

Abstract
Arterial and venous thrombosis are major contributors to coagulation-associated morbidity and mortality. Greater understanding of mechanisms leading to thrombus formation and stability is expected to lead to improved treatment strategies. Factor XIII (FXIII) is a transglutaminase found in plasma and platelets. During thrombosis, activated FXIII crosslinks fibrin and promotes thrombus stability. Recent studies have provided new information about FXIII activity during coagulation and its effects on clot composition and function. These findings reveal newly-recognized roles for FXIII in thrombosis. Herein, we review published literature on FXIII biology and effects on fibrin structure and stability, epidemiologic data associating FXIII with thrombosis, and evidence from animal models indicating FXIII has an essential role in determining thrombus stability, composition, and size.
Keywords: Factor XIII, myocardial infarction, venous thromboembolism, stroke, fibrinogen
INTRODUCTION
Intravascular coagulation, or thrombosis, is a primary contributor to coagulation-associated morbidity and mortality. Thrombosis can occur in the arterial or venous circulation, leading to myocardial infarction, stroke, deep vein thrombosis, and pulmonary embolism. Arterial thrombosis is usually associated with atherosclerotic plaque rupture that exposes subendothelial cells and procoagulant material (e.g., tissue factor, collagen) to blood, leading to platelet activation and aggregation in high shear rates and stress. Ultimately, platelet accumulation and fibrin deposition produce an occlusive platelet-rich “white thrombus” (reviewed in1). In contrast, venous thrombosis/thromboembolism is thought to be triggered by inappropriate expression of cell adhesion molecules on intact, but dysfunctional endothelium in the presence of plasma hypercoagulability in reduced blood flow (stasis). This group of abnormalities is known as Virchow’s triad.2 Because venous thrombi have high red blood cell (RBC) content, they are known as “red thrombi.” Fibrin formation is a central, etiologic component of both arterial and venous thrombosis, and is the ultimate target of thrombolytic enzymes used to treat both of these clinical presentations.
Factor XIII (FXIII) is a protransglutaminase found in cells and plasma. As the central driver of fibrin stability, the contribution of activated FXIII (FXIII[a]) to arterial and venous thrombosis has been investigated. However, although epidemiologic studies have associated abnormal FXIII levels and/or function with thrombosis risk, these associations are complex and the mechanisms by which FXIII contributes to thrombosis in vivo remain unclear.
Herein, we briefly review aspects of FXIII biology and activity. We then review recent findings on the contributions of these functions to thrombus formation, stability, and composition in vitro, and how these observations have exposed newly-recognized pathophysiologic roles for FXIII(a) during thrombosis in vivo. These discoveries may help reconcile apparently discordant findings from previous studies, and reveal new insight into the role of FXIII in arterial and venous thrombosis.
FXIII BIOLOGY
FXIII is one of 9 members of the transglutaminase superfamily found in both cellular (transglutaminases 1–7 and erythrocyte band 4.2) and plasma (pFXIII) compartments. The cellular forms of FXIII are present in multiple cell types, including monocytes, megakaryocytes, osteoblasts, and platelets (reviewed in3). Plasma and platelet FXIII are thought to contribute to blood coagulation.
FXIII in plasma
Plasma FXIII is unique because it circulates as two catalytic subunits (FXIII-A2) and two non-catalytic subunits (FXIII-B2) arranged in a non-covalent, heterotetramer (FXIII-A2B2, Mr 325-kDa, ~70 nM, 14–28 μg/mL).4 Mechanisms mediating the interactions between the A and B subunits are reviewed by Schroeder and Kohler in this issue of Seminars in Thrombosis and Hemostasis5. Essentially all FXIII-A2B2 zymogen circulates in complex with fibrinogen.6 Although older studies implicated the alternatively-spliced fibrinogen γ-chain (termed γ′) in FXIII-A2B2 binding7,8, a more recent study using recombinant fibrinogen suggested FXIII-A2B2 binds both γA- and γ′-containing molecules with similar affinity (KD ~40 nM)9. Both older10 and more recent11 studies identified regions in the fibrinogen α-chain that bind FXIII. Smith et al.11 found that FXIII-A2B2 binds a peptide containing amino acid residues 371–425 of the fibrinogen αC domain with high affinity (KD 5–30 nM). However, they did not demonstrate binding of FXIII-A2B2 to this region on full-length fibrinogen and suggested that the interaction between FXIII-A2B2 and the αC region may arise during FXIII activation, and that other fibrinogen residues bind zymogen FXIII-A2B2 in circulation.11 Our group showed that murine fibrin(ogen) containing alanine mutations within γ-chain residues γ390–396 (Fibγ390–396A) exhibits decreased binding of FXIII-A2B2, suggesting these residues mediate the carrier function.12 Since residues γ390–396 are immediately N-terminal to the fibrin γ-chain residues that are crosslinked by FXIII (glutamine residues 398/399 and lysine 406), localization of FXIII-A2B2 at this site would position FXIIIa for rapid translocation to its nearby substrate recognition residues during thrombus formation.
The interface of FXIII-A2B2 that interacts with fibrin(ogen) has not been determined. However, Souri et al.13 suggested the FXIII-B subunits augment the catalytic effect of fibrin(ogen) binding on FXIII activation. Further experiments using truncated FXIII-B subunits suggested sushi domains 1 and 10 mediate this interaction.13 More studies are needed to characterize the specific FXIII residues involved in the FXIII-fibrinogen interaction, and how these residues contribute to FXIII localization during thrombus formation.
FXIII in platelets
Platelet FXIII is a homodimer of catalytic subunits (FXIII-A2) present in high concentrations (~46–82 fg/platelet).14 Most platelet FXIII is derived from megakaryocytes during platelet production, but platelets may also uptake a minor fraction from plasma as fibrinogen-bound FXIII-A2B2. Platelets may also translate FXIII mRNA de novo.15 In unactivated platelets, FXIII-A2 is primarily associated with the cytoplasmic fraction.16
FXIII activation
During activation of plasma FXIII-A2B2, thrombin first catalyzes the cleavage of activation peptide(s) from the N-termini of the FXIII-A subunits (FXIII-A2′B2).17,18 Calcium then promotes dissociation of the FXIII-B subunits from FXIII-A2′, yielding activated FXIII-A2* (FXIIIa).19 Plasma FXIII activation occurs early during coagulation and is accelerated when it is bound to fibrinogen, which facilitates dissociation of the FXIII-B subunits.10,20,21 This function ascribes fibrin(ogen) with an important regulatory role during FXIII(a) activation and activity. Plasma from mice that have reduced binding of FXIII-A2B2 to fibrin(ogen) (Fibγ390–396A mice) exhibits delayed activation of FXIII-A2B2 and slower formation of fibrin γ- and α-chain crosslinks.12 In addition to demonstrating the catalytic role of FXIII-A2B2 binding to fibrinogen in plasma, these findings led to new observations on the impact of FXIII activation kinetics on thrombus formation in vivo (discussed below).
Activation of platelet FXIII-A2 can occur after thrombin- or calpain-mediated cleavage of the activation peptide(s).22 FXIII-A2 can also be activated without activation peptide cleavage, in the presence of high calcium concentrations.23 Following platelet activation, FXIII-A2* is exposed in protruding caps on the surface of phosphatidylserine-positive platelets, but is not released into the supernatant.16 The mechanisms mediating FXIII exposure on activated platelets are still poorly-understood.
