

This article summarizes the scientific review of the application leading to regulatory approval of carfilzomib in combination with lenalidomide and dexamethasone in the European Union.
Keywords: Carfilzomib, Multiple myeloma, European Medicines Agency
Abstract
On November 19, 2015, a marketing authorization valid through the European Union was issued for carfilzomib in combination with lenalidomide and dexamethasone for the treatment of adult patients with multiple myeloma (MM) who have received at least one prior therapy.
In a phase III trial in patients with relapsed MM, median progression‐free survival (PFS) for patients treated with carfilzomib in combination with lenalidomide and dexamethasone (CRd) was 26.3 months versus 17.6 months for those receiving lenalidomide and dexamethasone alone (hazard ratio = 0.69; 95% confidence interval, 0.57–0.83; one‐sided log‐rank p value < .0001). The most frequently observed toxicity (grade ≥3, treatment arm vs. control arm) in the phase III trial included neutropenia (29.6% vs. 26.5%), anemia (17.9% vs. 17.7%), thrombocytopenia (16.8% vs. 12.3%), pneumonia (12.5% vs. 10.5%), fatigue (7.7% vs. 6.4%), hypertension (4.6% vs. 2.1%), diarrhea (3.8% vs. 4.1%), and respiratory tract infection (4.1% vs. 2.1%).
The objective of this article is to summarize the scientific review of the application leading to regulatory approval in the European Union. The scientific review concluded that the gain in PFS of 8.7 months observed with the combination of CRd was considered clinically meaningful and was supported by a clear trend in overall survival benefit, although the data were not mature. The delay in disease progression appeared superior to available alternatives in the setting of relapsed MM at the time of the marketing authorization of carfilzomib. Therefore, given the overall accepted safety profile, which was considered manageable in the current context, the benefit risk for CRd was considered positive.
Implications for Practice.
Carfilzomib (Kyprolis) was approved in the European Union in combination with lenalidomide and dexamethasone for the treatment of adult patients with multiple myeloma who have received at least one prior therapy. The addition of carfilzomib to lenalidomide and dexamethasone resulted in a clinically meaningful and statistically significant improvement of progression‐free survival compared with lenalidomide and dexamethasone, which was supported by a clear trend in overall survival benefit, although the data were not mature. At the time of the marketing authorization of carfilzomib, the delay in disease progression appeared superior to available alternatives in the setting of relapsed multiple myeloma. In terms of safety, the overall accepted safety profile was considered manageable.
Introduction
Multiple myeloma (MM) is a clonal neoplastic proliferation of plasma cells in the bone marrow associated with the production of monoclonal immunoglobulins in the blood or urine [1]. The estimated incidence of MM was 35,309 cases in the European Union (EU) in 2015 [2]. Multiple myeloma is a disease of older adults, with a median age at diagnosis of 70 years, and it occurs slightly more often in males than in females [1], [3].
At the time of the marketing authorization of carfilzomib in the EU, therapies for myeloma consisted of the following main classes of agents: proteasome and histone deacetylase inhibitors (bortezomib and panobinostat, respectively), immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), corticosteroids, alkylators, anthracyclines, and nitrosoureas (to a lesser extent), plus high‐dose chemotherapy and autologous or allogeneic hematopoietic stem cell transplantation for those who are eligible. These agents have been combined in clinical practice in an attempt to prolong remission. Specifically, bortezomib has been combined with either dexamethasone and thalidomide (VTD) or dexamethasone and lenalidomide (VRD) for induction in young patients; thalidomide with melphalan and dexamethasone, bortezomib with melphalan and prednisolone, and lenalidomide with dexamethazone was used as induction regimen in elderly/frail patients. The second/third‐line treatment has varied according to the duration of the previous response and the drugs already given [4].
Response rates decrease with every successive relapse (defined by the International Multiple Myeloma Working Group as previously treated myeloma patients who, after a period of being off therapy, require salvage therapy): 58% at first relapse to 15% at fourth relapse. The prognosis is poor, with an expected survival of <1 year for relapsed and refractory disease (defined as relapse of disease in patients who achieve minor response or better, and then either become nonresponsive while on salvage therapy or progress within 60 days of last therapy) as compared with an expected survival of around 3 years with relapsed myeloma [5], [6], [7], [8].
The ubiquitin‐proteasome pathway is responsible for degradation of the majority of regulatory proteins in eukaryotic cells, and plays an essential role in maintaining normal cellular homeostasis. Proteasome inhibition has been extensively explored as a therapeutic strategy in MM, and proteasome inhibitors now form a cornerstone of antimyeloma therapy [9]. Carfilzomib is a tetrapeptide epoxyketone proteasome inhibitor that selectively and irreversibly binds to the N terminal threonine containing active sites of the 20S proteasome, the proteolytic core particle within the 26S proteasome, and displays little to no activity against other protease classes.
