with other solid tumors (mainly melanoma and NSCLC) receiving pembrolizumab 2 mg/kg Q3W, supporting a fixed 200 mg Q3W dose for the HNSCC indication. Based on the totality of data, FDA recommended pembrolizumab 200 mg fixed dose Q3W for the treatment of patients with metastatic NSCLC, irrespective of prior line of therapy.
Results
KEYNOTE‐024 randomized 305 patients with no prior systemic chemotherapy treatment for mNSCLC: 154 patients to the pembrolizumab arm and 151 to the chemotherapy arm. Population characteristics were as follows: median age of 65 years (range: 33–90), 54% age 65 or older; 61% male; 82% white and 15% Asian; 65% ECOG PS of 1; 18% with squamous histology and 82% with nonsquamous histology; and 9% with history of brain metastases. A total of 66 patients (44%) in the chemotherapy arm received pembrolizumab following disease progression.
KEYNOTE‐010 randomized 1,033 patients with disease progression on or after platinum‐containing chemotherapy. Population characteristics were as follows: median age of 63 years (range: 20–88), 42% age 65 or older; 61% male; 72% white and 21% Asian; 66% ECOG PS of 1; 21% with squamous histology, 70% with nonsquamous histology, and 8% with mixed, other, or unknown histology; 91% metastatic (M1) disease; 15% with history of brain metastases; and 8% and 1% with EGFR and ALK genomic aberrations, respectively. All patients received prior therapy with a platinum‐doublet regimen; 29% received two or more prior therapies for their metastatic disease. Programmed death‐ligand 1 tumor expression was low (TPS = 1%–49%) in 57% of the patients and high (TPS ≥50%) in 43% of the patients.
Efficacy
The primary efficacy endpoint of KEYNOTE‐024 was PFS as assessed by a BIRC according to RECIST 1.1. Secondary efficacy endpoints were OS and ORR as assessed by the BIRC according to RECIST 1.1. Key efficacy results are summarized in Table 1. There was a statistically significant and clinically meaningful improvement in PFS for patients in the pembrolizumab arm compared with those in the SOC arm. The Kaplan‐Meier curve for PFS is shown in Figure 1. At a prespecified interim analysis with 64% of the events needed for the final analysis, there was a statistically significant improvement in OS among patients randomized to receive pembrolizumab. At the time of data cutoff, median survival times had not been reached in either treatment arm. The OS and PFS improvements were similar across subgroups defined by age, gender, race, baseline PS, geographic region, and histology. The ORR was significantly higher in the pembrolizumab arm compared with the SOC arm.
Table 1. KEYNOTE‐024 summary of efficacy results.
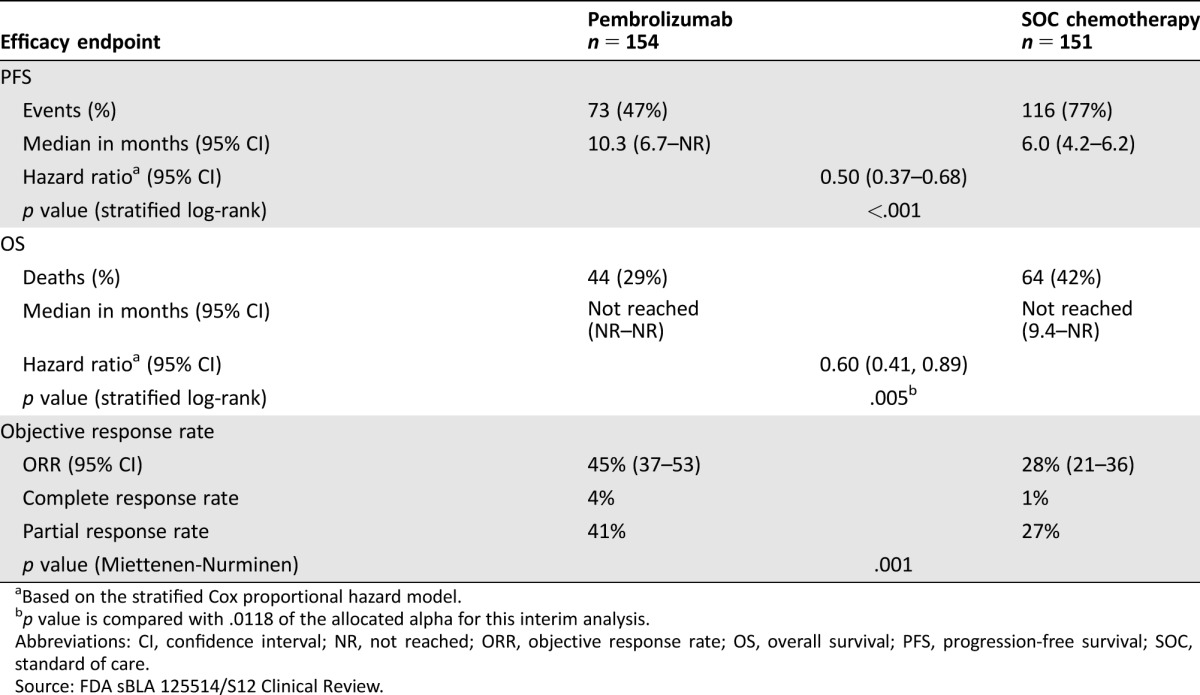
Based on the stratified Cox proportional hazard model.