Although early studies suggested the activation peptide(s) remain bound to FXIII-A2*, more recent studies indicate the activation peptide is released during FXIII activation18,24 and can be used as a biomarker of acute ischemic stroke.25
FXIIIa inactivation
FXIIIa activity is generated early during clot formation and can still be detected within experimental thrombi 6 or more hours after the onset of clotting.26 Consequently, mechanisms mediating FXIIIa inactivation are of substantial interest. FXIIIa inactivation has been attributed to reversible oxidation, proteolytic digestion by thrombin, and proteolytic enzymes released by granulocytes.27,28 Studies have examined the ability of plasmin to activate and inactivate FXIII(a)29,30, with conflicting conclusions. Recently, Hur et al. showed that both plasma- and platelet-derived FXIII-A2*, but not zymogen FXIII-A2B2, are cleaved by plasmin during fibrinolysis.31 The specificity of this mechanism for activated FXIII-A2* may stem from steric protection provided by the FXIII-B subunits when bound to FXIII-A2. This finding is interesting because it temporally-associates clot dissolution with a mechanism to inactivate any FXIIIa that may be released from the dissolving clot. Thus, this mechanism suggests the coagulation system maintains tight control over transglutaminase activity during hemostasis. It is therefore intriguing to speculate that reduced fibrinolytic activity could promote thrombosis by permitting prolonged transglutaminase activity and excessive crosslinking of proteins within the thrombus and the blood.
FXIIIA TRANSGLUTAMINASE ACTIVITY AND EFFECTS ON FIBRIN
Each FXIII-A subunit contains an active site (cysteine 314, histidine 373, and aspartate 396) that catalyzes the formation of intermolecular ε-N-(γ-glutamyl)-lysyl crosslinks. FXIII-A2B2 does not compete with FXIII-A2* for binding to fibrin21, indicating these interactions are mediated by unique binding sites. However, apart from the specific residues that are crosslinked, the nature of the intermolecular interaction between FXIII-A2* and fibrinogen has not been resolved. Smith et al. showed that the structural cleft on FXIII-A2* that is exposed by thrombin-mediated cleavage and subsequent release of the FXIII activation peptide contains a recognition site for the fibrin(ogen) α-chain.24 A consensus recognition motif for FXIIIa substrates has not been identified, suggesting the interaction between FXIIIa and its substrates is mediated by substrate secondary and/or tertiary structure rather than primary amino acid sequence, and may involve a distinct exosite on the FXIIIa molecule.32,33
Cellular FXIII(a) (cFXIII[a]) crosslinks cellular and plasma proteins and regulates a variety of physiologic functions including phagocytosis and cell locomotion, osteoblast differentiation and matrix formation, and preadipocyte maturation and proliferation. The contributions of FXIIIa activity to these and other non-classical functions are reviewed by Schroeder and Kohler5.
Although FXIIIa has over 140 potential substrates in plasma34, it actually displays high substrate specificity during clot formation and is best characterized for its role in fibrin crosslinking. Briefly, during coagulation, thrombin cleaves N-terminal fibrinopeptides from the Aα- and Bβ-chains to produce fibrin monomers that polymerize into fibrin fibers (Figure 1). FXIIIa catalyzes the formation of isopeptide bonds between glutamine residues 398/399 and lysine 406 in the fibrin γ-chain and subsequently between glutamine and lysine residues in the α-chain (Figure 1). It is likely that the preference of FXIIIa for fibrin is at least partly mediated by its pre-localization on fibrinogen in circulation, in that fibrin(ogen) “delivers” FXIII(a) to the nascent clot.
Figure 1. FXIIIa crosslinking during fibrin formation.
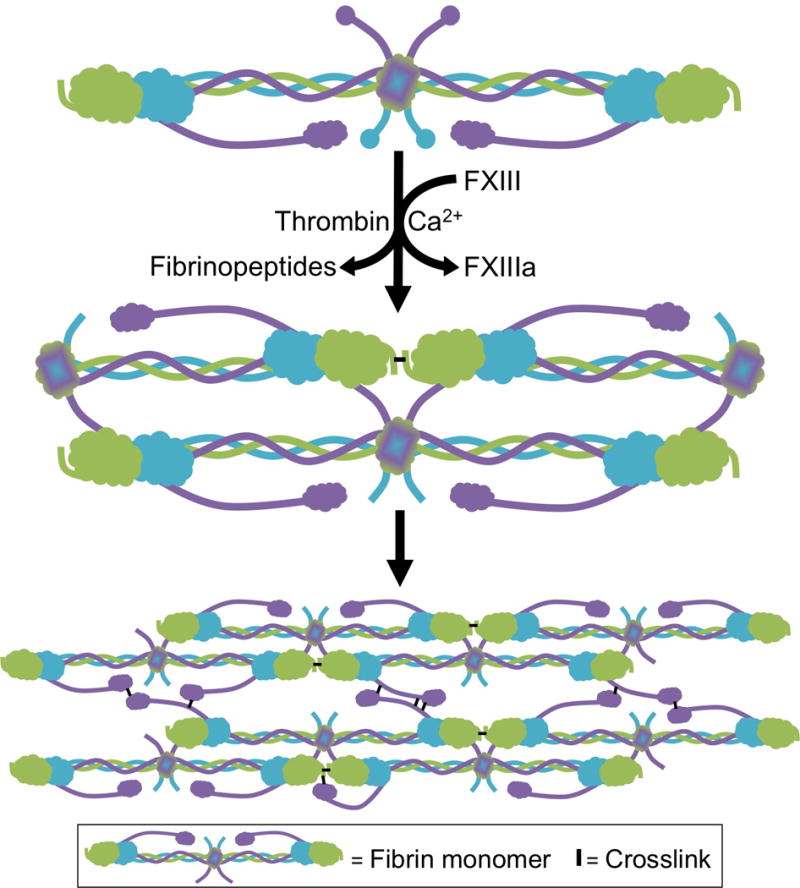
Fibrinogen is a hexamer composed of 2 Aα- (purple), 2 Bβ- (blue), and 2 γ-chains (green). During coagulation, thrombin cleaves N-terminal fibrinopeptides from the Aα- and Bβ-chains, producing fibrin monomers which polymerize into protofibrils and subsequently, fibers.110 FXIIIa increases clot stability by introducing ε-N-(γ-glutamyl)-lysyl crosslinks between residues in the γ- and α-chains of fibrin monomers within individual fibers. FXIIIa first introduces crosslinks between γ-chains (forming γ-γ dimers) and subsequently between γ- and α-chains (forming high molecular weight species [γ-multimers, α-polymers, and αγ-hybrids]).37,50–52
Fibrin crosslinking has minor effects on global network structure, but substantial effects on the structure and function of individual fibers, and consequently, clot biochemical and mechanical stability, and clot composition (Figure 2).
Figure 2. Contributions of FXIIIa to clot biochemical and mechanical stability.