The recommended starting dose of carfilzomib in combination with lenalidomide and dexamethasone is 20 mg/m2, which should be increased to 27 mg/m2 if it is tolerated. In combination with carfilzomib, lenalidomide is administered as 25 mg orally on days 1–21 and dexamethasone is administered as 40 mg orally or intravenously (IV) on days 1, 8, 15, and 22 of the 28‐day cycles. Treatment may be continued until disease progression or until unacceptable toxicity occurs; however, treatment for longer than 18 cycles should be based on an individual benefit‐risk assessment.
Carfilzomib entered clinical trials in September 2005 and received accelerated approval by the U.S. Food and Drug Administration in 2012 on the basis of the results of a single‐arm study [10].
The applicant submitted an application for marketing authorization to the European Medicines Agency in December 2015 requesting the approval for the following indication: “Kyprolis in combination with lenalidomide and dexamethasone is indicated for the treatment of adult patients with multiple myeloma who have received at least one prior therapy.” Table 1 displays a summary of regulatory steps in the evaluation of the marketing authorization for Kyprolis (carfilzomib; Amgen, Thousand Oaks, CA, http://www.amgen.com).
Table 1. Steps in the evaluation of the marketing authorization for Kyprolisa.
Kyprolis (carfilzomib; Amgen, Thousand Oaks, CA, http://www.amgen.com).
Abbreviations: CHMP, Committee for Medicinal Products for Human Use; PRAC, Pharmacovigilance Risk Assessment Committee; RMP, Risk Management Plan.
Before the start of the evaluation of Kyprolis, the applicant requested an accelerated assessment. Accelerated assessment means rapid assessment of medicines in the centralized procedure that are of major interest for public health, especially ones that are therapeutic innovations. Accelerated assessment usually takes 150 evaluation days, rather than 210.
The applicant's request for an accelerated assessment was accepted because the product was considered to be of major public health interest. This was based on the fact that the provided data indicated that the benefit of carfilzomib in combination with lenalidomide and dexamethasone in terms of progression‐free survival (PFS), in comparison with lenalidomide and dexamethasone, could be considered as a significant improvement for the treatment of relapsed MM patients. Even if some safety data had to be assessed with caution, and more mature overall survival (OS) data were awaited, it could be expected that carfilzomib represents a therapeutic option of major interest.
Nonclinical Aspects and Clinical Pharmacology
Carfilzomib is a proteasome inhibitor that selectively and irreversibly binds to the N terminal threonine containing active sites of the 20S proteasome, the proteolytic core particle within the 26S proteasome. It differs structurally and mechanistically from bortezomib. Carfilzomib is a tetrapeptide bearing an epoxyketone moiety and functions by irreversibly inhibiting chymotrypsin‐like activity of the proteasome, whereas bortezomib, a boronic acid dipeptide, inhibits the chymotrypsin‐like activity of the 26S proteasome in a reversible manner (Fig. 1).
Figure 1.
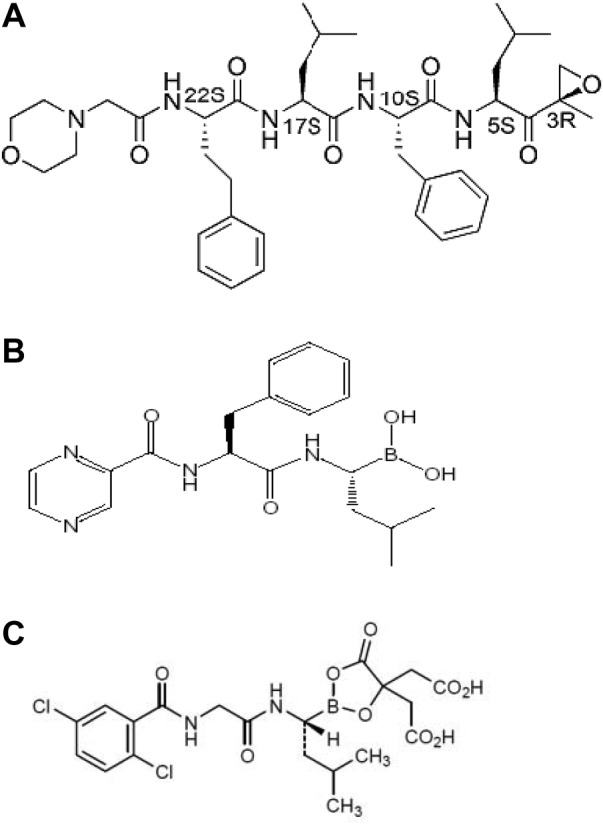
Molecular structure of carfilzomib, bortezomib, and ixazomib citrate. (A): Chemical name of the active substance carfilzomib: (2S)‐N‐((S)‐1‐((S)‐4‐methyl‐1‐((R)‐2‐methyloxiran‐2‐yl)‐1‐oxopentan‐2‐ylcarbamoyl)‐2‐phenylethyl)‐2‐((S)‐2‐(2‐morpholinoacetamido)‐4‐phenylbutanamido)‐4‐methylpentanamide. (B): Chemical name of the active substance bortezomib: {(1R)‐3‐methyl‐1‐[(2S)‐3‐phenyl‐2‐(pyrazin‐carboxamido)=propanamido]butyl}boronic acid. (C): Chemical name of the active substance ixazomib citrate: 2,2′‐{2‐[(1R)‐1‐({[(2,5 dichlorobenzoyl)amino]acetyl}amino)‐3‐methylbutyl]‐5‐oxo‐1,3,2‐ dioxaborolane‐4,4‐diyl}diacetic acid.
