

Summary
Systemic lupus erythematosus (SLE) is an autoimmune disease with unrestrained T‐cell and B‐cell activity towards self‐antigens. Evidence shows that apoptotic cells (ApoCells) trigger an autoreactive response against nuclear antigens in susceptible individuals. In this study, we focus on generating and characterizing tolerogenic dendritic cells (tolDCs) to restore tolerance to ApoCells. Monocyte‐derived dendritic cells (DCs) from healthy controls and patients with SLE were treated with dexamethasone and rosiglitazone to induce tolDCs. Autologous apoptotic lymphocytes generated by UV irradiation were given to tolDCs as a source of self‐antigens. Lipopolysaccharide (LPS) was used as a maturation stimulus to induce the expression of co‐stimulatory molecules and secretion of cytokines. TolDCs generated from patients with SLE showed a reduced expression of co‐stimulatory molecules after LPS stimulation compared with mature DCs. The same phenomenon was observed in tolDCs treated with ApoCells and LPS. In addition, ApoCell‐loaded tolDCs stimulated with LPS secreted lower levels of interleukin‐6 (IL‐6) and IL‐12p70 than mature DCs without differences in IL‐10 secretion. The functionality of tolDCs was assessed by their capacity to prime allogeneic T cells. TolDCs displayed suppressor properties as demonstrated by a significantly reduced capacity to induce allogeneic T‐cell proliferation and activation. ApoCell‐loaded tolDCs generated from SLE monocytes have a stable immature/tolerogenic phenotype that can modulate CD4+ T‐cell activation. These properties make them suitable for an antigen‐specific immunotherapy for SLE.
Keywords: autoimmunity, dendritic cells, systemic lupus erythematosus, tolerance/suppression/anergy
Abbreviations
- ApoCell‐tolDCs
apoptotic cell‐loaded tolerogenic dendritic cells
- CD
cluster of differentiation
- CFSE
carboxyfluorescein succinimidyl ester
- DCs
dendritic cells
- DEXA
dexamethasone
- FABP4
fatty acid binding protein
- GILZ
glucocorticoid‐induced leucine zipper
- HC
healthy controls
- HLA‐DR
human leucocyte antigen DR
- iDCs
immature dendritic cell
- IFN
interferon
- IL
interleukin
- LPS
lipopolysaccharide
- mDCs
mature dendritic cells
- MFI
mean fluorescence intensity
- MLR
mixed lymphocyte reaction assay
- PE
phycoerythrin
- PerCP
peridinin chlorophyll protein
- RGZ
rosiglitazone
- SLEDAI‐2K
Systemic Lupus Erythematosus Disease Activity Index
- SLE
systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that preferentially affects women of childbearing age, which is associated with higher morbidity and mortality than the general population.1 The hallmark of SLE is the production of autoantibodies against nuclear components, which leads to immune complex formation and deposition in blood vessels, activation of innate immune cells and complement with subsequent systemic tissue damage, including kidneys, lungs, skin and central nervous system.2 One of the key questions about SLE pathogenesis that has intrigued immunologists for decades is how intracellular antigens are targeted by cognate circulating autoantibodies. The answer to this fundamental question has led scientists to explore the contribution of cell death to the triggering of SLE immunopathogenesis.3 Indeed, it has been shown that ribonucleoproteins and other intracellular self‐antigens become available to circulating autoantibodies when cells undergo apoptosis, necrosis or other forms of cell death such as NETosis.4 Although the SLE mortality rate has decreased,5 available therapeutic approaches based on immunosuppressive drugs such as corticosteroids, cyclophosphamide, azathioprine and mycophenolate mofetil, have not undergone significant improvement. Furthermore, current treatments are not curative, have partial efficacy and are associated with considerable adverse effects, which predominantly comprise major risks of infections, gonadal failure and bone marrow suppression.6 Current research is focused on biological agents to improve efficacy and decrease the adverse effects of therapy. However, new drugs in development, as well as the US Food and Drug Administration‐approved Belimumab, block relevant cytokines and their receptors in order to interfere with B‐cell function and lifespan.7 Nevertheless, these new insightful approaches do not resolve the problem of non‐specific immunosuppression and therefore, associated infections remain as an unsolved major issue in clinical practice. Consequently, safer and more specific therapies are needed for patients with SLE.
There has been significant progress in understanding the underlying mechanisms of tolerance failure in autoimmune diseases. Dendritic cells (DCs) are known for their ability to orchestrate adaptive immune responses as well as self‐tolerance.8, 9, 10 DCs can be found in two different states, mature/immunogenic DCs (mDCs) that are able to activate naive T cells and immature/tolerogenic DCs (tolDCs) that modulate T‐cell activity through the induction of anergy and regulatory T cells.8, 11 Interestingly, it has been shown that DCs from patients with SLE have a mature phenotype characterized by higher expression of co‐stimulatory molecules CD86, CD80 and CD40 and a higher ratio of activating/inhibitory Fcγ receptors.12 In contrast, DCs that express low levels of co‐stimulatory molecules are able to induce immune tolerance by reducing T‐cell reactivity.13 Based on these observations, it is reasonable to propose that a novel therapeutic approach for treating and eventually curing autoimmune diseases may reside on autologous tolDC transfer (or re‐infusion). This interesting strategy would restore tolerance to specific autoantigens without any detrimental effect on protective immunity against pathogens and tumours.
