

Multiple myeloma is mostly an incurable disease. The U.S. Food and Drug Administration (FDA) granted daratumumab accelerated approval in November 2015 as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome and an immunomodulatory agent. This article describes the FDA review of the strength of evidence for this application and its clinical implications for the multiple myeloma population.
Keywords: Daratumumab, Multiple myeloma
Abstract
On November 21, 2016, the U.S. Food and Drug Administration granted regular approval to daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy. Approval was based on two randomized, open‐label trials in which daratumumab was added to these backbone therapies. The MMY3003 trial demonstrated substantial improvement in progression‐free survival (PFS) when daratumumab was added to lenalidomide and dexamethasone compared with lenalidomide and dexamethasone alone. The estimated median PFS had not been reached in the daratumumab arm and was 18.4 months in the control arm (hazard ratio [HR] = 0.37; 95% confidence interval [CI]: 0.27–0.52; p < .0001), representing a 63% reduction in the risk of disease progression or death. Similar results were observed in the MMY3004 trial comparing the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone. The estimated median PFS was not reached in the daratumumab arm and was 7.2 months in the control arm (HR = 0.39; 95% CI: 0.28–0.53; p < .0001), representing a 61% reduction in the risk of disease progression or death. The most frequently reported adverse reactions (greater than or equal to 20%) in MMY3003 were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions (greater than or equal to 20%) in MMY3004 were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, and peripheral sensory neuropathy. Neutropenia and thrombocytopenia have been added to the Warnings and Precautions of the drug label.
Implications for Practice.
Daratumumab, the first monoclonal antibody targeted against CD38, received U.S. Food and Drug Administration accelerated approval in 2015 based on data from single‐agent, single‐arm trials that provided response rate information. Results of the MMY3003 and MMY3004 trials established that daratumumab can be combined synergistically with some of the most highly active agents used to treat multiple myeloma, leading to daratumumab's regular approval in 2016. Daratumumab added to lenalidomide and dexamethasone, or bortezomib and dexamethasone, provides a substantial improvement in progression‐free survival in previously treated patients with multiple myeloma. These combinations will likely improve the survival outlook for patients with multiple myeloma.
Introduction
Multiple myeloma (MM) remains a mostly incurable disease. In 2017, it is estimated that there will be 30,280 new cases and 12,590 deaths from multiple myeloma [1]. Multiple myeloma is primarily a disease of the elderly, with a median age at diagnosis of 69 [2]. Treatment options have significantly improved over recent decades with the introduction of alkylating agents, the use of high‐dose therapy in combination with autologous stem cell rescue, and the introduction of new classes of agents such as immunomodulatory agents and proteasome inhibitors. The median overall survival for a cohort diagnosed between the years 2006–2010 was 6.1 years, compared with 4.6 years for the cohort diagnosed between the years 2001–2005 [3]. Despite these advances, patients with MM often relapse or develop refractory disease, underscoring the need for new therapies.
DARZALEX (daratumumab; Janssen, Beerse, Belgium, http://www.janssen.com) is a human IgG1κ monoclonal antibody against CD38 antigen. The U.S. Food and Drug Administration (FDA) granted daratumumab accelerated approval in November 2015 as monotherapy for patients with MM who have received at least three prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent, or who are double refractory to a PI and an immunomodulatory agent. The drug sponsor submitted two efficacy supplements for (a) daratumumab in combination with lenalidomide and dexamethasone for patients with MM who have received at least one prior therapy and (b) daratumumab in combination with bortezomib and dexamethasone for patients with MM who have received at least one prior therapy. These data were also submitted to support full approval of daratumumab. The FDA granted daratumumab breakthrough therapy and orphan drug designation, as well as priority review. The current approval was granted on November 21, 2016, nearly 3 months prior to the goal date. Herein, we describe the FDA review of the strength of evidence for this application, and its clinical implications for the multiple myeloma population.