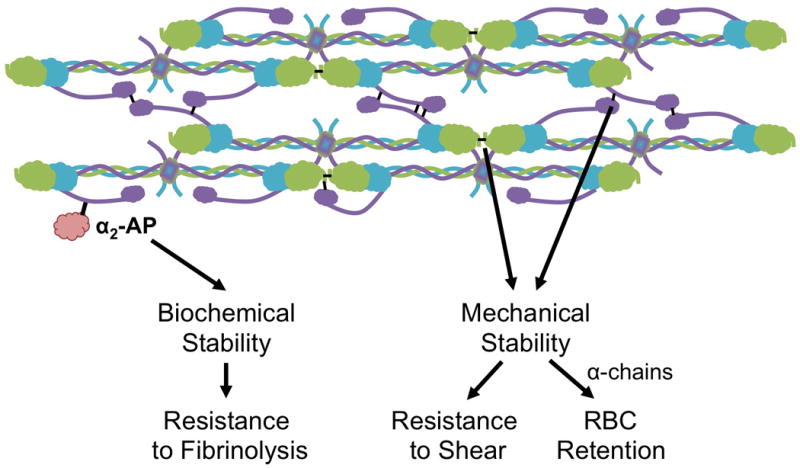
FXIIIa crosslinking of plasma proteins [i.e. α2-antiplasmin, (α2-AP)] increases the resistance of the clot to fibrinolysis. Crosslinking of the fibrin α- (purple) and γ-chains (green) stiffens fibrin fibers and increases the mechanical stability of the clot. Increased mechanical stability renders the clot more resistant to shear forces. α-chain crosslinking enables RBC retention during clot contraction.39
Effects of FXIIIa on fibrin structure
Crosslinking has little effect on gross fibrin network morphology, producing, at most, only a minor (~12%) increase in fibrin network density.35–39 In contrast, fibrin crosslinking substantially alters the structure of individual fibrin fibers by promoting protofibril coupling within the fiber, itself.40 This effect, which is specifically associated with the formation of α-chain-rich, high molecular weight (HMW) crosslinked species, causes fiber compaction. Compaction decreases the size of pores within individual fibers, with potentially important effects on the diffusion of molecules like tissue-type plasminogen activator (tPA) through the fibers and consequently, on fibrinolysis. Fiber compaction also promotes fiber stiffening, making fibers more resistant to deformation under low strain.40 Thus, the effect of FXIIIa on thrombus formation likely stems from its effects on the biochemical and biomechanical properties of individual fibrin fibers.
Effects of FXIIIa on fibrin resistance to fibrinolysis
FXIII(a) has critical anti-fibrinolytic functions during coagulation, mediated by its ability to crosslink antifibrinolytic proteins, including α2-antiplasmin41,42, thrombin activatable fibrinolysis inhibitor43, and type-2 plasminogen activator inhibitor44 to fibrin. This activity is mediated primarily by plasma FXIII-A2*. However, in situations in which the concentration of plasma FXIII is low (≤10%), platelet FXIII-A2* exposed on the platelet surface also promotes fibrin stability by crosslinking α2-antiplasmin to fibrin.16,45
Effects of FXIIIa on fibrin mechanical stability
FXIIIa-mediated crosslinking significantly increases the elastic modulus (stiffness) of both individual fibrin fibers46–48 and whole clot networks49, and is required to protect the clot against premature disruption or dissolution. Using a synthetic FXIII inhibitor and a patient-derived antibody to selectively inhibit the formation of HMW crosslinked species yet permit γ-γ dimer formation, Ryan et al.50 showed that γ-chain crosslinking, alone, is insufficient to stiffen fibrin networks, and that the increased stiffness is correlated with the formation of α-chain-rich, HMW crosslinked species.50 More recent studies using a recombinant fibrinogen mutated to eliminate γ-chain crosslinking sites confirmed that α-chain crosslinking is the primary contributor to clot stiffness and elasticity.37,51,52
Effects of FXIIIa on clot contraction
Several studies have examined the role of FXIII in platelet-mediated clot contraction, with discordant results. Whereas some investigators reported impaired retraction of FXIII-deficient plasma clots53–55, others observed normal or even increased contraction of plasma and whole blood clots from FXIII-deficient humans and mice12,39,56–58. Some of the discord may stem from the use of a transglutaminase inhibitor, cystamine, that also reduces thrombin activity59 and therefore, may have indirectly reduced thrombin-mediated platelet activation. Other experimental differences that may account for these discordant findings have not been identified.
Effects of FXIIIa on clot composition
Our group recently showed that FXIIIa-mediated crosslinking of fibrin is also a major determinant of thrombus RBC content.12,39 Briefly, reduced plasma FXIII or presence of a FXIII(a) inhibitor decreases RBC retention in contracted human whole blood clots.12 Whole blood from mice with full or partial deficiency in the FXIII catalytic A-subunit also show decreased retention of RBCs in contracted clots.12 These data are consistent with the “excessive red cell fallout” previously reported in a family with congenital FXIII deficiency56, and demonstrate a newly-recognized function for FXIII(a) during clot formation. Further work using recombinant fibrinogen variants that lack γ- or α-chain crosslinking residues, and FXIIIa inhibitor concentrations that preferentially block formation of α-chain-rich HMW crosslinked species versus γ-γ dimers associated this effect specifically with fibrin α-chain crosslinking.39 Given the effect of fibrin α-chain crosslinks on fibrin biophysical properties37,50–52, these data suggest FXIIIa promotes RBC retention in clots via its ability to increase fibrin elastic modulus and/or elasticity during clot contraction. Since clot contraction packs and deforms RBCs trapped within the clot and decreases clot permeability60, these data implicate FXIII activity as a major determinant of clot composition and function. The contribution of these effects to thrombosis in vivo is discussed below.
FXIII-ASSOCIATED ISOMERASE ACTIVITY
Some studies have suggested that members of the transglutaminase family are also associated with a protein disulphide isomerase (PDI) activity.61,62 The thiol isomerase family is part of the intracellular biosynthetic pathway during protein synthesis, but members of this family also have extracellular functions. Curiously, none of the transglutaminases carry the canonical isomerase catalytic motif (cysteine-X-X-cysteine, where X can be any one of a number of amino acids), suggesting the PDI-like activity may reside in a protein that associates with these transglutaminases. Since canonical PDIs contribute to thrombosis in mice and possibly humans (reviewed in63,64), the possibility that a FXIII-related thiol isomerase activity promotes thrombus formation warrants further investigation.
EPIDEMIOLOGIC DATA ON THE ROLE OF FXIII IN THROMBOSIS
Excellent reviews have summarized epidemiologic studies investigating the contribution of FXIII to arterial and venous thrombosis.65–67 In aggregate, findings suggest FXIII antigen, activity, and/or genotype influence thrombosis risk, at least in certain populations and clinical situations. Notably, however, these studies have shown considerable discordance. This discord is thought to reflect several as-yet incompletely-understood aspects of FXIII function that may have influenced conclusions. These potential aspects are discussed below.
First, early studies were confounded by the use of FXIII activity assays that were unknowingly influenced by the FXIII-A Val34Leu polymorphism present in ~25% of European Caucasians.68 This polymorphism causes accelerated release of the FXIII activation peptide, 2.5-fold earlier FXIII activation, and consequently, faster fibrin crosslinking in vitro.69–71 In certain assays, this accelerated activation erroneously appears as increased FXIIIa activity, leading to poor correlation between FXIII antigen and activity and overestimation of the normal range for FXIIIa activity.71 Notably, however, even after this effect was recognized, findings from studies evaluating the association of the Val34Leu polymorphism with venous and arterial thrombosis have been inconsistent.70,72–78 Meta-analyses of these studies suggest presence of the 34Leu allele offers significant, but modest protection against coronary artery disease (CAD, odds ratio (OR) 0.81, 95% confidence interval (95% CI) 0.70–0.92)79 and venous thromboembolism (OR 0.85, 95% CI 0.77–0.95).80
Second, effects of the Val34Leu polymorphism are modulated by complex gene-environment interactions that were not appreciated in early studies. For example, an analysis of 474 patients with deep vein thrombosis enrolled in the Leiden Thrombophilia Study (LETS) showed only a possible weak protective effect of Leu34 homozygosity in men (OR 0.7, 95% CI 0.4–1.3).81 However, a subsequent analysis of the same samples that considered plasma fibrinogen level suggested slightly stronger protection in both men and women, especially in those over 45 years old (OR 0.4, 95% CI 0.2–1.0).82 Similarly, in a large prospective study (Norfolk cohort of the European Prospective Investigation in Cancer and Nutrition study [EPIC-Norfolk]), the Val34Leu variant was not broadly associated with risk of CAD. However, when fibrinogen levels were considered in the analysis, Leu34 homozygotes in the lowest tertile of fibrinogen concentration had increased risk of developing CAD (OR 2.88, 95 CI 1.24–6.74, P=0.003), whereas those in the highest tertile of fibrinogen concentrations trended towards reduced risk of developing CAD (OR 0.47, 95% CI 0.18–1.17, P=0.1).83 Similarly, in a high-risk Hungarian population, although presence of the Leu34 allele did not appear to alter the risk of coronary sclerosis or myocardial infarction (MI) in the general population, when patients with fibrinogen levels in the upper quartile (>4.6 g/L) were analyzed, the Leu34 allele was associated with protection against MI (OR 0.41, 95% CI 0.18–0.93).84 The proposed mechanism for this effect is interesting. In plasmas with normal fibrinogen levels, the Leu34 allele produces clots with thinner fibers and decreased permeability, whereas in plasmas with high fibrinogen, it produces clots with thicker fibers, and increased permeability and susceptibility to fibrinolysis.85 Since abnormal fiber thickness and altered clot permeability and stability are associated with increased thrombosis risk (reviewed in86), these observations indicate that both FXIII genotype and plasma fibrinogen concentration should be considered when calculating thrombosis risk in population studies.