p value is compared with .0118 of the allocated alpha for this interim analysis.
Abbreviations: CI, confidence interval; NR, not reached; ORR, objective response rate; OS, overall survival; PFS, progression‐free survival; SOC, standard of care.
Source: FDA sBLA 125514/S12 Clinical Review.
Figure 1.

KEYNOTE‐024 Kaplan‐Meier curves of progression‐free survival by independent review committee (Intent to Treat population).
Abbreviation: SOC, standard of care.
Source: FDA sBLA 125514/S12 Statistical Review.
In KEYNOTE‐010, the coprimary efficacy endpoints were OS and PFS as determined by a BIRC in the subgroup of patients with TPS ≥50% and in the intent‐to‐treat (TPS ≥1%) population. Key efficacy results in the TPS ≥50% subgroup and in the overall population are summarized in Tables 2 and 3. In the overall population, there was a statistically significant improvement in OS in patients who received pembrolizumab 2 mg/kg or pembrolizumab 10 mg/kg compared with docetaxel. The Kaplan‐Meier curves for OS are shown in Figure 2. Due to coprimary endpoints and multiple arm comparisons of the trial, an alpha of .001 was allocated to the analysis of PFS endpoints, and the trial did not demonstrate a statistically significant improvement in PFS for patients randomized to receive pembrolizumab 10 mg/kg (stratified log‐rank p = .005) or pembrolizumab 2 mg/kg (stratified log‐rank p = .068) when compared with docetaxel. Objective response rates per RECIST 1.1 by a BIRC assessment were 18% for both pembrolizumab arms and 9% in the docetaxel arm.
Table 2. KEYNOTE‐010 efficacy results: tumor proportion score ≥50% stratum.
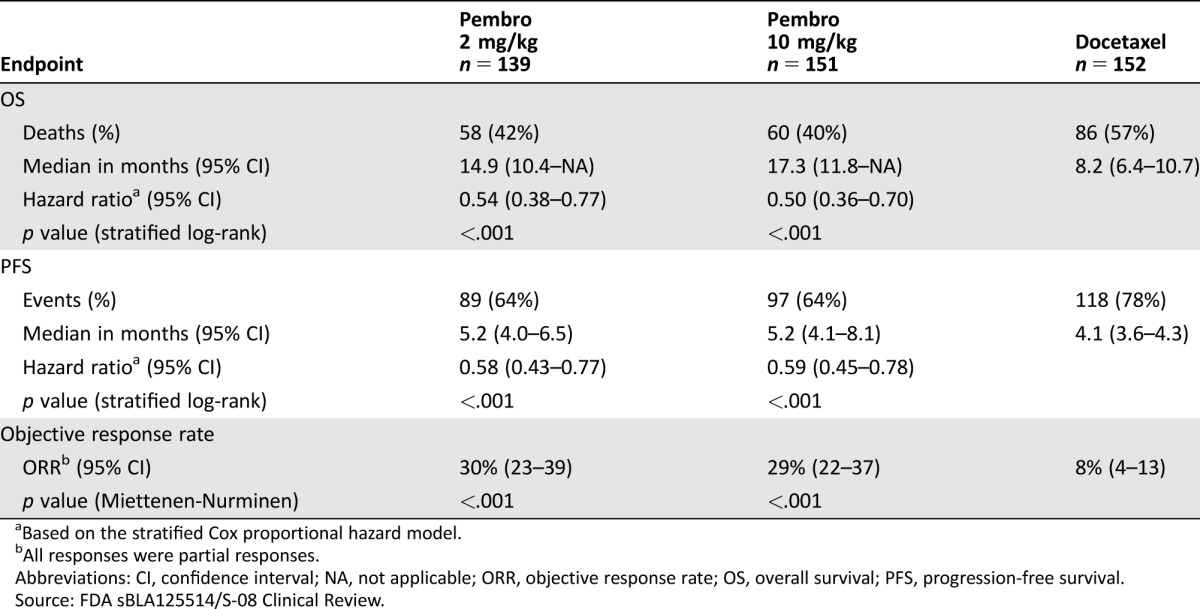
Based on the stratified Cox proportional hazard model.