Clinical Trial Designs
MMY3003 (ClinicalTrials.gov identifier NCT02076009) was a randomized, open‐label, phase 3 study comparing daratumumab added to lenalidomide and dexamethasone (DRd) versus lenalidomide and dexamethasone (Rd) in patients with relapsed or refractory MM. Eligible patients were those with relapsed or refractory MM who had received at least one prior line of therapy and had achieved at least a partial response to one or more of their prior therapies and had documented progressive disease on or after their last regimen. Approximately 560 subjects were planned to be randomized in a 1:1 ratio to DRd or Rd. Randomization was stratified by the following stratification factors: International Staging System (ISS; I, II, or III) at screening (based on central laboratory results), number of prior lines (1 vs. 2 or 3 vs. >3), and prior lenalidomide treatment (no vs. yes). Subjects were treated until disease progression, unacceptable toxicity, or other reason.
For subjects randomized to receive DRd, treatment consisted of daratumumab administered as an intravenous (IV) infusion at a dose of 16 mg/kg (the approved dose for daratumumab monotherapy) weekly for 8 weeks, then every 2 weeks for 16 weeks, and then every 4 weeks thereafter. Lenalidomide was to be administered at a dose of 25 mg orally on days 1–21 of each 28‐day cycle for subjects with creatinine clearance >60 mL/minute. For subjects with creatinine clearance between 30 and 60 mL/minute, the dose was 10 mg daily. Dexamethasone was to be administered at a total dose of 40 mg weekly. For subjects randomized to receive Rd, treatment consisted of lenalidomide 25 mg orally on days 1–21 of a 28‐day cycle and dexamethasone 40 mg weekly.
The primary endpoint was progression‐free survival (PFS) based on International Myeloma Working Group (IMWG) criteria, as assessed by a computerized algorithm (CA). Progression‐free survival analysis was to be based on a two‐sided (significance level of 0.05), stratified log‐rank test. A total of 295 PFS events out of 560 subjects were estimated to provide a power of 85% to detect a reduction of 30% in the risk of either progression or death (hazard ratio [HR; DRd vs. Rd] of 0.70).
MMY3004 (NCT02136134) was a randomized, open‐label, phase 3 study comparing daratumumab added to bortezomib and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients with relapsed or refractory MM. Eligible patients were those with relapsed or refractory MM who had received at least one prior line of therapy and had achieved at least a partial response to one or more of their prior therapies and had documented progressive disease on or after their last regimen. A total of 295 PFS events were estimated for the PFS analyses based on similar assumptions as shown in study MMY3003. Approximately 480 subjects were planned to be randomized in a 1:1 ratio to DVd or Vd. Randomization was stratified by the following stratification factors: ISS (I, II, or III) at screening (based on central laboratory results), number of prior lines (1 vs. 2 or 3 vs. >3), and prior bortezomib treatment (no vs. yes). Subjects in the DVd arm were treated until disease progression, unacceptable toxicity, or other reason. Subjects in the Vd arm were to receive a maximum of eight cycles of Vd followed by observation until disease progression or other reasons. For subjects randomized to receive DVd, treatment consisted of daratumumab administered as an IV infusion at a dose of 16 mg/kg weekly for the first 3 cycles, on day 1 of cycles 4–8, and then every 4 weeks thereafter. For both treatment groups, bortezomib was administered at a dose of 1.3 mg/m2 subcutaneously (SC) twice weekly on days 1, 4, 8, and 11 for eight 21‐day cycles (cycles 1–8). Dexamethasone was administered at a total dose of 80 mg weekly in 2 out of 3 weeks for cycles 1–8. The primary endpoint was PFS based on IMWG criteria, as assessed by a CA. For both studies, one efficacy interim analysis was planned at approximately 60% information level based on the O'Brien Fleming alpha spending function.