Because the process of cell death and deficient debris removal is thought to contribute to the development of SLE,3, 4, 14, 15, 16, 17 it is reasonable to hypothesize that loss of tolerance towards dead‐cell‐related epitopes is intimately linked to SLE onset. For this reason, restoring tolerance to autoantigens present in apoptotic cells using tolDCs would be a suitable strategy to treat SLE. Previous studies show that rosiglitazone (RGZ) treatment prevents kidney damage and antinuclear antibody production in lupus FcγRIIb−/− mice.18 Furthermore, splenic CD11c+ DCs from RGZ‐treated lupus mice express reduced levels of CD40 and CD86, compared with DCs from PBS‐treated mice.18 In addition, corticosteroids have been widely used to treat SLE, and in particular dexamethasone (DEXA) has been effectively used to generate human tolDCs for transplantation19, 20 and rheumatoid arthritis.21, 22 Based on the previous data, we decided to use RGZ and DEXA in our pre‐clinical studies aimed at generating tolDCs. Hence, we have produce autologous apoptotic cell‐loaded tolDCs from SLE patients and evaluated their phenotype stability and function. The ApoCell‐loaded tolDCs show immature DC (iDC)‐like phenotype expressing low levels of co‐stimulatory molecules, secreting lower levels of interleukin (IL‐6) and IL‐12p70 and modulating CD4+ T‐cell activation.
Materials and methods
Patients
Patients with SLE, according to American College of Rheumatology 1997 criteria, were recruited at the Hospital Clínico, Pontificia Universidad Católica de Chile (inclusion criteria: age between 18 and 65 years; exclusion criteria: concomitant diagnosis of other autoimmune diseases, concomitant diagnosis of severe chronic illness such as uncompensated diabetes mellitus type 2, end‐stage renal disease, liver cirrhosis, acquired immunodeficiency syndrome, solid tumours and/or haematological malignancies, heart failure, chronic airflow limitation or others, and pregnancy). Demographic and clinical characteristics of patients included in this study, including treatment at the time of blood sample withdrawal are shown in Table 1. Disease activity score was calculated using the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI‐2K). All individuals signed an informed consent form before enrolling in the study. The approval for this study was obtained from the Research Ethics Committee of Pontificia Universidad Católica de Chile.
Table 1.
Clinical characteristics of patients with systemic lupus erythematosus (SLE) recruited in the study including SLE‐related features and SLE Activity Disease Index (SLEDAI‐2K)
| Patient | Gender | Age (years) | SLEDAI | Treatment | Arthritis | Immune | NS | Kidney | Haem. | Serositis | MC | ANA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLE1 | F | 44 | 8 | HCQ, MMF | + | + | − | + | + | − | + | + |
| SLE2 | F | 30 | 2 | None | + | + | − | + | + | − | + | + |
| SLE3 | F | 23 | 8 | HCQ, PDN 15 mg, MMF | + | + | + | + | − | + | + | + |
| SLE4 | F | 65 | 10 | HCQ | − | + | − | − | + | − | + | + |
| SLE5 | F | 33 | 4 | HCQ | − | + | − | − | + | − | − | + |
| SLE6 | F | 30 | 10 | None | + | + | − | + | + | + | + | + |
| SLE7 | F | 46 | 4 | PDN 5 mg | − | + | − | − | + | − | + | + |
| SLE8 | F | 25 | 14 | HCQ, PDN 15 mg | + | + | − | − | − | − | + | + |
| SLE9 | F | 34 | 2 | HCQ, PDN 5 mg, MMF | + | + | − | + | + | − | + | + |
| SLE10 | F | 65 | 0 | HCQ, PDN 5 mg | − | + | − | + | + | − | − | + |
| SLE11 | M | 24 | 2 | HCQ, MMF | + | + | − | + | − | − | − | + |
| SLE12 | F | 24 | 0 | HCQ | + | − | − | − | + | − | + | + |
| SLE13 | F | 26 | 2 | HCQ | + | + | − | + | − | − | + | + |
| SLE14 | F | 36 | 0 | HCQ | + | + | − | − | + | − | − | + |
| SLE15 | F | 35 | 8 | HCQ | − | + | − | − | + | − | + | + |
| SLE16 | F | 26 | 6 | HCQ, PDN 10 mg, CYT | − | + | − | − | + | − | + | + |
| SLE17 | F | 47 | 2 | HCQ, PDN 5 mg, AZT | − | − | − | − | − | + | + | + |
| SLE18 | F | 49 | 4 | HCQ, PDN 10 mg, AZT | + | + | − | − | − | − | + | + |
| SLE19 | F | 45 | 2 | HCQ, PDN10 mg, MMF | + | + | − | + | + | − | + | + |
| SLE20 | F | 38 | 4 | HCQ, PDN 10 mg | − | + | − | − | + | − | + | + |
| SLE21 | M | 24 | 2 | HCQ, PDN 15 mg, MMF | + | + | − | + | − | − | + | + |
| SLE22 | F | 23 | 4 | HCQ, PDN 10 mg, MMF | + | + | − | + | + | − | + | + |
| SLE23 | F | 34 | 4 | HCQ, PDN 10 mg | + | + | + | + | + | − | + | + |
| SLE24 | F | 34 | 0 | HCQ | + | − | − | − | − | − | + | + |
| SLE25 | F | 24 | 2 | HCQ | + | + | − | − | − | − | + | + |
| SLE26 | F | 28 | 6 | None | + | + | − | − | − | − | + | + |
| SLE27 | F | 37 | 8 | PDN 7·5 mg MMF | + | + | − | + | + | − | + | + |
| SLE28 | F | 33 | 2 | HCQ, PDN 5 mg, MMF | − | + | − | + | − | − | − | + |
| SLE29 | F | 43 | 2 | HCQ, PDN 2·5 mg | − | + | − | + | − | − | + | + |
| SLE30 | F | 40 | 10 | HCQ, PDN 15 mg, MMF | − | − | − | + | − | − | − | + |
Art, arthritis; Immune, presence of anti‐DNA, anti‐Sm or anti‐cardiolipin antibodies; NS, nervous system compromise (seizures or psychosis); Kidney, renal compromise; Haem., haematological compromise; MC, mucocutaneous; ANA, anti‐nuclear antibodies; HCQ, hydroxychloroquine; PDN, prednisone; MMF, mycophenolate mofetil; AZT, azathioprine, CYT, cyclophosphamide.