All responses were partial responses.
Abbreviations: CI, confidence interval; NA, not applicable; ORR, objective response rate; OS, overall survival; PFS, progression‐free survival.
Source: FDA sBLA125514/S‐08 Clinical Review.
Table 3. KEYNOTE‐010 efficacy results: tumor proportion score ≥1% population (all randomized).
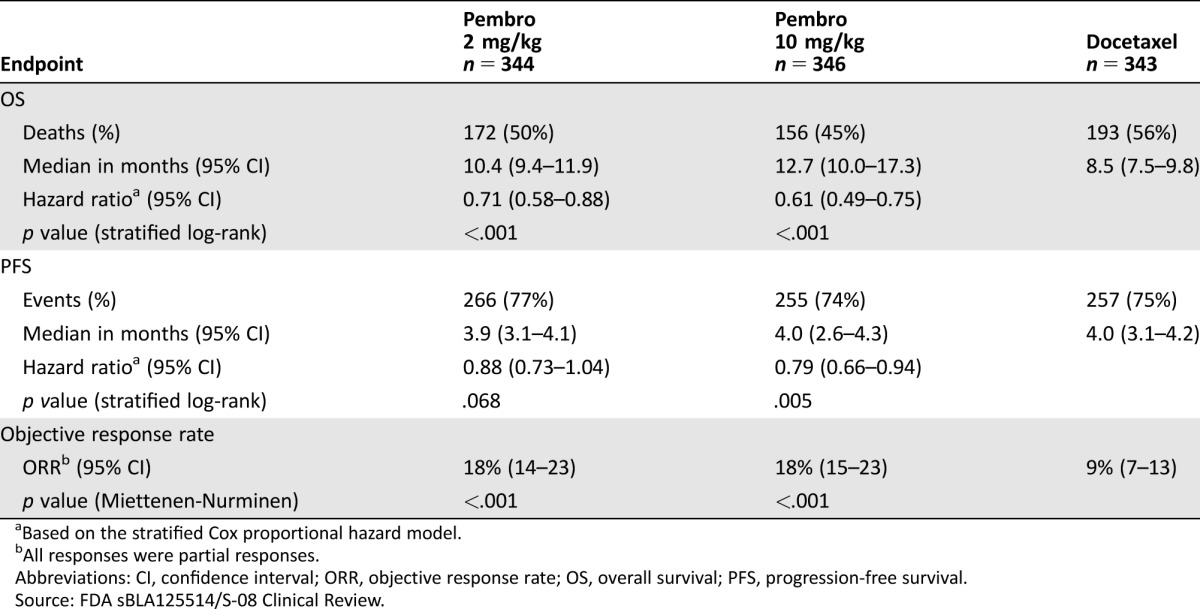
Based on the stratified Cox proportional hazard model.
All responses were partial responses.
Abbreviations: CI, confidence interval; ORR, objective response rate; OS, overall survival; PFS, progression‐free survival.
Source: FDA sBLA125514/S‐08 Clinical Review.
Figure 2.

KEYNOTE‐010: Kaplan‐Meier curves of overall survival (tumor proportion score ≥1%, Intent to Treat population).
Source: FDA sBLA125514/S‐08 Statistical Review.