Results
MMY3003 enrolled a total of 569 subjects (DRd: 286 subjects, Rd: 283 subjects). The median age was 65 years (range: 34–89). Most (69%) subjects were white, 18% were Asian, and 3% were black. The median number of prior lines was 1 (range: 1–11) and 19% of subjects had received ≥3 prior lines of therapy. In total, 86% of subjects had received a prior proteasome inhibitor (PI); 84% had received prior bortezomib; 55% had received a prior immunomodulatory agent (IMID), including 18% who had received prior lenalidomide. In total, 44% had received a prior PI and an IMID, and 27% were refractory to their last line. Per protocol definition, patients who were refractory to lenalidomide were to be excluded.
MMY3004 enrolled a total of 498 subjects (DVd: 251 subjects, Vd: 247 subjects). The median age was 64 years (range: 30–88). Most (87%) subjects were white, 5% were Asian, and 4% were black. The median number of prior lines was 2 (range: 1–10) and 24% of subjects had received ≥3 lines of prior therapy. In total, 69% of subjects had received a prior PI; 66% had received prior bortezomib; 76% had received a prior IMID; and 48% had received a prior PI and an IMID. In total, 36% of subjects were refractory to an IMID.
Efficacy
For MMY3003, as of the data cutoff date of March 7, 2016, the median follow‐up was 13.5 months for the intention‐to‐treat (ITT) population. A statistically significant improvement in PFS as assessed by the CA was observed for the DRd arm over the Rd arm at 57% information level, with an estimated median PFS not yet reached for the DRd arm and 18.43 months for the Rd arm (HR = 0.37, 95% CI: 0.27–0.52; p < .0001, which crossed the prespecified boundary at the first efficacy interim analysis). The Kaplan‐Meier plot for PFS is shown in Figure 1. No outliers were observed based on the results from sensitivity analyses and subgroup analyses (gender, race, age group, geographic region, and stratification factors). Of note, prior lenalidomide treatment was one of the subgroups evaluated. Prior lenalidomide subgroups (yes/no) had similar HR results as the overall group. These analyses support the primary analysis result. The analysis of overall response rate (ORR) also demonstrated a favorable result (91.3% [95% CI: 87.4%–94.3%] vs. 74.6% [95% CI: 69.1%–79.5%] for DRd vs. Rd, respectively; and a p value of <.0001 based on a Cochran Mantel Haenszel test). The best ORR of stringent complete response (sCR) + complete response (CR) in the Rd arm was 18.8% versus 42.3% in the DRd arm. Overall survival data at the time of the analysis were immature and did not allow for inferential conclusions to be made.
Figure 1.

Progression‐free survival Kaplan‐Meier plot for study MMY3003.
Abbreviations: DRd, daratumumab added to lenalidomide and dexamethasone; Rd, lenalidomide and dexamethasone.
Figure 2.

Study MMY3003 Kaplan‐Meier plot for overall survival.
Abbreviations: DRd, daratumumab added to lenalidomide and dexamethasone; Rd, lenalidomide and dexamethasone.
For MMY3004, as of the data cutoff date of January 11, 2016, the median follow‐up was 7.4 months for the ITT population. A statistically significant improvement in PFS as assessed by the CA was observed for the DVd arm over the Vd arm at 64% information level, with a median PFS not yet reached for the DVd arm and with 7.2 months for the Vd arm (HR = 0.39, 95% CI: 0.28–0.53; p < .0001, which crossed the prespecified boundary at the first efficacy interim analysis). The Kaplan‐Meier plot for PFS is shown in Figure 3. No outliers were observed based on the results from sensitivity analyses and subgroup analyses (gender, race, age group, geographic region, and stratification factors). The analysis of ORR also demonstrated a favorable result (79.3% [95% CI: 73.7%–84.1%] vs. 59.9% [95% CI: 53.5%–66.1%] for DVd vs. Vd, respectively; and a p value of <.0001 based on a Cochran Mantel Haenszel test). The rate of sCR + CR was 8.5% in the Vd arm versus 18.3% in the DVd arm. This supports the primary analysis results of PFS in favor of the DVd arm. Overall survival data at the time of the analysis were immature and did not allow for inferential conclusions to be made.