Third, other FXIII polymorphisms that are not frequently incorporated into epidemiological analyses may also modulate the association between FXIII and thrombosis risk in certain populations. For example, the FXIII-A Tyr204Phe polymorphism has been associated with high (9-to-11-fold) increased risk for MI and ischemic stroke in a Dutch cohort87,88, although there was no association seen in a Brazilian population89. The FXIII-B*2 polymorphism (His95Arg) primarily found in African populations is associated with accelerated dissociation of the B-subunit.90 This polymorphism was associated with mildly increased risk of venous thrombosis in Dutch Caucasians (OR 1.5, 95% CI 1.1–2.0)90, but not in Iranians91, and is not associated with CAD or MI in a Hungarian population92. The FXIII-B*3 polymorphism that predominantly occurs in Asians results in a novel splice acceptor site which replaces the final 10 residues of the C-terminus with 25 new residues containing one additional acidic and two extra basic residues.93 In a Hungarian population, this FXIII-B*3 polymorphism results in lower FXIII activity and antigen levels and is protective against CAD and MI, but only in the presence of both high fibrinogen levels and the FXIII-A Leu34 allele (OR 0.23, 95% CI 0.06–0.85 and OR 0.10, 95% CI 0.02–0.53, respectively, adjusted for synergy).92 Together, these studies suggest that incorporating information about these alleles may refine interpretations on the contribution of FXIII to thrombosis in certain populations.
Fourth, the effect of FXIII on thrombosis risk also appears to reflect sex-specific factors that have not been considered in all studies, many of which were not powered to detect a modulating effect of sex. For example, in a Hungarian population, FXIII antigen correlates with moderately increased risk of MI (OR 1.999, 95% CI 1.051–3.765, P=0.03)94 and peripheral artery disease (PAD, OR 2.000, 95% CI 0.943–4.240, P=0.07)95 in women, but not men. Moreover, although FXIII activity is slightly increased in both male and female PAD patients compared to controls, FXIII activity in the upper tertile (>120%) is correlated with increased risk of PAD in women (OR 2.316, 95% CI 1.157–4.635, P=0.02), but not men.95 The effect of the Val34Leu polymorphism on risk may also be sex-specific; female, but not male, homozygotes for the Leu34 allele have increased risk of fatal atherothrombotic stroke.96 The sex-specific factors that mediate these differences have not been identified.
Finally, but importantly, the possibility that publication bias has inflated the apparent impact of FXIII antigen, activity, and/or genotype on thrombosis risk must be considered when evaluating these data in aggregate. A meta-analysis of 16 studies on the Val34Leu polymorphism suggested that smaller studies with negative findings are less frequently published than studies with positive findings.79 Thus, the relationship between FXIII and thrombosis risk may be weaker than it appears.
ANIMAL MODELS INVESTIGATING THE ROLE OF FXIII IN THROMBOSIS
Animal models are a useful tool for studying thrombosis in physiologically-relevant settings that lack the heterogeneity of human populations. Studies using animal models have revealed early and rapid contributions of FXIII(a) activity to clot formation, stabilization, and composition during thrombosis in vivo.
Contribution of FXIII to arterial thrombosis
Mouse models have been used to extend clinical observations and examine the association of cellular FXIII with arterial thrombosis. For example, Mansfield et al. detected increased levels of circulating FXIII in patients with type 2 diabetes relative to healthy controls.97 Using mouse models of diabetes, Elgheznawy et al. subsequently associated calpain- and miR-223 dependent upregulation of platelet FXIII with increased ferric chloride-mediated carotid artery thrombosis.15 Although preliminary, these findings suggest FXIII is involved in thrombotic complications in diabetes. In addition, patients who experienced post-MI heart failure or death have lower FXIII levels at hospital admission and increased FXIII consumption 5 days after MI.98 Reduced FXIII levels are also associated with increased risk of ventricular rupture.99,100 Studies using mice to examine the mechanism of this association indicate the relationship between FXIII level and myocardial repair is causative; FXIII-deficient mice die from left ventricular rupture within 5 days after MI, whereas wild type mice or FXIII-deficient mice infused with FXIII replacement have normal survival rates.100 These data suggest FXIII plays a role in healing the infarcted area and that FXIII levels may be an early prognostic indicator during and immediately following acute MI. In addition, since reduced FXIII decreases survival after acute MI, this outcome may bias retrospective studies on the role of FXIII in MI.
Two studies used animal models to test the ability of FXIIIa inhibitors to reduce arterial thrombus stability. In a rabbit model of femoral artery thrombosis and tPA-mediated thrombolysis, administration of a FXIIIa inhibitor reduced thrombus weights more than administration of tPA, alone, and facilitated thrombolysis in 50% of treated rabbits.101 Similarly, in a canine model of coronary artery thrombosis, FXIIIa inhibition accelerated reperfusion and reduced residual thrombus mass following tPA administration.102 Notably, in both models this effect was limited to animals pretreated with the FXIIIa inhibitor, and was not observed in animals infused with FXIIIa inhibitor after thrombus induction.101,102 This finding suggests FXIII activation and crosslinking activity occur early during thrombogenesis. Accordingly, using a fluorescent peptide derived from α2-antiplasmin to probe for FXIIIa activity in mice, Jaffer et al.103 detected signal enhancement within 30 minutes of thrombus induction, indicating early generation of FXIIIa activity during thrombogenesis.
Contribution of FXIII to venous thrombosis
Our group used mice to investigate the effect of FXIII on venous thrombosis in vivo. Consistent with ex vivo observations of reduced RBC retention in FXIII-deficient clots12,39,56, thrombi from FXIII-deficient mice are smaller than thrombi from wild type mice and have reduced RBC content.12 These findings indicate a central role for FXIII activity during venous thrombosis in vivo. Intriguingly, mice with normal levels of FXIII, but reduced binding of plasma FXIII-A2B2 to fibrinogen (Fibγ390–396A mice) phenocopy FXIII-deficient mice, producing smaller venous thrombi with reduced RBC content.12 Notably, the loss of FXIII binding to fibrinogen in Fibγ390–396A mice delays, but does not abolish, FXIII activation and fibrin crosslinking12; Fibγ390–396A thrombi are fully-crosslinked 24 hours after thrombus induction (M.M. Aleman and A.S. Wolberg, unpublished observation). These observations in mice suggest biological rationales for FXIII binding to circulating fibrinogen in vivo: 1) binding and localization to fibrin(ogen) facilitates delivery of FXIII to the nascent thrombus, 2) localization of FXIII on fibrin(ogen) accelerates FXIIIa activation during thrombogenesis, and 3) FXIII activation at the thrombus site spatially and temporally associates FXIIIa activity with an essential function thrombus contraction. These data suggest not only the generation, but also the timing, of FXIIIa activity is a critical determinant of venous thrombus composition and consequently, size.