In the TPS ≥50% stratum, there was a statistically significant improvement in OS in patients who received pembrolizumab 2 mg/kg or pembrolizumab 10 mg/kg compared with docetaxel, which was larger in magnitude than that for the overall population. Progression‐free survival and ORR were also statistically significantly improved for both pembrolizumab 2 mg/kg and pembrolizumab 10m/kg arms compared with docetaxel.
To assess whether the OS benefit in the overall population in KEYNOTE‐010 was not driven by patients with high PD‐L1 expression (TPS ≥50%), FDA conducted an exploratory analysis in the subgroup of patients with “low” PD‐L1 tumor expression, defined as TPS ≥1% but <50%. In the low PD‐L1 subgroup, the HR for OS was 0.79 (95% CI: 0.61–1.04), p = .088 for pembrolizumab 2 mg/kg versus docetaxel and 0.71 (95% CI: 0.53–0.94), p = .015 for pembrolizumab 10 mg/kg versus docetaxel, indicating that patients with low PD‐L1 expression also derive benefit from pembrolizumab 2 mg/kg. The magnitude of the improvements in PFS and in ORR in the low PD‐L1 expression subgroup was similar regardless of the pembrolizumab dose. The HR for PFS was 1.07 (95% CI: 0.85–1.34), p = .718 for pembrolizumab 2 mg/kg versus docetaxel and 0.99 (95% CI: 0.78–1.25), p = .464 for pembrolizumab 10 mg/kg versus docetaxel. Objective response rate was 10.5% (95% CI: 6.5–15.7) in the docetaxel arm compared with 9.8% (95% CI: 6.1–14.7) and 10.3% (95% CI: 6.4–15.4) in the pembrolizumab 2 mg/kg and 10 mg/kg arms, respectively.
Safety
The safety profile of pembrolizumab is well characterized in patients with melanoma, NSCLC, and HNSCC, as described in the Keytruda product label [11]. No new safety signals were identified during review of these sBLAs. The major concerns with pembrolizumab are immune‐related adverse events (irAEs). In KEYNOTE‐024, irAEs were observed in 29.2% (9.7% grade 3–4) of patients in the pembrolizumab arm. Immune‐related adverse events observed in more than two patients (≥1%) in the pembrolizumab arm were hypothyroidism (9.1%), hyperthyroidism (7.8%), pneumonitis (5.8%), infusion reactions (4.5%), skin reactions (3.9%), thyroiditis (2.6%), colitis (1.9%), and myositis (1.9%). Pneumonitis was the most common serious AE associated with pembrolizumab in this trial and other lung cancer trials, and is the leading AE resulting in pembrolizumab discontinuation. Because respiratory function in lung cancer patients is frequently compromised due to the underlying tumor, prior surgery, or radiation therapy to the thoracic region, as well as comorbid conditions such as chronic obstructive pulmonary disease, health care providers need to maintain a high index of suspicion when patients develop worsening of respiratory symptoms and take appropriate measures to diagnose, initiate corticosteroids, and withhold or discontinue pembrolizumab when indicated. This information is addressed in the Warnings and Precautions section of the Keytruda product label.
In KEYNOTE‐024, the nature, incidence, and severity of AEs in the control arm were consistent with the known toxicities of the chemotherapy agents. Common AEs occurring at >10% incidence in the chemotherapy arm when compared with the pembrolizumab arm included the following: oral/gastrointestinal (nausea, vomiting, decreased appetite, dysgeusia), myelosuppression (anemia, neutropenia, and thrombocytopenia), and fatigue. More patients in the chemotherapy arm experienced a grade 3–4 toxicity compared with the pembrolizumab arm (72.7% vs. 53.2%) and more patients discontinued treatment due to an AE in the chemotherapy arm (14.0% vs. 9.1%). Serious AEs, defined as AEs that resulted in death, were life threatening, resulted in persistent or significant disability/incapacity, or resulted in or prolonged inpatient hospitalization, occurred with a similar incidence between the treatment arms (44.0% and 44.2%). Death attributed to an AE occurred in 10 patients in the pembrolizumab arm and 8 patients in the SOC arm.