Figure 3.

Progression‐free survival Kaplan‐Meier plot for study MMY3004.
Abbreviations: DVd, daratumumab added to bortezomib and dexamethasone; Vd, bortezomib and dexamethasone.
Figure 4.

Study MMY3004 Kaplan‐Meier plot of overall survival.
Abbreviations: DVd, daratumumab added to bortezomib and dexamethasone; Vd, bortezomib and dexamethasone.
Minimal residual disease (MRD) was assessed in both pivotal trials, MMY3003 (DRd vs. Rd) and MMY3004 (DVd vs. Vd). Minimal residual disease was performed using next‐generation sequencing (NGS) methodology. Minimal residual disease was assessed in bone marrow samples in all patients suspected of having a CR in both studies. Additional evaluations were performed at 3 months and 6 months after CR (in subjects who maintained CR) in the MMY3003 study. In the MMY3004 study, additional evaluations were performed at 6 months after the first dose and 12 months after the first dose (in subjects who were initially determined to be MRD negative). An MRD threshold of 10−4 was used for these trials. A baseline diagnostic sample from each subject was used to identify the myeloma clone for subsequent MRD testing. In the MMY3003 study, MRD‐negative status was obtained in 29% of patients compared with 7.8% of patients in the Rd arm, irrespective of CR status. The test failed to identify a baseline MM clone in 24.7% of tests. In the MMY3004 study, MRD‐negative status was obtained in 13.6% of patients treated with DVd compared with 2.8% treated with Vd, irrespective of CR status. The test failed to identify a baseline MM clone in 22.7% of tests.
Safety
The safety review of MMY3003 was based on data from 283 subjects treated with DRd and 281 subjects treated with Rd. The study population was monitored for deaths, serious adverse events (SAEs), adverse events (AEs) of special interest, common AEs, and common laboratory tests. Four percent of subjects treated with DRd died within 30 days of the last dose of study drug. The most common fatal AEs were due to infection. There was a similar rate of death within 30 days of last dose in the Rd arm. An SAE was reported for 49% of subjects in the DRd arm and 42% of subjects in the Rd arm. The most common SAEs in the DRd arm were pneumonia (12%), upper respiratory tract infection (7%), and febrile neutropenia (4%). There was no difference between study arms in the rate of discontinuation of at least one study drug (13% on DRd vs. 13% on Rd). The most common preferred term resulting in lenalidomide discontinuation was pneumonia (four subjects on DRd and two subjects on Rd). The assessment of AEs of special interest in MMY3003 revealed a trend for increased rates of neutropenia and infections in the DRd arm. Infusion‐related reactions (IRRs) occurred in 48% of subjects treated with daratumumab. The majority occurred in the first cycle because only 2% of the DRd safety population had an IRR after the first infusion. Fifteen subjects (5%) of the DRd arm had a grade 3 IRR and the remaining subjects had grade 1 or 2 IRRs. The most common IRRs were cough, dyspnea, and vomiting. The most common AEs occurring in the DRd arm were upper respiratory tract infection (65%), neutropenia (59%), diarrhea (43%), and fatigue (35%). Events with a risk difference >10% in the DRd arm in comparison with the Rd arm were diarrhea, neutropenia, cough, upper respiratory tract infection, and vomiting. Subgroups of subjects treated with DRd who had an increased risk of anemia included those who were ≥65 years of age or who had ISS stage III disease.