Generation of monocyte‐derived DCs and induction of tolerogenic phenotype
Peripheral blood mononuclear cells were isolated as previously described.12 Briefly, blood was obtained by peripheral venous puncture, except for the experiments shown in Fig. 3 where buffy coats from 450 ml of blood donation were used, and peripheral blood mononuclear cells were separated from 50 ml of heparinized blood, by density gradient centrifugation using Ficoll‐Paque (GE Healthcare, Pittsburgh, PA, USA), then 1 × 106 cells/cm2 were cultured on six‐well plates for 2 hr at 37° in 5% CO2 to allow adherence of monocytes. Culture dishes were washed to remove lymphocytes. Whole lymphocyte fraction was collected and maintained for 6 days in complete RPMI‐1640 medium before manipulation. The adhered monocytes were incubated in serum‐free AIM‐V medium (Life Technologies, Grand Island, NY, USA) and differentiated into DCs by addition of recombinant human IL‐4 (1000 UI/ml) (ProSpec, Rehovot, Israel) and recombinant human granulocyte–macrophage colony‐stimulating factor (1000 UI/ml) (ProSpec) at days 1, 3 and 5. To generate tolDCs, RGZ (10 μm) (Cayman Chemical, Ann Arbor, MI, USA) and/or DEXA (1 μm) (Tocris, Bristol, UK) was added to culture at day 6. In some experiments, DCs were treated with lipopolysaccharide (LPS; 1 μg/ml) for 48 hr as a maturation stimulus. DC immunophenotypes were analysed by flow cytometry using a FACSCanto II™ system (BD Biosciences, San Jose, CA, USA) with specific antibodies against surface markers. Viability assays were performed on tolDCs using an XTT Cell Viability Assay Kit (Biotium, Hayward, CA, USA).
Figure 3.
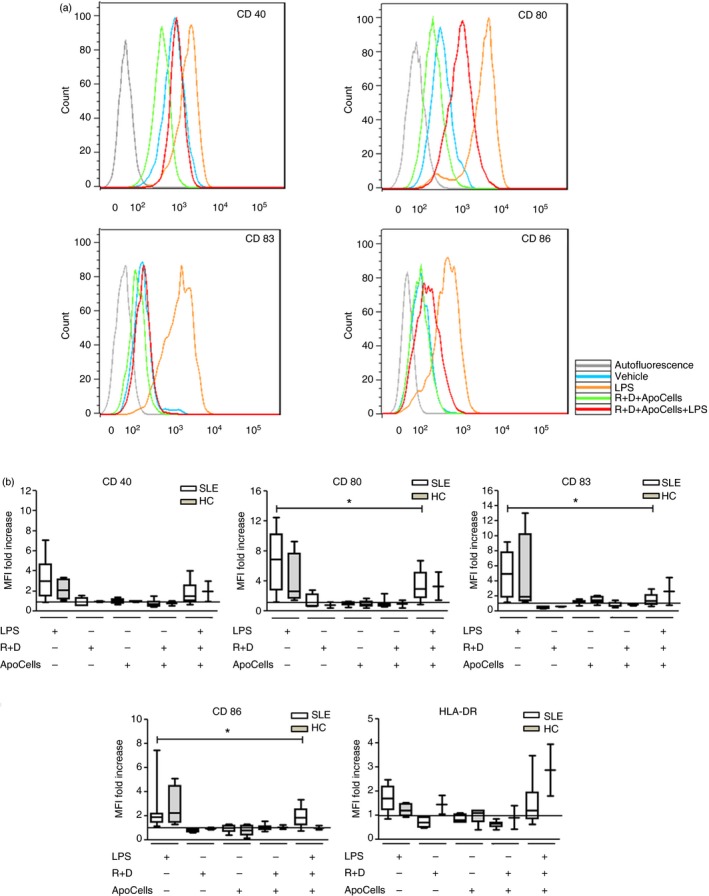
Tolerogenic dendritic cells (tolDCs) from patients with systemic lupus erythematosus (SLE) and healthy controls (HC) retain an immature phenotype when cultured in the presence of apoptotic cells (ApoCells). No differences between SLE patients and HC were found. Monocyte‐derived DCs were differentiated into tolDCs by immunosuppressive‐treatment and co‐cultured with autologous ApoCells and challenged with lipopolysaccharide (LPS). The expression of maturation markers CD40, CD80, CD83, CD86 and HLA‐DR was evaluated by flow cytometry. (a) Representative histograms for maturation marker expression of SLE DCs. (b) MFI values were normalized with respect of DCs treated with vehicle represented by the horizontal black line. SLE patients (SLE6–SLE23, Table 1) n = 18; *P = 0·0026 for CD80, *P = 0·0004 for CD83 and *P = 0·0038 for CD86 markers. HCs (see Supplementary material, Table S2) n = 4, Friedman test. Graphs represent box‐and‐whisker plots showing the medians of each group. [Colour figure can be viewed at wileyonlinelibrary.com]
Generation of apoptotic lymphocytes
Whole lymphocyte fractions were treated with UV‐B radiation (1·8–2·5 mW/cm2 for 1·5 hr) to induce apoptosis,14 which was confirmed by flow cytometry using AnnexinV and propidium iodide staining (BD Bioscience). Apoptotic cells (ApoCells) were collected by centrifugation for 10 min (500 g), and DNA concentration was determined by silica‐based DNA purification (Qiagen, Hilden, Germany). The pellet was resuspended in PBS and DCs were co‐cultured with ApoCells to a final concentration of 12·5 μg/ml of DNA content. Staurosporine (Sigma‐Aldrich, St. Lous, MO, USA) and heat‐shock treatment were used as controls for cell death induction (see Supplementary material, Fig. S1).