Ixazomib is a dipeptide boronic acid proteasome inhibitor. Whereas carfilzomib binds irreversibly to the proteasome's catalytic enzyme active sites, ixazomib binds reversibly to the proteasome's catalytic enzyme active sites. This difference between ixazomib (reversible) and carfilzomib (irreversible) translates into clearly quantifiable differences in the preclinical pharmacology of the two drugs, both in vitro and in vivo, in rodent models.
In the in vitro studies, carfilzomib showed to be a potent, selective, and irreversible inhibitor of the chymotrypsin‐like activity of purified human 20S proteasome and was 50‐ to 300‐fold selective over the other proteasome catalytic activities caspase‐like and trypsin‐like. In animals, carfilzomib inhibited proteasome activity in blood and tissue and delayed tumor growth in models of MM. In vivo, carfilzomib showed more potent cytotoxic activity against both hematological and solid tumor cell lines than caspase‐like inhibitor bortezomib. In vitro, carfilzomib was cytotoxic to bortezomib‐resistant cells.
A combined safety pharmacology study conducted with carfilzomib in telemetered monkeys after IV administration showed no effect on central nervous system or respiratory parameters at doses up to 3 mg/kg.
Single bolus IV doses of carfilzomib at 3 mg/kg in monkeys were associated with hypotension, increased heart rate, and increased serum levels of troponin T. This is a relevant effect because this dose corresponds to the recommended dose in humans of 27 mg/m2. Repeated bolus IV administration of carfilzomib at ≥2 mg/kg per dose in rats and 2 mg/kg per dose in monkeys, using dosing schedules similar to those used clinically, resulted in fatal cardiovascular, gastrointestinal, renal, and pulmonary toxicity. The dose of 2 mg/kg per dose in rats is approximately half the recommended dose in humans.
Carfilzomib was not mutagenic. However, in the presence and absence of metabolic activation, an increase of clastogenic activity in human peripheral blood lymphocytes was observed in vitro.
No effects on reproductive tissues were observed in chronic toxicity studies. Embryo‐fetal toxicity was reported in rabbits at doses that were lower than in patients receiving the recommended dose. Carfilzomib was not teratogenic when administered to pregnant rats during the period of organogenesis, although carfilzomib exposure in animals was below the exposure achieved in humans. Carfilzomib should not be used during pregnancy unless the potential benefit outweighs the potential risk for the fetus.
The pharmacokinetics of IV administration of carfilzomib has been studied in 483 subjects, across multiple phase Ib/II and phase III trials. The median maximum concentration (Cmax) and area under the curve (AUC) following a 2‐ to 10‐minute IV infusion of 27 mg/m2 was 4,232 ng/mL and 379 ng/mL per hour, respectively. At the same dose level, the terminal half‐life (t½) was 0.35 hours. Peptidase cleavage and epoxide hydrolysis were the principal pathways of metabolism. The metabolites are likely systemically formed. Carfilzomib is eliminated primarily via metabolism with subsequent excretion of its metabolites in urine.