For MMY3004, the safety review was based on data from 243 subjects treated with DVd and 237 subjects treated with Vd. The study population was monitored for deaths, SAEs, AEs of special interest, common AEs, and common laboratory tests. Five percent of subjects treated with DVd or Vd died within 30 days of the last dose of study drug. The most common fatal AEs in the DVd arm were contained within cardiac, respiratory, or nervous system disorders system organ class (SOC). An SAE was reported for 42% of subjects in the DVd arm and 34% of subjects in the Vd arm. There were similar rates of SAEs experienced by system organ class. The most common SAEs in the DVd arm were pneumonia (11%), anemia (3%), bronchitis (2%), and thrombocytopenia (3%). There was no difference between study arms in the rate of discontinuation of at least one study drug (15% on DVd vs. 17% on Vd). The most common cause was a grade 3 or 4 AE. The most common preferred term resulting in bortezomib discontinuation in both arms was peripheral sensory neuropathy. Infusion‐related reactions occurred in 45% of subjects receiving daratumumab infusions. The majority occurred during the first cycle because only 2% of the DVd safety population had an IRR after the first infusion. Twenty‐one subjects (9%) in the DVd arm had a grade 3 IRR and the remaining subjects had a grade 1 or 2 IRR. The most common IRRs were dyspnea, bronchospasm, cough, and throat irritation. The most common AEs occurring in the DVd arm were thrombocytopenia (59%), peripheral sensory neuropathy (47%), upper respiratory infection (44%), and diarrhea (32%). Events with a risk difference >10% in the DVd arm in comparison with the Vd arm were thrombocytopenia, upper respiratory tract infection, and cough. Subgroups of subjects treated with DVd who had an increased risk of thrombocytopenia included those who were ≥65 years or who had ISS stage III disease. In a comparison of AEs between cycles 1–3 and cycles 4–8 in subjects treated with DVd, there was a higher incidence of peripheral sensory neuropathy in later cycles. The rate of peripheral sensory neuropathy was 32% in later cycles versus 19% in the early cycles. There was a higher incidence of grade 3 or higher lymphocytopenia (51%) and thrombocytopenia (48%) in the DVd arm. Given the higher rates of thrombocytopenia and neutropenia, new warnings and precautions were warranted for these AEs.
Clinical Pharmacology
The approved monotherapy dose is 16 mg/kg as an IV infusion weekly on weeks 1–8 (8 doses), every 2 weeks on weeks 9–24 (8 doses), and every 4 weeks thereafter. The same dose and schedule was used for the DRd regimen. For the DVd regimen, the daratumumab dosing was modified to improve patient convenience. See the clinical trial design section above for a description of the dosing schedule for both the DRd and DVd regimens.
The exposure‐response results support the proposed dosing regimen for daratumumab when it is administered with Rd and Vd.
No dose adjustments appeared warranted for several intrinsic and extrinsic factors. Population analyses showed that age (31–84 years), sex, mild (total bilirubin 1 to 1.5 times upper limit of normal [ULN] and any alanine transaminase [ALT]) or moderate (total bilirubin 1.5 to 3 times ULN and any ALT) hepatic impairment, or renal impairment (creatinine clearance 15–89 mL/minute) had no observed effect on daratumumab pharmacokinetics (PK). Additional analyses showed that the PK of daratumumab was not affected by coadministration of Rd or Vd, and the PK of bortezomib was not affected by daratumumab.
Discussion
When combined with Rd or Vd, daratumumab demonstrated a statistically significant and clinically meaningful improvement in PFS and response rate. In the MMY3003 trial, there was a 63% reduction in the risk of disease progression or death. Similar results were observed in the MMY3004 trial, in which there was a 61% reduction in the risk of disease progression or death. The safety profile of daratumumab was similar to that already known with daratumumab monotherapy or the backbone therapies with which daratumumab was combined. The magnitude of effect resulted in submission of the trial data at the interim analysis stage of both trials. Given the strength of results and wide applicability within a noncurable disease, the FDA reviewed this application in an expedited fashion. This approval serves as an example of a drug with substantial clinical benefit moving swiftly through the drug development process.
The FDA approval and the results of these clinical trials provide clinicians with two additional triplet regimens for the treatment of patients whose disease progressed after one course of treatment or does not respond to treatment. As a result, patients could receive daratumumab earlier in their course of treatment, added on to therapies already known to provide clinical benefit.