Evaluation of phagocytosis of apoptotic cells by tolDCs
ApoCells were stained with carboxyfluorescein succinimidyl ester (CFSE) dye 1·5 μm (Life Technologies) according to the manufacturer's instructions. TolDCs were co‐cultured with stained ApoCells for 24 hr, then collected and stained with BODIPY TR Ceramide 5 μm (Life Technologies) according the manufacturer's instructions. Stained samples were analysed with a Nikon C2si+ confocal microscope (Nikon Instruments, Melville, NY, USA).
Antibodies used for flow cytometry
FITC‐conjugated anti‐human/mouse CD40 (clone 5C3), phycoerythrin (PE) ‐conjugated anti‐human CD80 (clone L307.4), allophycocyanin‐conjugated anti‐human CD86 (clone FUN‐1), PE‐conjugated anti‐human CD83 (clone HB15e), FITC‐conjugated anti‐human HLA‐DR (clone TU36), purified anti‐human CD3 (clone OKT3), FITC‐conjugated anti‐human CD25 (clone M‐A251), purified anti‐human CD28 (clone CD28.2), Peridinin chlorophyll protein (PerCP)/Cy5.5‐conjugated anti‐human CD69 (clone FN50) and PE‐conjugated anti‐human CD71 (clone M‐A712) monoclonal antibodies were all purchased from BD Biosciences. PerCP/Cy5.5‐conjugated anti‐human CD11c (clone 3.9) was purchased from BioLegend (San Diego, CA, USA).
Immunostaining and flow cytometry analysis
Dendritic cells and T cells were harvested from culture plates, incubated in PBS/fetal bovine serum 2% for 10 min and then incubated with fluorochrome‐conjugated monoclonal antibodies for 40 min at 4°, washed with PBS/fetal bovine serum 2%, resuspended in FACSFlow buffer and acquired by FACS Canto II. Flow cytometry data were analysed by flowjo 7.6.1 (Ashland, OR, USA) and facs diva 6.0 software FACS DIVA, BD Bioscience).
Enzyme‐linked immunosorbent assay
Protein concentrations of IL‐6, IL‐10, IL‐12p70 and interferon‐γ (IFN‐γ) in DCs culture supernatants were measured using Ready‐Set‐GO! ELISA kits (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions.
RNA extraction and quantitative PCR
Total RNA was extracted from DCs using TRIzol (Invitrogen, Carlsbad, CA, USA) and cDNAs were generated from 1 μg RNA using an ImProm‐II™ Reverse Transcription System (Promega, Madison, WI, USA) according to the manufacturer's instructions. One microlitre of cDNA product was used as template for quantitative PCR experiments performed in a StepOnePlus™ Real‐Time PCR system (Applied Biosystems, Foster City, CA, USA) using Fast SYBR® Green Master Mix (Applied Biosystems) to measure relative expression of the genes of interest through the ΔΔC t method using the β‐actin gene as reference. Amplification was carried out with the following primers (5′→3′): fatty acid binding protein (FABP4): forward: GCAGCTTCCTTCTCACCTTG – reverse: ACTTTCCTGGTGGCAAAGC; glucocorticoid‐induced leucine zipper (GILZ): forward: TCTGCTTGGAGGGGATGTGG – reverse: ACTTGTGGGGATTCGGGAGC; β‐actin: forward: GTCCTCTCCCAAGTCCACAC – reverse: GGGAGACCAAAAGCCTTCAT. ΔΔC t was defined as (ΔC t GOI treatment − ΔC t GOI control) − (ΔC t reference treatment − ΔC t reference control).
Mixed lymphocyte reaction assay
Allogeneic CD4+ T cells were purified from the peripheral blood of healthy donors and one SLE patient by MACS using a CD4+ T‐cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) as indicated by the manufacturer. Purified allogeneic CD4+ T cells were co‐cultivated with mDCs or tolDCs (5 : 1 ratio) in complete RPMI‐1640 medium for 5 days. In some experiments, allogeneic CD4+ T cells were stained with CFSE. Incubations with plate‐bound anti‐CD3 (10 μg/ml) and soluble anti‐CD28 (2 μg/ml) was used as positive controls of activation and proliferation. Co‐cultures with iDCs and mDCs were used as additional controls.
Statistical analyses
Data and statistical analyses were performed using spss‐17 statistical software (SPSS Inc., Chicago, IL) and graph pad prism 5 software (Graph Pad Software, Inc., San Diego, CA). For comparison between SLE and healthy control (HC) groups, Mann–Whitney U‐test was used. For statistical analysis of the interest group (SLE) between treatments, paired one‐way Friedman test was used with Bonferroni post hoc test over the ranked data. For comparisons between two treatments [cytokine levels in supernatants and mixed lymphocyte reaction (MLR) assays], Wilcoxon signed‐rank test was used. P‐values below 0·05 were considered statistically significant.
Results
Generation of immature DCs for SLE
Immunotherapy based on autologous DCs for SLE may require the induction of a stable immature phenotype in order to prevent the circulating danger signals of patients enhancing the expression of co‐stimulatory molecules with subsequent maturation of administered DCs.