Carfilzomib is not expected to inhibit the metabolism of CYP3A4/5 substrates and is not a CYP3A4 inducer in humans. However, because it is unknown whether carfilzomib is an inducer of CYP1A2, 2C8, 2C9, 2C19 and 2B6 at therapeutic concentrations, caution should be observed when carfilzomib is combined with medicinal products that are substrates of these enzymes, such as oral contraceptives. At the time of the initial marketing authorization, one study (PX‐171‐005), conducted in 50 MM patients with normal renal function (n = 12), mild (n = 12), moderate (n = 10), and severe (n = 8) renal impairment, and patients on chronic dialysis (n = 8) was submitted. A second renal impairment study (Study CFZ001) conducted in 23 relapsed MM patients with creatinine clearance (CrCL) ≥75 mL/minute (n = 13) and patients with end‐stage renal disease (ESRD) requiring dialysis (n = 10) was submitted post‐approval. In study CFZ001, a trend toward an increased AUC, Cmax, t½, and slower clearance (CL) was seen for ESRD patients compared with subjects with normal renal function. However, results from both studies showed that renal function status had no marked effect on the exposure of carfilzomib following single‐ or repeat‐dose administration. Therefore, no starting dose adjustments are required in patients with baseline mild, moderate, or severe renal impairment or patients on chronic dialysis. However, because the incidence of adverse events (AEs) of acute renal failure was higher in patients with lower baseline CrCL than that among patients with higher baseline CrCL. Renal function should be assessed at treatment initiation and monitored at least monthly or in accordance with accepted clinical practice guidelines, particularly in patients with lower baseline CrCL (CrCL < 30 mL/minute), and appropriate dose modifications based on toxicity should be made.
No dedicated pharmacokinetic (PK) studies had been completed in patients with hepatic impairment at the time of the initial marketing authorization; however, the results of a hepatic impairment study (Study CFZ002) have been submitted post‐approval, indicating that no starting dose adjustment is recommended in patients with mild or moderate hepatic impairment. However, because higher subject incidence of hepatic function abnormalities, ≥grade 3 AEs, and serious AEs (SAEs) have been reported in patients with mild or moderate hepatic impairment compared with patients with normal hepatic function, liver enzymes and bilirubin should be assessed at treatment initiation and monitored during treatment with carfilzomib and appropriate dose modifications should be considered.
Clinical Efficacy
The pivotal efficacy study was PX‐171‐009 (ASPIRE), a randomized, multicenter, phase III study to compare the efficacy and safety of CRd versus Rd in patients with relapsed MM [11].
Eligibility criteria included symptomatic and measurable MM; at least one prior treatment but no more than three protocol‐defined MM regimens, with documented relapsed or progressive disease (PD) on or after any regimen (subjects refractory to the most recent line of therapy were eligible); achieved a response to at least one prior regimen; Eastern Cooperative Oncology Group (ECOG) performance status 0–2. Patients were excluded in case of progression during treatment (if previously treated with bortezomib alone or in combination) or progression during the first 3 months of initiating treatment or at any time during treatment if the Rd combination was the patient's most recent line of therapy (if previously treated with a Rd combination).
Carfilzomib was administered at an initial dose of 20 mg/m2, which was increased to 27 mg/m2 on cycle 1, day 8, twice weekly for 3 out of 4 weeks as a 10‐minute infusion for a maximum of 18 cycles unless discontinued early for disease progression or unacceptable toxicity. Lenalidomide and dexamethasone administration could continue until progression or unacceptable toxicity. The dose selection of carfilzomib was based on PX‐171‐006, a phase Ib dose‐escalation study of CRd in patients with relapsed MM [12]. The stepped‐up dosing regimen was based on the PX‐171‐002 study, a phase I dose‐escalation study in subjects with hematologic malignancies [13].
The primary endpoint was PFS assessed by independent review committee (IRC), defined as the duration in months from the date of randomization to the date of confirmed PD or death due to any cause, whichever was earlier, according to the International Myeloma Working Group Uniform Response Criteria. Main secondary endpoints included OS, the rate of overall response (ORR), and health‐related quality of life (HRQoL). The secondary efficacy endpoints were to be tested sequentially—OS, ORR, DCR, and HRQOL (as measured by EORTC QLQ‐C30 [European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core Module] in order to control the family‐wise type I error rate. Because the interim analysis of OS did not cross the prespecified early stopping boundary, the results presented for treatment comparisons, including p values, are for descriptive purposes only.
A total of 792 patients were randomly assigned (1:1 ratio) to receive either CRd or Rd and they were stratified by β2 microglobulin levels (<2.5 mg/L vs. ≥2.5 mg/L), prior bortezomib (yes vs. no), and prior lenalidomide (yes vs. no).
In the PFS analysis, which was carried out with 82% of the planned events, the median PFS in the CRd group was 26.3 months compared with 17.6 months for the Rd group (HR = 0.69; 95% CI, 0.57–0.83; one‐sided log‐rank p value < .0001; Fig. 2, as reported in reference [11]).