This is the first report of the use of MRD in the relapsed or refractory disease setting, but minimal residual disease was not added to the prescribing information due to the high rate of inability to detect an MM clone using NGS, 24.7% and 22.7%, respectively, in studies MMY3003 and MMY3004. It is unclear why the test was unable to identify an MM clone, but it is possible that the test performs differently in this disease population compared with those with newly diagnosed disease. The inability to identify an MM clone could also be due to assay performance issues or issues related to sample procurement and processing. Ultimately, the relatively high failure rate for clone detection limited the interpretability of the MRD results and posed a regulatory challenge. One could consider use of a second MRD assay to be used in cases of NGS calibration failure. However, this approach should be prespecified and may not completely resolve issues with missing data if the reasons for missing data are related to sample procurement and processing.
Conclusion
The efficacy and safety results of studies MMY3003 and MMY3004 demonstrated an acceptable benefit‐risk profile for daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy (Table 1). Ultimately, the benefit‐risk assessment supported regular approval for the proposed indication.
Table 1. FDA benefit‐risk assessment.
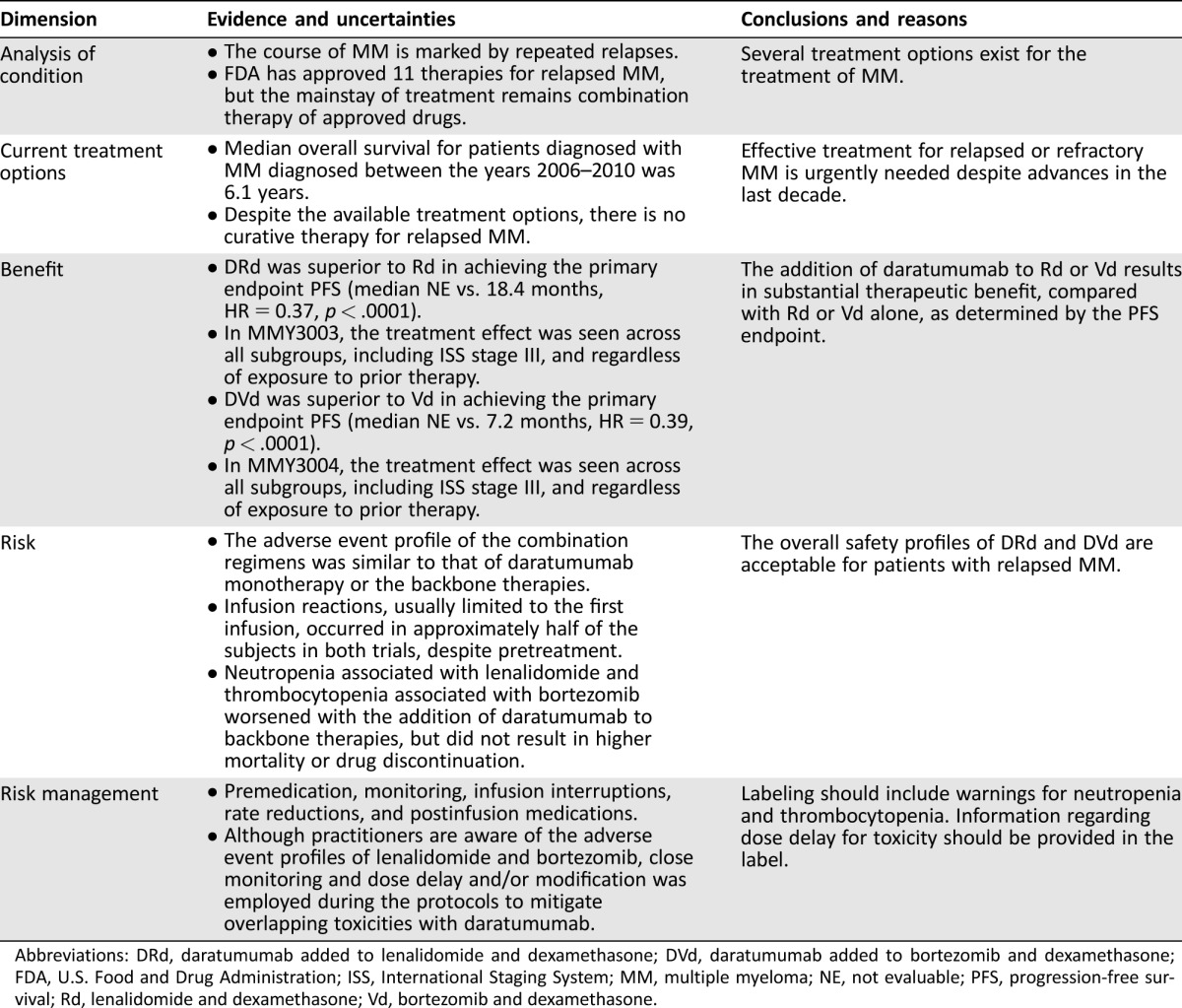
Abbreviations: DRd, daratumumab added to lenalidomide and dexamethasone; DVd, daratumumab added to bortezomib and dexamethasone; FDA, U.S. Food and Drug Administration; ISS, International Staging System; MM, multiple myeloma; NE, not evaluable; PFS, progression‐free survival; Rd, lenalidomide and dexamethasone; Vd, bortezomib and dexamethasone.
Footnotes
For Further Reading: Saad Usmani, Tahamtan Ahmadi, Yvette Ng et al. Analysis of Real‐World Data on Overall Survival in Multiple Myeloma Patients With ≥ Prior Lines of Therapy Including a Proteasome Inhibitor (PI) and an Immunomodulatory Drug (IMiD), or Double Refractory to a PI and an IMiD The Oncologist 2016;21:1355‐1361.