Contribution of FXIII to thrombus stability
FXIIIa activity may also influence risk of embolization of both arterial and venous thrombi. Platelet FXIII-A mRNA is significantly lower in patients with non-valvular atrial fibrillation and thrombus embolization than in similar patients with left atrial appendage thrombus and no history of embolization.104 Accordingly, in mice, FXIIIa-mediated crosslinking of plasma fibronectin into thrombi formed in mesenteric arterioles enhances platelet aggregation and increases the stability of platelet-rich thrombi.105–107 In a ferric chloride model of venous thrombosis, FXIII-deficient mice show increased embolization compared to wild type mice.108
Together, these observations demonstrate critical FXIIIa contributions at several steps during thrombosis.
CONCLUSIONS AND REMAINING QUESTIONS
FXIII is unique among coagulation proteins, not only in the nature of its enzymatic activity that crosslinks, rather than proteolyzes proteins, but also in its ability to directly impact both biochemical and biophysical properties of thrombi. In the almost 70 years since its first description as a “fibrin stabilizing factor109”, defining the role of FXIII in coagulation has been a fundamental goal. Growing data from biochemical, epidemiological, and animal model studies suggest FXIII(a) is an important determinant of thrombus composition and stability.
Recent findings have resolved several long-standing questions, but also exposed new ones. For example, studies examining the contributions of the Val34Leu polymorphism to clotting have revealed complex gene-environment interactions that mediate this activity. However, these studies used purified proteins and cell-free plasma systems. Given the newly-recognized effect of FXIII activation kinetics on whole blood clot composition (RBC retention)12,39, what is the effect of the Val34Leu polymorphism in more physiologically-relevant whole blood systems? Studies have shown that both plasma and platelet FXIII can contribute to clot stability in vitro.16,41–45 Do both of these pools contribute to clot composition and stability in vivo? Data from animal models show FXIII(a) contributes to thrombus size and stability and subsequent tissue repair. Is there a potential role for FXIII(a) inhibitors as a primary or adjunct approach for anticoagulation? Continued investigations of both basic FXIII biology and the effect of FXIII on thrombosis in humans and animal models are expected to uncover central information about these (patho)physiologic mechanisms.
Acknowledgments
FUNDING
ASW is supported by a research grant from the National Institutes of Health (R56HL094740). JRB is supported by a National Science Foundation Graduate Research Fellowship (DGE-1144081).
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
References
- 1.Wolberg AS, Aleman MM, Leiderman K, Machlus KR. Procoagulant activity in hemostasis and thrombosis: Virchow’s triad revisited. Anesth Analg. 2012;114(2):275–285. doi: 10.1213/ANE.0b013e31823a088c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolberg AS, Rosendaal FR, Weitz JI, et al. Venous thrombosis. Nature Reviews Disease Primers. 2015;1:1–17. doi: 10.1038/nrdp.2015.6. [DOI] [PubMed] [Google Scholar]
- 3.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91(3):931–972. doi: 10.1152/physrev.00016.2010. [DOI] [PubMed] [Google Scholar]
- 4.Fickenscher K, Aab A, Stuber W. A photometric assay for blood coagulation factor XIII. Thromb Haemost. 1991;65(5):535–540. [PubMed] [Google Scholar]
- 5.Schroeder V, Kohler HP. Factor XIII, structure and function. Sem Thromb Haemost. 2016 doi: 10.1055/s-0036-1571341. In Press. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg CS, Shuman MA. The zymogen forms of blood coagulation factor XIII bind specifically to fibrinogen. J Biol Chem. 1982;257(11):6096–6101. [PubMed] [Google Scholar]
- 7.Siebenlist KR, Meh DA, Mosesson MW. Plasma factor XIII binds specifically to fibrinogen molecules containing γ chains. Biochemistry. 1996;35(32):10448–10453. doi: 10.1021/bi9606206. [DOI] [PubMed] [Google Scholar]
- 8.Moaddel M, Farrell DH, Daugherty MA, Fried MG. Interactions of human fibrinogens with factor XIII: roles of calcium and the γ′ peptide. Biochemistry. 2000;39(22):6698–6705. doi: 10.1021/bi000098u. [DOI] [PubMed] [Google Scholar]
- 9.Gersh KL, Lord ST. An investigation of factor XIII binding to recombinant γ′/γ′ and γ/γ′ fibrinogen. Blood. 2006;108(11) Abstract 1705. [Google Scholar]
- 10.Credo RB, Curtis CG, Lorand L. α-chain domain of fibrinogen controls generation of fibrinoligase (coagulation factor XIIIa). Calcium ion regulatory aspects. Biochemistry. 1981;20(13):3770–3778. doi: 10.1021/bi00516a016. [DOI] [PubMed] [Google Scholar]
- 11.Smith KA, Adamson PJ, Pease RJ, et al. Interactions between factor XIII and the αC region of fibrinogen. Blood. 2011;117(12):3460–3468. doi: 10.1182/blood-2010-10-313601. [DOI] [PubMed] [Google Scholar]
- 12.Aleman MM, Byrnes JR, Wang JG, et al. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124(8):3590–3600. doi: 10.1172/JCI75386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Souri M, Osaki T, Ichinose A. The non-catalytic B subunit of coagulation factor XIII accelerates fibrin cross-linking. J Biol Chem. 2015;290(19):12027–12039. doi: 10.1074/jbc.M114.608570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katona EE, Ajzner E, Toth K, Karpati L, Muszbek L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates. J Immunol Methods. 2001;258(1–2):127–135. doi: 10.1016/s0022-1759(01)00479-3. [DOI] [PubMed] [Google Scholar]
- 15.Elgheznawy A, Shi L, Hu J, et al. Dicer cleavage by calpain determines platelet microRNA levels and function in diabetes. Circ Res. 2015;117(2):157–165. doi: 10.1161/CIRCRESAHA.117.305784. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell JL, Lionikiene AS, Fraser SR, et al. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124(26):3982–3990. doi: 10.1182/blood-2014-06-583070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takagi T, Doolittle RF. Amino acid sequence studies on factor XIII and the peptide released during its activation by thrombin. Biochemistry. 1974;13(4):750–756. doi: 10.1021/bi00701a018. [DOI] [PubMed] [Google Scholar]
- 18.Schroeder V, Vuissoz JM, Caflisch A, Kohler HP. Factor XIII activation peptide is released into plasma upon cleavage by thrombin and shows a different structure compared to its bound form. Thromb Haemost. 2007;97(6):890–898. [PubMed] [Google Scholar]
- 19.Hornyak TJ, Shafer JA. Role of calcium ion in the generation of factor XIII activity. Biochemistry. 1991;30(25):6175–6182. doi: 10.1021/bi00239a014. [DOI] [PubMed] [Google Scholar]