Figure 2.

Kaplan‐Meier plot of progression‐free survival as determined by the Independent. Review Committee (ITT Population, PX‐171‐009).
Abbreviations: CI, confidence interval; CRd, carfilzomib with Revlimid (lenalidomide; Celgene, Durham, NC, http://www.celgene.com) with low‐dose dexamethasone; HR, hazard ratio; ITT, intention to treat; PFS, progression‐free survival; Rd: Revlimid (lenalidomide) with low‐dose dexamethasone.
Based on an interim analysis carried out with 60% of final number of events required, the median OS was not reached in either group and did not cross the prespecified early stopping boundary for the interim analysis (HR = 0.79; 95% CI, 0.63–0.99; one‐sided log‐rank p = .0182). In the CRd group, the ORR was 87.1%, compared with 66.7% in the Rd group, and more patients had a stringent complete response (sCR; 14.1% vs. 4.3%), complete response (CR; 17.7% vs. 5.1%), and very good partial response (VGPR; 38.1% vs. 31.1%) in the CRd group compared with those in the Rd group (Table 2).
Table 2. Effects table for carfilzomib (study PX‐171‐009).

Table includes treatment‐emergent grade 3 or higher adverse events. Treatment‐emergent adverse events are defined as any adverse event with an onset date between the date of first dose and 30 days after the date of last dose of any study drug. Adverse events were coded using MedDRA version 15.1 and graded using NCI‐CTCAE version 4.0.
Abbreviations: CR, complete response; CRd, carfilzomib with Revlimid (lenalidomide; Celgene, Durham, NC, http://www.celgene.com) with low‐dose dexamethasone; HR, hazard ratio; NE, not estimable; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; PR, partial response; Rd: Revlimid (lenalidomide) with low‐dose dexamethasone; sCR, stringent complete response; VGPR, very good partial response.
PX‐171‐009 assessed HRQOL using the EORTC QLQ‐C30 and EORTC QLQ‐MY20. Questionnaire completion at baseline was very similar between the study arms, with 94.9% of subjects in the CRd arm versus 93.2% in the Rd arm completing the questionnaires. Just under half of subjects completed the QLQ‐C30 questionnaire (47.3%) at cycle 18. A higher proportion of subjects randomized to CRd completed the QLQ‐C30 questionnaire at each cycle compared with Rd subjects. This difference was largest at cycle 18, with 57.3% in the CRd arm versus 37.4% in the Rd arm, respectively. Using a restricted maximum likelihood‐based mixed model for repeated measures analysis under the assumption of missing at random, subjects treated with CRd reported improved global health status with higher QLQ‐C30 Global Health Status/QoL scores compared with Rd over 18 cycles of treatment (p value = .0001). The minimal important difference for between‐group differences on the QLQ‐C30 Global Health Status/QoL was met at cycle 12 (5.56) and approached at cycle 18 (4.81) when comparing CRd versus Rd.
Two sensitivity analyses were performed that confirmed the findings of the main HRQL analysis. The first sensitivity analysis based on a pattern mixture model using an ancillary variable to account for missingness showed CRd consistently improved global health status based on the QLQ‐C30 Global Health Status/QoL compared with Rd over 18 cycles of treatment (p = .0006). The second sensitivity analysis used a pattern mixture model with the missing patterns being defined by the timing of the last assessment (p < .0001).
Supportive study PX 171 011 was an open‐label, randomized, phase III study that evaluated carfilzomib monotherapy versus low‐dose corticosteroids with optional cyclophosphamide in patients with relapsed and refractory (≥3 prior therapies) MM. Patients enrolled to study (n = 315) were more heavily pretreated and with lower organ and marrow function as compared with those enrolled in the pivotal study (PX 171 009). No significant difference was observed in the primary analysis of OS (HR = 0.98; 95% CI, 0.76–1.25; p = .4172). Median PFS was 3.7 months (95% CI, 2.8–4.2) in the carfilzomib group compared with 3.3 months (95% CI, 2.2–5.2) in the control group (HR 1.091; 95% CI, 0.843–1.410; one‐sided p = .2479). Overall response was higher with carfilzomib (19.1 vs. 11.4%) [14], [15].
Supportive study PX 171 003A1 was a single‐arm phase II study in patients (n = 266) with relapsed and refractory MM who had received at least two prior therapies and were previously treated with both bortezomib and either of two immunomodulatory drugs (IMiDs) (thalidomide or lenalidomide). The IRC‐assessed ORR on study was 22.9% (95% CI, 18.0–28.5) [16].