Implications for Practice: This real‐world retrospective study of electronic health records examines the survival outcomes of patients with multiple myeloma who are heavily pretreated or highly refractory to currently approved treatments, including recently approved proteasome inhibitors and immunomodulatory drugs. This survival analysis showed that outcomes for these patients remain poor despite the availability of newer agents, with median overall survival of approximately 8 months. These findings highlight a critical need to develop novel therapies for these patients and also serve as a reference point against which emerging agents for heavily pretreated or highly refractory disease may be evaluated.
Author Contributions
Conception/design: Vishal Bhatnagar, Nicole J. Gormley, Lola Luo, Yuan Li Shen, Rajeshwari Sridhara, Sriram Subramaniam, Guoxiang Shen, Lian Ma, Stacy Shord
Collection and/or assembly of data: Vishal Bhatnagar, Nicole J. Gormley, Lola Luo, Yuan Li Shen, Rajeshwari Sridhara, Sriram Subramaniam, Guoxiang Shen, Lian Ma, Stacy Shord
Data analysis and interpretation: Vishal Bhatnagar, Nicole J. Gormley, Lola Luo, Yuan Li Shen, Rajeshwari Sridhara, Sriram Subramaniam, Guoxiang Shen, Lian Ma, Stacy Shord, Ann T. Farrell
Manuscript writing: Vishal Bhatnagar, Nicole J. Gormley, Kirsten B. Goldberg, Ann T. Farrell, Amy E. McKee, Richard Pazdur
Final approval of manuscript: Vishal Bhatnagar, Nicole J. Gormley, Lola Luo, Yuan Li Shen, Kirsten B. Goldberg, Ann T. Farrell, Amy E. McKee, Richard Pazdur
Disclosures
The authors indicated no financial relationships.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017:67:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Howlader N, Noone AM, Krapcho M et al. SEER Cancer Statistics Review, 1975–2013. Available at http://seer.cancer.gov/csr/1975_2013/. Accessed August 2, 2017.
- 3. Kumar SK, Dispenzieri A, Lacy MQ et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014:28:1122–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]