For the second‐line indication, FDA's safety review focused on 991 patients randomized in KEYNOTE‐010 who received at least one dose of study drug and the pooled safety data from a total of 2,799 patients with melanoma and NSCLC enrolled in studies previously submitted to support melanoma and NSCLC approvals: KEYNOTE‐001, KEYNOTE‐002, and KEYNOTE‐006, exposed to at least one dose of pembrolizumab. In the pembrolizumab pooled group, 46% of the patients experienced a grade 3–5 AE, compared with 56% in the docetaxel arm. The incidence of serious AEs was similar (36.1% vs. 34.6%). More patients in the docetaxel arm discontinued study drug due to an AE than in the pembrolizumab arms (13.6% vs. 7.9%). In the P010 trial, irAEs were observed in 20.4% (6% grade 3–5) of patients in the pembrolizumab 2 mg/kg arm, 18.7% (5% grade 3–5) in the 10 mg/kg arm, and 4.2% in the docetaxel arm. The incidence and severity of irAEs were similar between the two pembrolizumab arms, regardless of PD‐L1 expression. Immune‐related adverse events that were observed in more than five patients (≥0.7%) in the pembrolizumab pooled group (2 mg/kg and 10 mg/kg arms) included adrenal insufficiency (0.7%), hyperthyroidism (4.7%), hypothyroidism (8.2%), colitis (0.9%), and pneumonitis (4.1%).
Discussion
On October 24, 2016, the FDA approved pembrolizumab for treatment of patients with mNSCLC whose tumors express PD‐L1 as determined by an FDA‐approved test. Table 4 summarizes the FDA benefit‐risk analysis. The results of KEYNOTE‐010 verified the clinical benefit (improved survival) of pembrolizumab for second‐line treatment of mNSCLC, leading to full approval for this indication. The application for the first‐line indication was approved 2 months prior to the goal date required by the Prescription Drug User Fee Act.
Table 4. FDA benefit‐risk assessment.
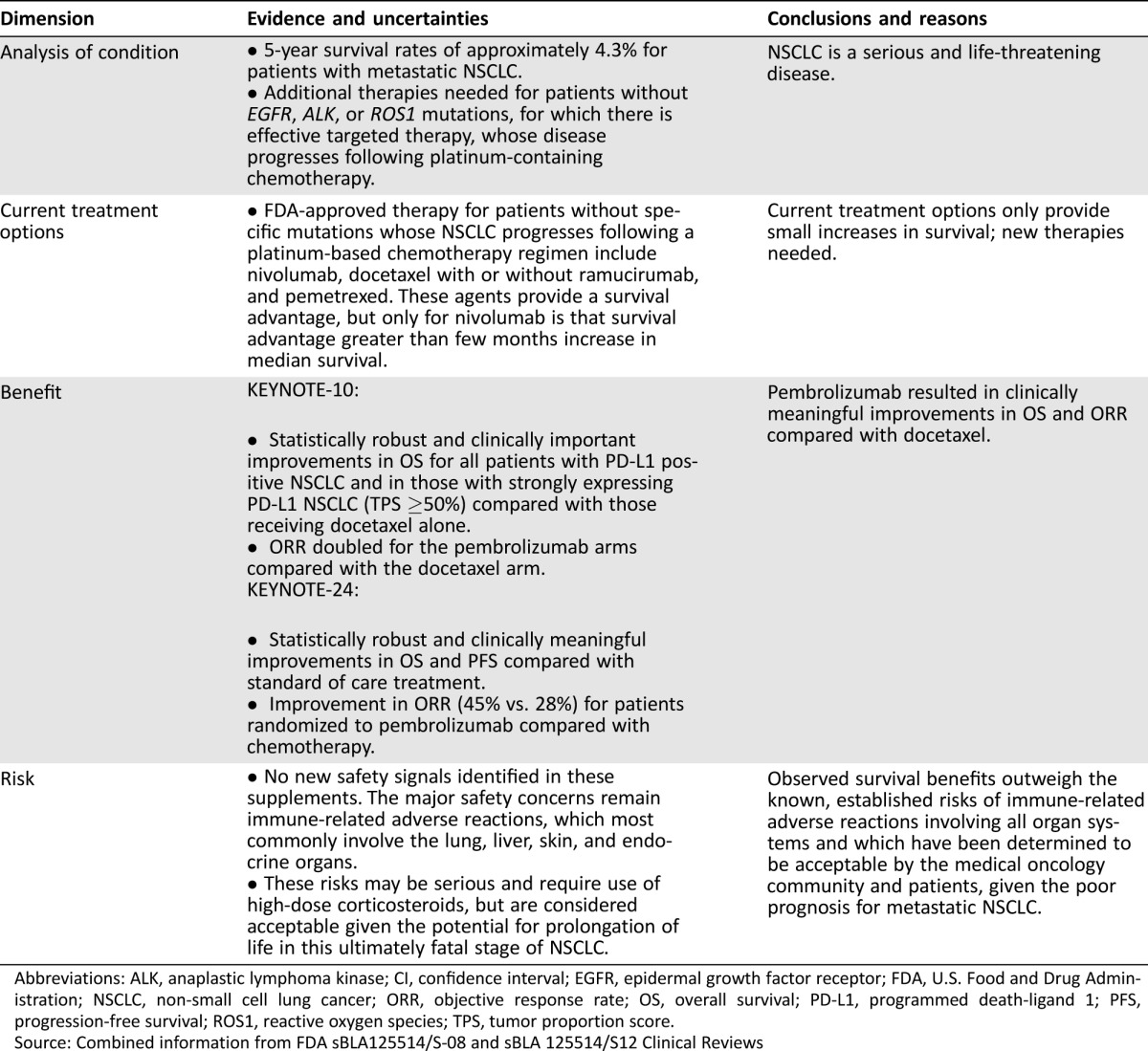
Abbreviations: ALK, anaplastic lymphoma kinase; CI, confidence interval; EGFR, epidermal growth factor receptor; FDA, U.S. Food and Drug Administration; NSCLC, non‐small cell lung cancer; ORR, objective response rate; OS, overall survival; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival; ROS1, reactive oxygen species; TPS, tumor proportion score.
Source: Combined information from FDA sBLA125514/S‐08 and sBLA 125514/S12 Clinical Reviews