Clinical Safety
Eleven completed clinical studies with 2,123 patients formed the primary basis for evaluation of carfilzomib safety. Patients in study PX‐171‐006 received carfilzomib for a median duration of 72 weeks (range: 1‐93.1), and the median number of carfilzomib‐containing cycles initiated was 18 (range: 1–18). In study PX‐171‐009, the median relative dose intensity was 96% (CRd group) for carfilzomib, 91% and 92% for lenalidomide (CRd and Rd group, respectively), and 95% and 95% for dexamethasone.
The most common adverse reactions (occurring in >20% of patients) observed in patients receiving carfilzomib were anemia, fatigue, diarrhea, thrombocytopenia, nausea, pyrexia, dyspnea, respiratory tract infection, cough, and peripheral edema (Table 1).
In study PX‐171‐009, 59.9% and 54.0% of patients experienced at least one SAE in the CRd and Rd arms, respectively, with pneumonia, respiratory tract infection, pyrexia, pulmonary embolism, deep vein thrombosis, anemia, bronchitis, and febrile neutropenia being the most frequently observed. The incidence of treatment‐related grade ≥3 AEs was 67.1% in the CRd group and 60.2% in the Rd group. There were no grade ≥3 treatment‐related AEs with a ≥5% difference between study arms. Grade ≥3 AEs with a ≥2% to <5% difference between study arms included neutropenia, thrombocytopenia, pneumonia, and hypophosphatemia.
Thirty (7.7%) patients in the CRd arm and 33 (8.5%) patients in the Rd arm died on study (within 30 days after their last dose of any study treatment: carfilzomib, lenalidomide, or dexamethasone) with AEs of infection being the most common cause of on‐study deaths. No deaths were considered to be related specifically to carfilzomib alone. Two of the deaths in the CRd arm were considered to be related to both carfilzomib and lenalidomide.
In clinical studies, cardiac failure (reported in approximately 7% of subjects), myocardial infarction (reported in approximately 2% of subjects), and myocardial ischaemia (reported in approximately 1% of subjects) typically occurred early in the course of carfilzomib therapy (<5 cycles). Approximately 65% of cardiac failure events, 75% of myocardial infarction events, and 83% of myocardial ischaemia events were grade ≥3 events. In pivotal study PX‐171‐009, there were more cardiac failure events (6.4% vs. 4.1%), grade ≥3 cardiac failure (3.8% vs. 1.8%), ischemic heart disease (5.9% vs. 4.6%), grade ≥3 ischemic heart disease (3.3% vs. 2.1%), cardiac arrhythmias (16.6% vs. 15.2%), and cardiomyopathy (1.0% vs. 0.3%) in the CRd arm than in the Rd group. In addition, cardiotoxicity was reported as primary cause of death in 10 patients in CRd group vs. 7 patients in the Rd arm (see benefit‐risk assessment and discussion).
Other important identified risks included pulmonary toxicities, pulmonary hypertension, dyspnea, hypertension, acute renal failure, tumor lysis syndrome, infusion reactions, thrombocytopenia, hepatic toxicity, thrombotic microangiopathy, posterior reversible encephalopathy syndrome, and febrile neutropenia.
Missing information included safety in patients with hepatic impairment, safety in patients with clinically significant cardiovascular disease, and safety in pregnant or breastfeeding women. Ongoing and planned studies in the pharmacovigilance development plan included a phase I study in subjects with relapsed MM and ESRD, and a phase I study of the pharmacokinetics and safety of carfilzomib in subjects with advanced malignancies and varying degrees of hepatic impairment.
Benefit‐Risk Assessment and Discussion
During the initial evaluation, the concerns were raised about the indication, with reference to subjects who have progressed while on bortezomib or Rd therapy who were excluded from the pivotal study. Additional subgroup analyses of PFS provided by the applicant company showed that for patients refractory to prior bortezomib (they did not progress during treatment), the median PFS was 22.3 months versus 19.4 months in CRd and Rd arms, respectively (HR 0.799; 95% CI, 0.492–1.297). For patients refractory to prior lenalidomide (they did not progress during the first 3 months of therapy, or at any time on therapy if it was the last regimen prior to study entry, or discontinued due to intolerance), the median PFS was 11.3 months versus 9.0 months in CRd and Rd arms, respectively (HR 0.637; 95% CI, 0.333–1.219). Furthermore, similar results were observed in patients double‐refractory to lenalidomide and dexamethasone in the same regimen, with a median PFS of 10.3 months versus 8.8 months in CRd and Rd arms, respectively (HR 0.602; 95% CI, 0.275–1.318). In light of these results, it was considered that benefits of the CRd combination had been established in the group of patients with refractory disease (defined as they met any of the following three criteria: nonresponsive to any regimen; progression during any regimen; or progression within 60 days of completion of any regimen).