- 20.Credo RB, Curtis CG, Lorand L. Ca2+-related regulatory function of fibrinogen. Proc Natl Acad Sci U S A. 1978;75(9):4234–4237. doi: 10.1073/pnas.75.9.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornyak TJ, Shafer JA. Interactions of factor XIII with fibrin as substrate and cofactor. Biochemistry. 1992;31(2):423–429. doi: 10.1021/bi00117a017. [DOI] [PubMed] [Google Scholar]
- 22.Ando Y, Imamura S, Yamagata Y, et al. Platelet factor XIII is activated by calpain. Biochem Biophys Res Commun. 1987;144(1):484–490. doi: 10.1016/s0006-291x(87)80535-1. [DOI] [PubMed] [Google Scholar]
- 23.Muszbek L, Polgar J, Boda Z. Platelet factor XIII becomes active without the release of activation peptide during platelet activation. Thromb Haemost. 1993;69(3):282–285. [PubMed] [Google Scholar]
- 24.Smith KA, Pease RJ, Avery CA, et al. The activation peptide cleft exposed by thrombin cleavage of FXIII-A(2) contains a recognition site for the fibrinogen α chain. Blood. 2013;121(11):2117–2126. doi: 10.1182/blood-2012-07-446393. [DOI] [PubMed] [Google Scholar]
- 25.Ortner E, Schroeder V, Walser R, Zerbe O, Kohler HP. Sensitive and selective detection of free FXIII activation peptide: a potential marker of acute thrombotic events. Blood. 2010;115(24):5089–5096. doi: 10.1182/blood-2009-11-253062. [DOI] [PubMed] [Google Scholar]
- 26.Finlayson JS, Aronson DL. Crosslinking of rabbit fibrin in vivo. Thromb Diath Haemorrh. 1974;31(3):435–438. [PubMed] [Google Scholar]
- 27.Robinson BR, Houng AK, Reed GL. Catalytic life of activated factor XIII in thrombi. Implications for fibrinolytic resistance and thrombus aging. Circulation. 2000;102(10):1151–1157. doi: 10.1161/01.cir.102.10.1151. [DOI] [PubMed] [Google Scholar]
- 28.Bagoly Z, Haramura G, Muszbek L. Down-regulation of activated factor XIII by polymorphonuclear granulocyte proteases within fibrin clot. Thromb Haemost. 2007;98(2):359–367. [PubMed] [Google Scholar]
- 29.Hedner U, Johansson L, Nilsson IM. Effects of porcine plasmin on the coagulation and fibrinolytic systems in humans. Blood. 1978;51(1):157–164. [PubMed] [Google Scholar]
- 30.Rider DM, McDonagh J. Resistance of factor XIII to degradation or activation by plasmin. Biochim Biophys Acta. 1981;675(2):171–177. doi: 10.1016/0304-4165(81)90223-3. [DOI] [PubMed] [Google Scholar]
- 31.Hur WS, Mazinani N, Lu XJ, et al. Coagulation factor XIIIa is inactivated by plasmin. Blood. 2015;126(20):2329–2337. doi: 10.1182/blood-2015-07-650713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doiphode PG, Malovichko MV, Mouapi KN, Maurer MC. Evaluating factor XIII specificity for glutamine-containing substrates using a matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay. Anal Biochem. 2014;457:74–84. doi: 10.1016/j.ab.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cleary DB, Maurer MC. Characterizing the specificity of activated Factor XIII for glutamine-containing substrate peptides. Biochim Biophys Acta. 2006;1764(7):1207–1217. doi: 10.1016/j.bbapap.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Nikolajsen CL, Dyrlund TF, Poulsen ET, Enghild JJ, Scavenius C. Coagulation factor XIIIa substrates in human plasma: identification and incorporation into the clot. J Biol Chem. 2014;289(10):6526–6534. doi: 10.1074/jbc.M113.517904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophys J. 1999;77(5):2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collet JP, Moen JL, Veklich YI, et al. The αC domains of fibrinogen affect the structure of the fibrin clot, its physical properties, and its susceptibility to fibrinolysis. Blood. 2005;106(12):3824–3830. doi: 10.1182/blood-2005-05-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Standeven KF, Carter AM, Grant PJ, et al. Functional analysis of fibrin γ-chain cross-linking by activated factor XIII: determination of a cross-linking pattern that maximizes clot stiffness. Blood. 2007;110(3):902–907. doi: 10.1182/blood-2007-01-066837. [DOI] [PubMed] [Google Scholar]
- 38.Hethershaw EL, Cilia La Corte AL, Duval C, et al. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J Thromb Haemost. 2014;12(2):197–205. doi: 10.1111/jth.12455. [DOI] [PubMed] [Google Scholar]
- 39.Byrnes JR, Duval C, Wang Y, et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin α-chain crosslinking. Blood. 2015;126(16):1940–1948. doi: 10.1182/blood-2015-06-652263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurniawan NA, Grimbergen J, Koopman J, Koenderink GH. Factor XIII stiffens fibrin clots by causing fiber compaction. J Thromb Haemost. 2014;12(10):1687–1696. doi: 10.1111/jth.12705. [DOI] [PubMed] [Google Scholar]
- 41.van Giezen JJ, Minkema J, Bouma BN, Jansen JW. Cross-linking of α2-antiplasmin to fibrin is a key factor in regulating blood clot lysis: species differences. Blood Coagul Fibrinolysis. 1993;4(6):869–875. [PubMed] [Google Scholar]
- 42.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through α(2)-antiplasmin cross-linking. Blood. 2011;117(23):6371–6374. doi: 10.1182/blood-2011-02-333203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. 1998;273(42):27220–27224. doi: 10.1074/jbc.273.42.27220. [DOI] [PubMed] [Google Scholar]
- 44.Jensen PH, Lorand L, Ebbesen P, Gliemann J. Type-2 plasminogen-activator inhibitor is a substrate for trophoblast transglutaminase and factor XIIIa. Transglutaminase-catalyzed cross-linking to cellular and extracellular structures. Eur J Biochem. 1993;214(1):141–146. doi: 10.1111/j.1432-1033.1993.tb17906.x. [DOI] [PubMed] [Google Scholar]
- 45.Reed GL, Matsueda GR, Haber E. Platelet factor XIII increases the fibrinolytic resistance of platelet-rich clots by accelerating the crosslinking of α2-antiplasmin to fibrin. Thromb Haemost. 1992;68(3):315–320. [PubMed] [Google Scholar]
- 46.Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW. The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci U S A. 2005;102(26):9133–9137. doi: 10.1073/pnas.0504120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu W, Jawerth LM, Sparks EA, et al. Fibrin fibers have extraordinary extensibility and elasticity. Science. 2006;313(5787):634. doi: 10.1126/science.1127317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Houser JR, Hudson NE, Ping L, et al. Evidence that αC region is origin of low modulus, high extensibility, and strain stiffening in fibrin fibers. Biophys J. 2010;99(9):3038–3047. doi: 10.1016/j.bpj.2010.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glover CJ, McIntire LV, Brown CH, 3rd, Natelson EA. Rheological properties of fibrin clots. Effects of fibrinogen concentration, factor XIII deficiency, and factor XIII inhibition. J Lab Clin Med. 1975;86(4):644–656. [PubMed] [Google Scholar]