We first standardized a protocol to generate tolerogenic DCs both in HC and patients with SLE. In these samples, parameters such as phenotype, number of cells seeded per well, levels of differentiation and incubation times were determined. Hence, autologous tolDCs from five patients with SLE (SLE1–SLE5, Table 1; average age: 39 ± 16·4 years; SLEDAI‐2K: 6 ± 3·3) and six HC (see Supplementary material, Table S1; average age: 45 ± 8·3 years) were generated. After the differentiation process monocyte‐derived DCs were HLA‐DR+ CD11c+ CD14− (data not shown). After stimulation of DCs with LPS, HC and SLE mDCs showed a comparable increase in the expression of maturation markers (see Supplementary material, Fig. S2). In the next step, tolDCs were generated in vitro using RGZ and DEXA as immunomodulatory drugs and stimulated with LPS. Our preliminary data show that, under our experimental settings, either RGZ or DEXA alone failed to prevent the maturation of HC or SLE DCs (see Supplementary material, Fig. S3). The expression of maturation markers on tolDCs was measured to characterize the resulting phenotype of DCs (expression of CD40, CD80, CD83, CD86 and HLA‐DR) (Fig. 1). Furthermore, to evaluate the stability of the tolDC phenotype, we challenged these cells with LPS and the expression of co‐stimulatory molecules and HLA‐DR was measured by flow cytometry. As shown in Fig. 1, treatment with RGZ and DEXA successfully prevented LPS‐induced maturation in DCs from patients with SLE characterized by reduced expression of CD40 and CD83. Furthermore, a reduction of CD80, CD86 and HLA‐DR expression was observed for tolDCs, although it did not reach statistical significance. There were no statistically significant differences between HC and patients with SLE in any of the conditions evaluated (Fig. 1). To corroborate that RGZ and DEXA were in fact exerting a biological effect on DCs, the expression of the target genes FABP4 and GILZ was determined by quantitative RT‐PCR (see Supplementary material, Fig. S4). As expected, RGZ and DEXA induced and increase 10‐ and 6‐fold mRNA expression of FABP4 and GILZ, respectively.
Figure 1.
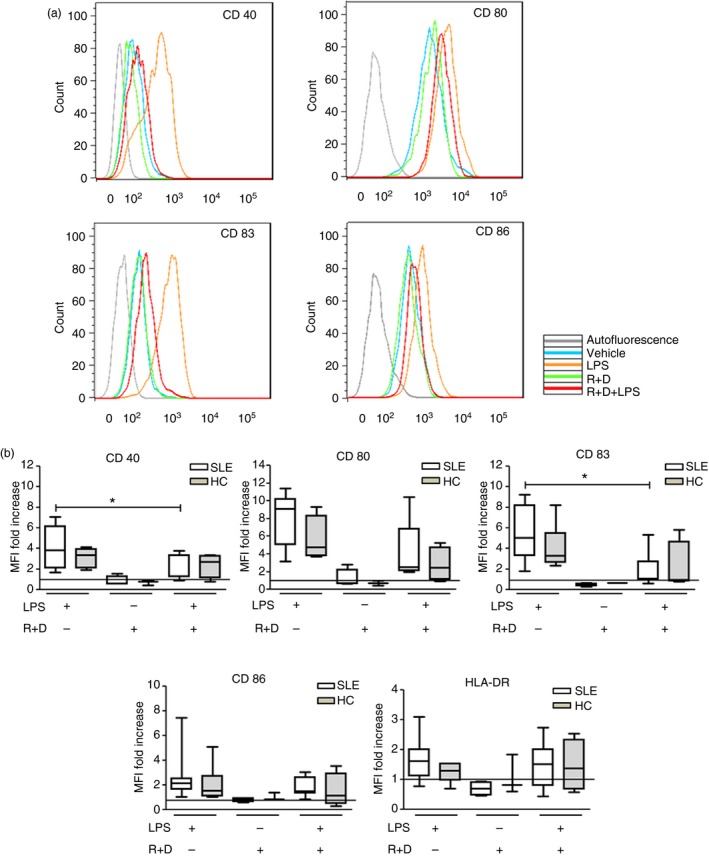
Systemic lupus erythematosus (SLE) dendritic cells (DCs) acquire resistance to full maturation upon immunosuppressive treatment with rosiglitazone (RGZ) and dexamethasone (DEXA). Monocytes from healthy controls (HC) and patients with SLE were differentiated into DCs, treated with RGZ and DEXA and challenged with lipopolysaccharide (LPS). We used DCs stimulated with LPS alone to generate mature DCs (mDCs) as control. The expression of maturation markers CD40, CD80, CD83, CD86 and HLA‐DR was evaluated by flow cytometry. (a) Representative histograms for maturation marker expression of DCs from SLE patients. (b) MFI values were normalized with respect of DCs treated with vehicle represented by the horizontal black line. SLE patients (SLE1–SLE5, Table 1) n = 5, *P = 0·036 for CD40 and *P = 0·024 for CD83 markers, Friedman test, DCs+LPS versus DCs+R+D+LPS. Healthy controls (see Supplementary material, Table S1) n = 6, P = NS, Friedman test, DC+LPS versus DCs+R+D+LPS. Graphs represent box‐and‐whisker plots showing the medians of each group. [Colour figure can be viewed at wileyonlinelibrary.com]
Developing autologous tolerogenic DCs loaded with specific SLE auto‐antigens
It is well known that immune tolerance must be defined relative to a specific antigen. This requirement has delayed development of more effective and safer therapies in several autoimmune diseases including SLE. As a strategy to overcome this obstacle and based on the extensively described role of ApoCells in SLE pathogenesis, we decided to use autologous ApoCells as a source of autoantigens to load tolDCs. For the generation of ApoCells, autologous non‐adherent peripheral blood lymphocytes were treated with different agents to induce cellular death and analysed by flow cytometry. The results show that treatment of peripheral blood lymphocytes with UV‐B light is a secure and non‐chemical protocol to obtain ApoCells as a source of autoantigens (see Supplementary material, Fig. S1). Furthermore, UV‐B light exposure is directly involved in the pathogenesis of SLE,3 and so constitutes a more physiological manner of exposing autoantigens. Staurosporine and heat‐shock treatment were used as controls for cell death induction.