Taking into consideration the efficacy and safety data submitted in both sBLAs, FDA concluded that the results of KEYNOTE‐024 support a positive benefit‐risk assessment of pembrolizumab for first‐line treatment of patients with mNSCLC whose tumors have high PD‐L1 expression (TPS ≥50%), with no EGFR or ALK genomic tumor aberrations. Despite the high proportion (44%) of patients in the SOC chemotherapy arm who crossed over to receive pembrolizumab upon disease progression, the trial demonstrated an improvement in OS for the investigational arm. As observed in other randomized trials with large treatment effects on survival, patient crossover from the control arm to the investigational arm did not obscure detection of a treatment effect in KEYNOTE‐024; however, the confounding effect of this crossover on the true treatment effect cannot be estimated. The incidence and severity of AEs observed in KEYNOTE‐024 are consistent with the known safety profile of pembrolizumab and are acceptable in view of the serious and life‐threatening nature of mNSCLC.
The approval of pembrolizumab for the first‐line treatment of patients with mNSCLC with strong PD‐L1 expression impacts about a quarter of patients with mNSCLC. This is comparable to the proportion of patients with mNSCLC whose tumors harbor sensitizing mutations in EGFR, BRAF, or ALK/reactive oxygen species (ROS1) rearrangements. Therefore, about half of patients with mNSCLC are eligible to receive either an immune checkpoint inhibitor or a targeted therapy in lieu of first‐line platinum doublet chemotherapy.
Similarly, FDA concluded that the results of KEYNOTE‐010 support a favorable benefit‐risk assessment for pembrolizumab for the treatment of patients with PD‐L1 expressing (TPS ≥1%) mNSCLC who have progressed following platinum‐containing chemotherapy, and if appropriate, targeted therapy for ALK or EGFR mutations. Although the risk‐benefit assessment was clearly favorable in the subgroup of patients with high PD‐L1 tumor expression regardless of the pembrolizumab dose, the risk‐benefit assessment was more challenging for patients with low PD‐L1 expression, because an improvement in PFS was not demonstrated in the overall population. However, given the meaningful improvement in OS and the doubling of the ORR observed in both 2 mg/kg and 10 mg/kg pembrolizumab arms as compared with docetaxel arm, approval was granted for the overall population for the 2 mg/kg and 10 mg/kg regimens.