- 50.Ryan EA, Mockros LF, Stern AM, Lorand L. Influence of a natural and a synthetic inhibitor of factor XIIIa on fibrin clot rheology. Biophys J. 1999;77(5):2827–2836. doi: 10.1016/S0006-3495(99)77114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Helms CC, Ariens RA, Uitte de Willige S, Standeven KF, Guthold M. α-α cross-links increase fibrin fiber elasticity and stiffness. Biophys J. 2012;102(1):168–175. doi: 10.1016/j.bpj.2011.11.4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duval C, Allan P, Connell SD, et al. Roles of fibrin α- and γ-chain specific cross-linking by FXIIIa in fibrin structure and function. Thromb Haemost. 2014;111(5):842–850. doi: 10.1160/TH13-10-0855. [DOI] [PubMed] [Google Scholar]
- 53.Cohen I, Gerrard JM, White JG. Ultrastructure of clots during isometric contraction. J Cell Biol. 1982;93(3):775–787. doi: 10.1083/jcb.93.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Endo Y, Takahashi K, Mamiya S, Satoh M, Matsuda M. Factor XIII deficiency associated with Klippel-Weber disease, platelet dysfunction and cryofibrinogenemia. Acta Haematol. 1983;69(6):398–403. doi: 10.1159/000206928. [DOI] [PubMed] [Google Scholar]
- 55.Kasahara K, Souri M, Kaneda M, et al. Impaired clot retraction in factor XIII A subunit-deficient mice. Blood. 2010;115(6):1277–1279. doi: 10.1182/blood-2009-06-227645. [DOI] [PubMed] [Google Scholar]
- 56.Hanna M. Congenital deficiency of factor 13: report of a family from Newfoundland with associated mild deficiency of factor XII. Pediatrics. 1970;46(4):611–619. [PubMed] [Google Scholar]
- 57.Rao KM, Newcomb TF. Clot retraction in a factor XIII free system. Scand J Haematol. 1980;24(2):142–148. doi: 10.1111/j.1600-0609.1980.tb02358.x. [DOI] [PubMed] [Google Scholar]
- 58.Jelenska M, Kopec M, Breddin K. On the retraction of collagen and fibrin induced by normal, defective and modified platelets. Haemostasis. 1985;15(3):169–175. doi: 10.1159/000215140. [DOI] [PubMed] [Google Scholar]
- 59.Aleman MM, Holle LA, Stember KG, et al. Cystamine preparations exhibit anticoagulant activity. PLoS One. 2015;10(4):e0124448. doi: 10.1371/journal.pone.0124448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cines DB, Lebedeva T, Nagaswami C, et al. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014;123(10):1593–1603. doi: 10.1182/blood-2013-08-523860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasegawa G, Suwa M, Ichikawa Y, et al. A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem J. 2003;373(Pt 3):793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lahav J, Karniel E, Bagoly Z, et al. Coagulation factor XIII serves as protein disulfide isomerase. Thromb Haemost. 2009;101(5):840–844. [PubMed] [Google Scholar]
- 63.Furie B, Flaumenhaft R. Thiol isomerases in thrombus formation. Circ Res. 2014;114(7):1162–1173. doi: 10.1161/CIRCRESAHA.114.301808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiu J, Passam F, Butera D, Hogg PJ. Protein disulfide isomerase in thrombosis. Semin Thromb Hemost. 2015;41(7):765–773. doi: 10.1055/s-0035-1564047. [DOI] [PubMed] [Google Scholar]
- 65.Muszbek L, Bagoly Z, Bereczky Z, Katona E. The involvement of blood coagulation factor XIII in fibrinolysis and thrombosis. Cardiovasc Hematol Agents Med Chem. 2008;6(3):190–205. doi: 10.2174/187152508784871990. [DOI] [PubMed] [Google Scholar]
- 66.Bereczky Z, Muszbek L. Factor XIII and venous thromboembolism. Semin Thromb Hemost. 2011;37(3):305–314. doi: 10.1055/s-0031-1273094. [DOI] [PubMed] [Google Scholar]
- 67.Bagoly Z, Koncz Z, Harsfalvi J, Muszbek L. Factor XIII, clot structure, thrombosis. Thromb Res. 2012;129(3):382–387. doi: 10.1016/j.thromres.2011.11.040. [DOI] [PubMed] [Google Scholar]
- 68.Muszbek L. Deficiency causing mutations and common polymorphisms in the factor XIII-A gene. Thromb Haemost. 2000;84(4):524–527. [PubMed] [Google Scholar]
- 69.Wartiovaara U, Mikkola H, Szoke G, et al. Effect of Val34Leu polymorphism on the activation of the coagulation factor XIII-A. Thromb Haemost. 2000;84(4):595–600. [PubMed] [Google Scholar]
- 70.Balogh I, Szoke G, Karpati L, et al. Val34Leu polymorphism of plasma factor XIII: biochemistry and epidemiology in familial thrombophilia. Blood. 2000;96(7):2479–2486. [PubMed] [Google Scholar]
- 71.Ariens RA, Philippou H, Nagaswami C, et al. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96(3):988–995. [PubMed] [Google Scholar]
- 72.Catto AJ, Kohler HP, Coore J, et al. Association of a common polymorphism in the factor XIII gene with venous thrombosis. Blood. 1999;93(3):906–908. [PubMed] [Google Scholar]
- 73.Franco RF, Reitsma PH, Lourenco D, et al. Factor XIII Val34Leu is a genetic factor involved in the etiology of venous thrombosis. Thromb Haemost. 1999;81(5):676–679. [PubMed] [Google Scholar]
- 74.Margaglione M, Bossone A, Brancaccio V, Ciampa A, Di Minno G. Factor XIII Val34Leu polymorphism and risk of deep vein thrombosis. Thromb Haemost. 2000;84(6):1118–1119. [PubMed] [Google Scholar]
- 75.Kohler HP, Stickland MH, Ossei-Gerning N, et al. Association of a common polymorphism in the factor XIII gene with myocardial infarction. Thromb Haemost. 1998;79(1):8–13. [PubMed] [Google Scholar]
- 76.Aleksic N, Ahn C, Wang YW, et al. Factor XIIIA Val34Leu polymorphism does not predict risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) Study. Arterioscler Thromb Vasc Biol. 2002;22(2):348–352. doi: 10.1161/hq0202.102874. [DOI] [PubMed] [Google Scholar]
- 77.Reiner AP, Heckbert SR, Vos HL, et al. Genetic variants of coagulation factor XIII, postmenopausal estrogen therapy, and risk of nonfatal myocardial infarction. Blood. 2003;102(1):25–30. doi: 10.1182/blood-2002-07-2308. [DOI] [PubMed] [Google Scholar]
- 78.Atherosclerosis Thrombosis Vascular Biology Italian Study Group. No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation. 2003;107(8):1117–1122. doi: 10.1161/01.cir.0000051465.94572.d0. [DOI] [PubMed] [Google Scholar]
- 79.Voko Z, Bereczky Z, Katona E, Adany R, Muszbek L. Factor XIII Val34Leu variant protects against coronary artery disease. A meta-analysis. Thromb Haemost. 2007;97(3):458–463. [PubMed] [Google Scholar]
- 80.Wells PS, Anderson JL, Scarvelis DK, Doucette SP, Gagnon F. Factor XIII Val34Leu variant is protective against venous thromboembolism: a HuGE review and meta-analysis. Am J Epidemiol. 2006;164(2):101–109. doi: 10.1093/aje/kwj179. [DOI] [PubMed] [Google Scholar]
- 81.Van Hylckama Vlieg A, Komanasin N, Ariens RA, et al. Factor XIII Val34Leu polymorphism, factor XIII antigen levels and activity and the risk of deep venous thrombosis. Br J Haematol. 2002;119(1):169–175. doi: 10.1046/j.1365-2141.2002.03797.x. [DOI] [PubMed] [Google Scholar]
- 82.Vossen CY, Rosendaal FR. The protective effect of the factor XIII Val34Leu mutation on the risk of deep venous thrombosis is dependent on the fibrinogen level. J Thromb Haemost. 2005;3(5):1102–1103. doi: 10.1111/j.1538-7836.2005.01312.x. [DOI] [PubMed] [Google Scholar]