We evaluated whether tolDCs were able to phagocytose ApoCells. As shown in Fig. 2(a–c) using confocal microscopy, ApoCells are located inside tolDC‐derived SLE monocytes, suggesting that SLE tolDCs are able to capture ApoCell‐derived antigens in vitro under our culture conditions. After 24 hr of incubation, CFSE‐stained ApoCells were detected within the cell membrane boundaries, suggesting that ApoCells are not attached to the outer cell membrane. The capture of ApoCells by tolDCs was additionally confirmed by flow cytometry (Fig. 2d‐e), we observed that 39·5% of CD11c+ cells captured CFSE‐stained ApoCells, indicating that tolDCs were able to phagocytose ApoCells. Additionally, we analysed the phenotype of these tolDCs from patients with SLE (SLE6–SLE23, Table 1; n = 18; average age: 35 ± 11·5 years; SLEDAI‐2K: 4 ± 3·8) and HC (HC7–HC10, see Supplementary material, Table S2) when they were cultured in the presence of ApoCells and challenged with LPS and there were no changes in the phenotype of DCs when ApoCells were added to cultures (Fig. 3). Surface expression of CD80, CD83 and CD86 was significantly decreased in SLE tolDCs co‐cultured with ApoCells and this decreased expression was sustained after LPS treatment, indicating that tolDCs keep an immature phenotype despite treatment with ApoCells and LPS (Fig. 3). No differences were observed for surface expression of CD40 and HLA‐DR in tolDCs under these conditions. Furthermore, no differences were detected between tolDCs derived from HC and SLE monocytes when loaded with ApoCells and stimulated with LPS. Decreased expression of maturation markers on tolDCs loaded with ApoCell antigens pose this therapy as a suitable strategy to restore immune tolerance in SLE.
Figure 2.
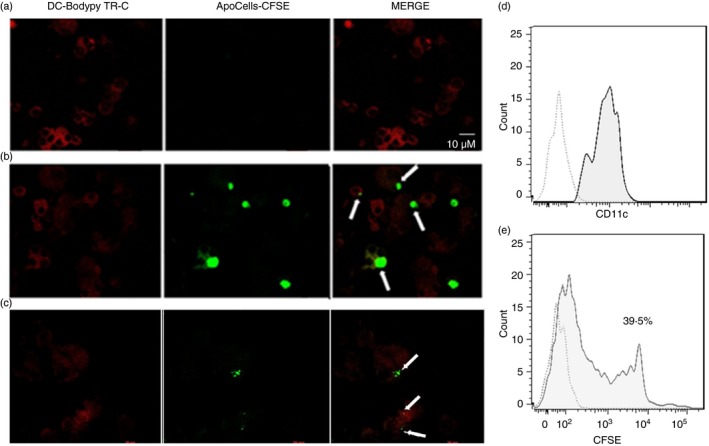
Systemic lupus erythematosus (SLE) dendritic cells (DCs) capture autologous apoptotic cells. Monocyte‐derived DCs from patients with SLE were treated with rosiglitazone (RGZ) and dexamethasone (DEXA) and then co‐cultured in the presence of autologous apoptotic cells (ApoCells). BODIPY TR Ceramide was used as contrast staining. (a,b) Representative images of tolerogenic DCs (tolDCs) from a patient with SLE pulsed with no‐stained ApoCells (a) and CFSE‐stained ApoCells (b). White arrows indicate capture of ApoCell‐loaded tolDCs. (b and c) Representative histograms of tolDC populations. Dotted line indicates autofluorescence in the histograms. Solid line indicates CD11c+ cells in DCs not treated (d) or treated (e) with CFSE‐stained ApoCells and percentage of +/+ cells.
ApoCell‐loaded tolDCs secrete low levels of pro‐inflammatory cytokines
To further characterize ApoCell‐loaded tolDCs, we determined the levels of cytokines found in the supernatants of DCs and in supernatants of ApoCell‐loaded‐tolDC co‐cultures. We measured the presence of pro‐inflammatory cytokines IL‐6 and IL‐12p70 and the levels of the anti‐inflammatory cytokine IL‐10 by ELISA. As shown in Fig. 4, tolDCs loaded with ApoCells and stimulated with LPS secreted significantly lower amounts of IL‐6 and IL‐12p70, with no changes in IL‐10 production when compared with mDCs. These data suggest that, in addition to a lower expression of maturation markers and co‐stimulatory molecules, our ex vivo procedure is also capable of decreasing the secretion of pro‐inflammatory cytokines, interfering as well with the third signal provided by LPS‐challenged DCs to promote T‐cell activation.
Figure 4.