Patients with New York Heart Association (NYHA) class III or IV heart failure were excluded from the pivotal study protocol (PX‐171‐009). Cardiovascular events were, however, commonly reported in the enrolled population with lower cardiovascular risk. There were more cardiac failure events (6.4% vs. 4.1%), grade ≥3 cardiac failure (3.8% vs. 1.8%), ischemic heart disease (5.9% vs. 4.6%), grade ≥3 ischemic heart disease (3.3% vs. 2.1%), cardiac arrhythmias (16.6% vs. 15.2%), and cardiomyopathy (1.0% vs. 0.3%) in the CRd group than in the Rd group. There were concerns about the cardiotoxicity because the mechanism behind this was not totally clear and patients at particular risk could not be identified based on the data available. Although the mechanism for the cardiac effects was unclear, based on the on‐target specificity of the carfilzomib molecule and the similarity of the cardiac effects observed preclinically and clinically with other proteasome inhibitors such as bortezomib (acute development or exacerbation of congestive heart failure, and/or new onset of decreased left ventricular ejection fraction), the effects are likely pharmacological. Therefore, excluding patients with higher cardiovascular risk from the target population for carfilzomib was not supported because these patients could benefit from treatment. Patients with signs or symptoms of NYHA class III or IV cardiac failure and/or recent history of myocardial infarction (in the last 4 months) and patients with uncontrolled angina or arrhythmias should have a comprehensive medical assessment prior to starting treatment with carfilzomib. Routine risk minimization measures to manage this risk include close monitoring of this population as described in the product information. In addition, cardiac toxicity (cardiac failure, myocardial ischemia, myocardial infarction, and cardiac arrest) has been classified as an identified risk in the Risk Management Plan.
Conclusion
The addition of carfilzomib to Rd resulted in a clinically meaningful and statistically significant improvement in the primary endpoint of PFS compared with Rd. The primary efficacy results were supported by increased response rates (sCR, CR, and VGPR). Although OS data were not mature, the results pointed out a clear positive trend in favor of CRd. The delay in disease progression observed with CRd was clinically relevant and appeared superior (albeit based on indirect comparisons) to available triple‐combinations in the setting of relapsed MM, such as VTD, VRD, or panobinostat, bortezomib, and dexamethasone [17], [18]. Although the interim analysis of OS did not cross the prespecified early stopping boundary, they are only descriptive; the use of the combination of CRd seem to provide a positive benefit in terms of ORR (CRd 87.1%; Rd 66.7%; p < .0001), duration of response (CRd 28.6 months; Rd 21.2 months), and QoL.
Regarding the toxicity associated with CRd, there is a pattern of more frequent AEs and more frequent grade 3–4 AEs in patients treated with CRd (83.7%) compared with those treated with Rd (81.2%). However, in light of the data related to discontinuations, the combination seems tolerable, given the comparable rates of discontinuation between arms in the main study (26% vs. 25.2%).
Overall, the scientific review concluded that the benefit‐risk balance of carfilzomib was positive in the treatment of adult patients with MM who have received at least one prior therapy.
Acknowledgments
The scientific assessment summarized in this report is based on important contributions from the rapporteur and co‐rapporteur assessment teams, CHMP members, and additional experts following the application for a marketing authorization from the company. Disclaimer: This publication is a summary of the European Public Assessment Report, the summary of product characteristics, and other product information as published on the EMA website (www.ema.europa.eu). For the most current information on this marketing authorization, please refer to the EMA website. The authors remain solely responsible for the opinions expressed in this publication.
Footnotes
For Further Reading: Zahra Hanaizi, Beatriz Flores, Robert Hemmings et al. The European Medicines Agency Review of Pomalidomide in Combination With Low‐Dose Dexamethasone for the Treatment of Adult Patients WithMultipleMyeloma: Summary of the Scientific Assessment of the Committee forMedicinal Products for Human Use. The Oncologist 2015;20:329‐334; first published on February 11, 2015.
Implications for Practice: Pomalidomide in combination with low‐dose dexamethasone has been approved in the European Union to treat adult patients with relapsed and refractory multiple myeloma. The approval is based on efficacy data in 455 adults with relapsed and refractory multiple myeloma who have received at least two prior treatment regimens, including both lenalidomide and bortezomib, and have demonstrated disease progression on the last therapy (Study CC‐4047‐MM‐003). In this study, pomalidomide/low‐dose dexamethasone was associated with a median progression‐free survival of 16 weeks compared to 8 weeks for high‐dose dexamethasone alone. The most common side effects associated with pomalidomide/low‐dose dexamethasone were anemia, neutropenia, thrombocytopenia, fatigue and pyrexia. A teratogenic effect of pomalidomide in humans is expected.