- 83.Boekholdt SM, Sandhu MS, Wareham NJ, et al. Fibrinogen plasma levels modify the association between the factor XIII Val34Leu variant and risk of coronary artery disease: the EPIC-Norfolk prospective population study. J Thromb Haemost. 2006;4(10):2204–2209. doi: 10.1111/j.1538-7836.2006.02154.x. [DOI] [PubMed] [Google Scholar]
- 84.Bereczky Z, Balogh E, Katona E, et al. Modulation of the risk of coronary sclerosis/myocardial infarction by the interaction between factor XIII subunit A Val34Leu polymorphism and fibrinogen concentration in the high risk Hungarian population. Thromb Res. 2007;120(4):567–573. doi: 10.1016/j.thromres.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 85.Lim BC, Ariens RA, Carter AM, Weisel JW, Grant PJ. Genetic regulation of fibrin structure. Lancet. 2003;361(9367):1424–1431. doi: 10.1016/S0140-6736(03)13135-2. [DOI] [PubMed] [Google Scholar]
- 86.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 87.Pruissen DM, Slooter AJ, Rosendaal FR, van der Graaf Y, Algra A. Coagulation factor XIII gene variation, oral contraceptives, and risk of ischemic stroke. Blood. 2008;111(3):1282–1286. doi: 10.1182/blood-2007-08-110254. [DOI] [PubMed] [Google Scholar]
- 88.Siegerink B, Maino A, Algra A, Rosendaal FR. Hypercoagulability and the risk of myocardial infarction and ischemic stroke in young women. J Thromb Haemost. 2015;13(9):1568–1575. doi: 10.1111/jth.13045. [DOI] [PubMed] [Google Scholar]
- 89.Landau MB, Renni MS, Zalis MG, Spector N, Gadelha T. Coagulation factor XIII Tyr204Phe gene variant and the risk of ischemic stroke. J Thromb Haemost. 2013;11(7):1426–1427. doi: 10.1111/jth.12260. [DOI] [PubMed] [Google Scholar]
- 90.Komanasin N, Catto AJ, Futers TS, et al. A novel polymorphism in the factor XIII B-subunit (His95Arg): relationship to subunit dissociation and venous thrombosis. J Thromb Haemost. 2005;3(11):2487–2496. doi: 10.1111/j.1538-7836.2005.01624.x. [DOI] [PubMed] [Google Scholar]
- 91.Pourgheysari B, Drees F, Hashemzadeh-Chaleshtori M. Factor XIIIA-V34L and factor XIIIB-H95R in venous thromboembolism in central Iran: protective and neutral. Blood Coagul Fibrinolysis. 2014;25(5):439–443. doi: 10.1097/MBC.0000000000000073. [DOI] [PubMed] [Google Scholar]
- 92.Mezei ZA, Bereczky Z, Katona E, et al. Factor XIII B subunit polymorphisms and the risk of coronary artery disease. Int J Mol Sci. 2015;16(1):1143–1159. doi: 10.3390/ijms16011143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iwata H, Kitano T, Umetsu K, et al. Distinct C-terminus of the B subunit of factor XIII in a population-associated major phenotype: the first case of complete allele-specific alternative splicing products in the coagulation and fibrinolytic systems. J Thromb Haemost. 2009;7(7):1084–1091. doi: 10.1111/j.1538-7836.2009.03443.x. [DOI] [PubMed] [Google Scholar]
- 94.Bereczky Z, Balogh E, Katona E, et al. Elevated factor XIII level and the risk of myocardial infarction in women. Haematologica. 2007;92(2):287–288. doi: 10.3324/haematol.10647. [DOI] [PubMed] [Google Scholar]
- 95.Shemirani AH, Szomjak E, Csiki Z, et al. Elevated factor XIII level and the risk of peripheral artery disease. Haematologica. 2008;93(9):1430–1432. doi: 10.3324/haematol.12708. [DOI] [PubMed] [Google Scholar]
- 96.Shemirani AH, Antalfi B, Pongracz E, et al. Factor XIII-A subunit Val34Leu polymorphism in fatal atherothrombotic ischemic stroke. Blood Coagul Fibrinolysis. 2014;25(4):364–368. doi: 10.1097/MBC.0000000000000055. [DOI] [PubMed] [Google Scholar]
- 97.Mansfield MW, Kohler HP, Ariens RA, McCormack LJ, Grant PJ. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care. 2000;23(5):703–705. doi: 10.2337/diacare.23.5.703. [DOI] [PubMed] [Google Scholar]
- 98.Gemmati D, Zeri G, Orioli E, et al. Factor XIII-A dynamics in acute myocardial infarction: a novel prognostic biomarker? Thromb Haemost. 2015;114(1):123–132. doi: 10.1160/TH14-11-0952. [DOI] [PubMed] [Google Scholar]
- 99.Nahrendorf M, Hu K, Frantz S, et al. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation. 2006;113(9):1196–1202. doi: 10.1161/CIRCULATIONAHA.105.602094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nahrendorf M, Aikawa E, Figueiredo JL, et al. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: implications for FXIII therapy and antithrombin use in myocardial infarction. Eur Heart J. 2008;29(4):445–454. doi: 10.1093/eurheartj/ehm558. [DOI] [PubMed] [Google Scholar]
- 101.Leidy EM, Stern AM, Friedman PA, Bush LR. Enhanced thrombolysis by a factor XIIIa inhibitor in a rabbit model of femoral artery thrombosis. Thromb Res. 1990;59(1):15–26. doi: 10.1016/0049-3848(90)90267-g. [DOI] [PubMed] [Google Scholar]
- 102.Shebuski RJ, Sitko GR, Claremon DA, et al. Inhibition of factor XIIIa in a canine model of coronary thrombosis: effect on reperfusion and acute reocclusion after recombinant tissue-type plasminogen activator. Blood. 1990;75(7):1455–1459. [PubMed] [Google Scholar]
- 103.Jaffer FA, Tung CH, Wykrzykowska JJ, et al. Molecular imaging of factor XIIIa activity in thrombosis using a novel, near-infrared fluorescent contrast agent that covalently links to thrombi. Circulation. 2004;110(2):170–176. doi: 10.1161/01.CIR.0000134484.11052.44. [DOI] [PubMed] [Google Scholar]
- 104.Gosk-Bierska I, McBane RD, Wu Y, et al. Platelet factor XIII gene expression and embolic propensity in atrial fibrillation. Thromb Haemost. 2011;106(1):75–82. doi: 10.1160/TH10-11-0765. [DOI] [PubMed] [Google Scholar]
- 105.Ni H, Yuen PS, Papalia JM, et al. Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proc Natl Acad Sci U S A. 2003;100(5):2415–2419. doi: 10.1073/pnas.2628067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cho J, Mosher DF. Impact of fibronectin assembly on platelet thrombus formation in response to type I collagen and von Willebrand factor. Blood. 2006;108(7):2229–2236. doi: 10.1182/blood-2006-02-002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, Reheman A, Spring CM, et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014;124(10):4281–4293. doi: 10.1172/JCI74630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shaya SA, Saldanha LJ, Vaezzadeh N, et al. Comparison of the effect of dabigatran and dalteparin on thrombus stability in a murine model of venous thromboembolism. J Thromb Haemost. 2015 doi: 10.1111/jth.13182. In Press. [DOI] [PubMed] [Google Scholar]
- 109.Laki K, Lorand L. On the solubility of fibrin clots. Science. 1948;108(2802):280. doi: 10.1126/science.108.2802.280. [DOI] [PubMed] [Google Scholar]
- 110.Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011;31(3):494–499. doi: 10.1161/ATVBAHA.110.213389. [DOI] [PMC free article] [PubMed] [Google Scholar]