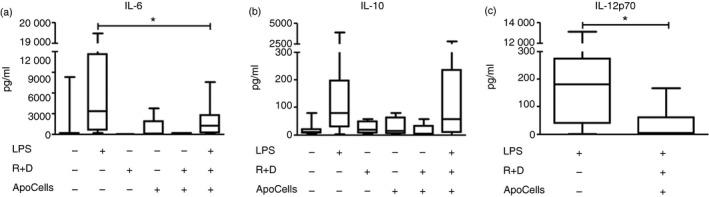
Apototic cell‐loaded tolerogenic dendritic cells (ApoCell‐loaded tolDCs) secrete low levels of pro‐inflammatory cytokines. Graphics show concentration of cytokines measured by ELISA in the supernatants of DC cultures of patients with systemic lupus erythematosus (SLE) in the indicated conditions. (a) interleukin‐6 (IL‐6) (n = 15; *P = 0·0009, Wilcoxon test). (b) IL‐10 (n = 15). (c) IL‐12p70 (n = 10; *P = 0·009, Wilcoxon test).
ApoCell‐loaded tolDCs impair the activation of allogeneic CD4+ T cells
With the purpose of evaluating the ability of tolDCs to suppress the activation of T cells, we performed MLRs. Allogeneic purified CD4+ T cells from HC were cultured in the presence of ApoCell‐loaded tolDCs from patients with SLE (5 : 1 ratio; SLE23‐SLE30, Table 1; n = 7; average age: 33 ± 7·8 years; SLEDAI‐2K: 5 ± 3·2) for 5 days and the expression of surface activation markers on CD4+ T cells was assessed by flow cytometry (Fig. 5). Remarkably, allogeneic CD4+ T cells cultured in the presence of ApoCell‐loaded tolDCs from patients with SLE show reduced expression of activation markers such as CD25, CD71 and CD69 (Fig. 5a–c) as compared with CD4+ T cells co‐cultured with iDCs or mDCs. Furthermore, we also assessed whether ApoCell‐loaded tolDCs were able to prevent allogeneic T‐cell expansion. Interestingly, ApoCell‐loaded tolDCs significantly decrease T‐cell proliferation, as determined by dilution of CFSE measured by flow cytometry (Fig. 5d). In addition, we measured the production of IFN‐γ in the supernatants obtained after MLR assays. We observed that in MLRs set up with ApoCell‐loaded tolDCs, IFN‐γ secretion was decreased compared with MLRs in which mDCs were used to stimulate allogeneic T cells (Fig. 5e). MLR assays show that ApoCell‐loaded tolDCs suppress the activation, proliferation and cytokine secretion of CD4+ T cells in an allogeneic response context. Furthermore, this suppression is also observed using lymphocytes from an SLE patient (see Supplementary material, Fig. S5).
Figure 5.
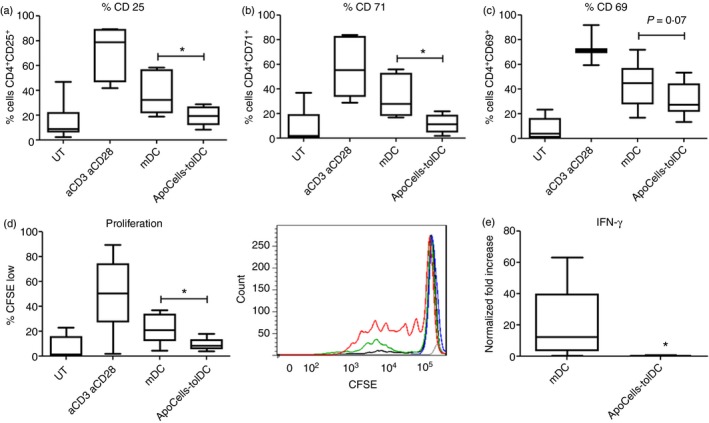
Apototic cell‐loaded tolerogenic dendritic cells (ApoCell‐loaded tolDCs) modulate allogeneic CD4+ T‐cell activation in a mixed lymphocyte antigen (MLR) assay. CD4+ T cells were co‐cultured with allogeneic ApoCell‐loaded tolDCs in a 5 : 1 ratio for 5 days to induce activation of T cells. Untreated T cells (UT) were used as control.38 Percentage of activation markers on CD4+ T cells is shown as a percentage of CD4+ gated cells (n = 7; Wilcoxon test). (a) CD25 (*P = 0·0156); (b) CD71 (*P = 0·0156); (c) CD69 (P = 0·0781). (d) Left, CFSE dilution of CD4+ T cells cultured in the presence of allogeneic tolDCs. CFSE low gate was defined for cells with lower fluorescence than untreated stained T cells. n = 8; *P = 0·0234, Wilcoxon test. (d) Right, representative histograms of CFSE dilution peaks; blue line: lymphocytes without DCs; black line: lymphocytes co‐cultured with immature DCs; red line lymphocytes in presence of aCD3 + aCD28 antibodies (control of proliferation); green line lymphocytes co‐cultured with mature DCs; grey line basal fluorescence. (e) Levels of interferon‐γ (IFN‐γ) were measured in supernatants of MLR assay. The graphic represents the fold increase of IFN‐γ levels related to ApoCell‐loaded tolDCs condition (n = 7; *P = 0·0313, Wilcoxon test). Box‐and‐whisker plots show the medians of each group. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Current therapeutic strategies to treat autoimmune diseases have failed in meeting the coveted goal of reducing disease activity while avoiding deleterious inhibition of protective immunity. In an effort to make progress towards achieving this critical goal we focused on tolDC development with the long‐term goal of producing antigen‐specific immunotherapy for SLE. We have generated for the first time tolDCs directed towards relevant self‐antigens involved in lupus pathogenesis. The use of tolDCs to restore immune tolerance has been under intense evaluation during the past years due to their potential role in transplant rejection23, 24 and also as a possible treatment for autoimmunity.8, 9
Our work and that of others has shown that pharmacological modulation of monocyte‐derived DCs with immunosuppressant drugs provides a successful method to generate tolDCs with a low expression of maturation markers and a stable phenotype that is even maintained after stimulation with a strong pro‐inflammatory stimulus such as LPS.25, 26, 27, 28, 29 In addition, RZG and DEXA have been used in a variety of clinical settings, which helps to mitigate concerns regarding the administration of cell therapy to immunosuppressed patients, which is an especially pertinent safety question when strategies like viral transduction and gene therapy approaches are used. Under our culture settings, we were able to generate DCs from patients with SLE with a tolerogenic‐like immunophenotype using peripheral blood as a source of precursors. Most reports on tolDC generation procedures are performed on HC DCs and, to our knowledge, no previous study has developed SLE monocyte‐derived tolDCs. Because of the differences between DCs from HC and patients with SLE,12, 13, 14 it could have been possible for SLE DCs to be resistant to the induction of the tolerogenic‐like phenotype and that would become a major drawback. No differences between tolDCs from HC and tolDCs from patients with SLE were found, which could be of benefit as it would mean that SLE tolDC had not lost the ability to be rendered tolerogenic.