Author Contributions
Interpretation of data: Jorge Camarero Jiménez, Isabel Garcia, Arantxa Sancho‐López, Marc Martin, Alexandre Moreau, Pierre Demolis
Manuscript writing: Kyriaki Tzogani, Jorge Camarero Jiménez, Isabel Garcia, Arantxa Sancho‐López, Marc Martin, Alexandre Moreau, Pierre Demolis, Francesco Pignatti
Final approval of manuscript: Kyriaki Tzogani, Jorge Camarero Jiménez, Isabel Garcia, Arantxa Sancho‐López, Marc Martin, Alexandre Moreau, Pierre Demolis, Tomas Salmonson, Jonas Bergh, Edward Laane, Heinz Ludwig, Christian Gisselbrecht, Francesco Pignatti
Disclosures
Heinz Ludwig: Amgen, Takeda (RF), Amgen, Takeda, Janssen‐Cilag, Celgene, Bristol‐Myers Squibb (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Kyle RA, Gertz MA, Witzig TE et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003;78:21–33. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Steliarova‐Foucher E, Lortet‐Tieulent J et al. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer 2013;49:1374–1403. [DOI] [PubMed] [Google Scholar]
- 3. Bray F, Ren JS, Masuyer E et al. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 2013;132:1133–1145. [DOI] [PubMed] [Google Scholar]
- 4. Moreau P, San Miguel J, Ludwig H et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up†. Ann Oncol 2013;24(suppl 6):vi133–vi137. [DOI] [PubMed] [Google Scholar]
- 5. Anderson KC, Kyle RA, Rajkumar SV et al. Clinically relevant end points and new drug approvals for myeloma. Leukemia 2007;22:231–239. [DOI] [PubMed] [Google Scholar]
- 6. van de Donk NW, Lokhorst HM, Dimopoulos M et al. Treatment of relapsed and refractory multiple myeloma in the era of novel agents. Cancer Treat Rev 2011;37:266–283. [DOI] [PubMed] [Google Scholar]
- 7. Kumar S, Lee JH, Lahuerta JJ et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012;26:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Durie BGM. Multiple Myeloma. International Myeloma Foundation. 2015 Edition. https://www.myeloma.org/sites/default/files/images/publications/concise_review.pdf.
- 9. Moreau P. Death of frontline allo‐SCT in myeloma. Blood 2012;119:6178–6179. [DOI] [PubMed] [Google Scholar]
- 10. Herndon TM, Deisseroth A, Kaminskas E et al. U.S. Food and Drug Administration approval: Carfilzomib for the treatment of multiple myeloma. Clin Cancer Res 2013;19:4559–4563. [DOI] [PubMed] [Google Scholar]
- 11. Stewart AK, Rajkumar SV, Dimopoulos MA et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–152. [DOI] [PubMed] [Google Scholar]
- 12. Wang M, Martin T, Bensinger W et al. Phase 2 dose‐expansion study (PX‐171‐006) of carfilzomib, lenalidomide, and low‐dose dexamethasone in relapsed or progressive multiple myeloma. Blood 2013;122:3122–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alsina M, Trudel S, Vallone M et al. Phase 1 single agent antitumor activity of twice weekly consecutive day dosing of the proteasome inhibitor carfilzomib (PR‐171) in hematologic malignancies. Blood 2007;110:411a. [Google Scholar]
- 14. Hájek R, Bryce R, Ro S et al. Design and rationale of FOCUS (PX‐171‐011): A randomized, open‐label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer 2012;12:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hájek R, Masszi T, Petrucci MT et al. A randomized phase III study of carfilzomib vs low‐dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia 2017;31:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siegel DS, Martin T, Wang M et al. A phase 2 study of single‐agent carfilzomib (PX‐171–003‐A1) in patients with relapsed and refractory multiple myeloma. Blood 2012;120:2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richardson PG, Xie W, Jagannath S et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood 2014;123:1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garderet L, Iacobelli S, Moreau P et al. Superiority of the triple combination of bortezomib‐thalidomide‐dexamethasone over the dual combination of thalidomide‐dexamethasone in patients with multiple myeloma progressing or relapsing after autologous transplantation: The MMVAR/IFM 2005‐04 Randomized Phase III Trial From the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2012;30:2475–2482. [DOI] [PubMed] [Google Scholar]