Merck agreed to postmarketing commitments (PMCs) to further assess the efficacy of pembrolizumab for first‐line treatment of mNSCLC. These PMCs are to submit final study reports and datasets for KEYNOTE‐024 [12] and KEYNOTE‐042 [13] to FDA to further characterize the OS, PFS, and ORR benefits in this population.
Conclusion
The approval of pembrolizumab represents the first FDA approval of a checkpoint inhibitor for the treatment of patients with PD‐L1 expressing mNSCLC. Notably, it is the first time that any therapy has displaced platinum‐doublet chemotherapy for the first‐line indication for patients with mNSCLC without EGFR or ALK genomic tumor aberrations, and provides a new treatment option in an area of unmet medical need.
Author Contributions
Conception/design: Lee Pai‐Scherf, Gideon M. Blumenthal, Kirsten B. Goldberg
Collection and/or assembly of data: Lee Pai‐Scherf, Gideon M. Blumenthal, Hongshan Li, Sriram Subramaniam, Pallavi S. Mishra‐Kalyani
Data analysis and interpretation: Lee Pai‐Scherf, Gideon M. Blumenthal, Hongshan Li, Sriram Subramaniam, Pallavi S. Mishra‐Kalyani, Kun He, Hong Zhao, Patricia Keegan
Manuscript writing: Lee Pai‐Scherf, Gideon M. Blumenthal, Kirsten B. Goldberg, Kun He, Hong Zhao, Patricia Keegan
Final approval of manuscript: Lee Pai‐Scherf, Gideon M. Blumenthal, Hongshan Li, Sriram, Subramaniam, Pallavi S. Mishra‐Kalyani, Kun He, Hong Zhao, Kirsten B. Goldberg, Amy E. McKee, Patricia Keegan, Richard Pazdur
Disclosures
The authors indicated no financial relationships.
References
- 1. D'Addario G, Pintilie M, Leighl NB et al. Platinum‐based versus non‐platinum based chemotherapy in advanced non‐small‐cell lung cancer: A meta‐analysis of the published literature. J Clin Oncol 2005;23:2926–2936. [DOI] [PubMed] [Google Scholar]
- 2.Taxotere Prescribing Information . Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020449s075lbl.pdf. Accessed July 19, 2017.
- 3.Erlotinib Prescribing Information . Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021743s027lbl.pdf. Accessed July 19, 2017.
- 4.National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology : Non‐small cell lung cancer. Available at https://www.nccn.org/professionals/physician_gls/pdf/nscl_blocks.pdf. Accessed December 12, 2016.
- 5.National Cancer Institute : Lung Cancer. Available at https://www.cancer.gov/types/lung/hp/non-small-cell-lung-treatment-pdq. Accessed December 12, 2016.
- 6. Schiller JH, Harrington D, Belani CP et al. Comparison of four chemotherapy regimens for advanced non‐small cell lung cancer. N Engl J Med 2002;346:92–98. [DOI] [PubMed] [Google Scholar]
- 7. Scagliotti GV, De Marinis F, Rinaldi M et al. Phase III randomized trial comparing three platinum‐based doublets in advanced non‐small cell lung cancer. J Clin Oncol 2002;20:4285–4291. [DOI] [PubMed] [Google Scholar]
- 8. Sul J, Blumenthal GM, Jiang X et al. FDA approval summary: Pembrolizumab for the treatment of patients with metastatic non‐small cell lung cancer whose tumors express programmed death‐ligand 1. The Oncologist 2016;21:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reck M, Rodríguez‐Abreu D, Robinson AG et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med 2016;375:1823–1833. [DOI] [PubMed] [Google Scholar]
- 10. Herbst RS, Baas P, Kim DW et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): A randomised controlled trial. Lancet 2016;387:1540–1550. [DOI] [PubMed] [Google Scholar]
- 11.Keytruda Prescribing Information . Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125514s012lbl.pdf. Accessed July 19, 2017.
- 12.Clinicaltrials.gov, Trial NCT02142738. Available at https://clinicaltrials.gov/ct2/show/NCT02142738. Accessed July 19, 2017.
- 13.Clinicaltrials.gov, Trial NCT02220894. Available at https://clinicaltrials.gov/ct2/show/NCT02220894. Accessed July 19, 2017.