To our knowledge, only three clinical trials exploring safety, biological and clinical effect of tolDCs in autoimmune diseases have been conducted. The first (Giannoukakis et al.) was performed in type 1 diabetes using intradermic injections of DCs that were not exposed to a self‐antigen.30 Although the primary outcome of Giannoukakis et al. was safety, they found that DCs up‐regulated the frequency of B220+ CD11c− B cells, which may be of clinical benefit in this disease. In addition, DCs were well tolerated by insulin‐requiring individuals with type 1 diabetes. However, unlike SLE subjects, patients with type 1 diabetes do not require immunosuppressant therapy. Two other clinical trials (by Bell et al. and Benham et al.) tested tolDCs in patients with rheumatoid arthritis, who are more comparable to individuals with SLE regarding their therapy and the physiopathology of disease.31, 32 Moreover, these authors used antigen‐loaded DCs as we reported here; Bell et al. used synovial fluid as a source of autoantigens and Benham et al. pulsed DCs with citrullinated peptide antigens. Both trials proved that DCs are safe in patients with rheumatoid arthritis either injected in the joint or using intradermic injections. Strikingly, no flares of rheumatoid arthritis were observed in any of these studies and Benham et al. reported that patients with active rheumatoid arthritis treated with DCs showed signs of recovery. In addition, treated patients had a reduction in effector T cells and an increased ratio of regulatory to effector T cells along with a reduction in circulating pro‐inflammatory cytokines such as serum IL‐15, IL‐29, CX3CL1 and CXCL11.31, 32 This observation suggests that optimal tolDC function depends on the capability of tolDCs to suppress the activity of autoantigen‐specific T cells. Also, complex autoantigen sources like rheumatoid arthritis synovial fluid can be useful for the induction of tolerance when the specific autoimmunity‐inducing antigen is unknown, which is precisely the case of SLE.
To provide tolDCs with specificity, we used ApoCells as a source of self‐antigens. Using two different approaches we observed that tolDCs were capable of capturing ApoCells. Several studies suggest that antigens and signals derived from dead cells, either apoptotic or necrotic cells, contribute to SLE pathogenesis.15, 16, 33 Furthermore, phagocytosis of these cell remnants could be part of the driving force behind the onset of disease.16 It is noteworthy that we did not observe a significant increase in the levels of CD80, CD83 or CD86 on SLE tolDCs after being pulsed with ApoCells even after stimulation with LPS, suggesting that ApoCell‐loaded tolDCs are stable after stimulation with LPS. As mentioned above, we did not find any significant difference in the expression of maturation markers on SLE tolDCs compared with HC tolDCs co‐cultured with ApoCells, suggesting that SLE tolDCs may have a normal function.
Moreover, ApoCell‐loaded tolDCs derived from SLE monocytes secreted lower levels of IL‐6 and IL‐12p70 than controls after LPS challenge. Conversely, ApoCell‐loaded tolDCs stimulated with LPS showed no differences in IL‐10 production compared with mDCs, which is not consistent with previous reports.11, 34, 35 However, there are data showing that functional tolDCs do not secrete higher levels of IL‐10, but the production of pro‐inflammatory cytokines is significantly impaired instead.26, 36 In fact, the decrease of IL‐12p70 production is a crucial factor in the induction of tolerance mediated by DCs.37, 38 We demonstrated that tolDCs generated from patients with SLE present an iDC‐like phenotype, although they do not secrete higher levels of IL‐10. These observations suggest that it is probably the reduction of immunogenic signals, instead of the up‐regulation of suppressive molecules, that is the key to achieving a tolerogenic DC phenotype. Although monocytes from patients with SLE have abnormal properties, our results suggest that functional tolDCs can be obtained from them and autologous immunotherapy for SLE may in fact be plausible, even with the described particular characteristics of this cell type in patients with SLE.16, 17, 39, 40, 41, 42
One of the critical steps of pre‐clinical studies in DC therapy is to demonstrate that tolDCs are able to maintain an immature phenotype. Indeed, Voigtländer et al.43 previously reported that DCs treated with tumour necrosis factor show a semi‐mature phenotype and they induced tolerance after intravenous injection in a murine model. However, these DCs were not terminally differentiated and remain responsive to further signals in vitro and in vivo, which can convert them from tolerogenic to immunogenic DCs. Accordingly, we evaluated the phenotype stability of tolDCs in vitro using LPS, a well‐known strong pro‐inflammatory challenge. We observed that ApoCell‐loaded tolDCs were refractory to maturation with LPS and failed to increase their secretion of IL‐6 and IL‐12p70 in response to LPS, suggesting that our cells are well suited for the treatment of a disease with several circulating inflammatory mediators, such as SLE.
Furthermore, we carried out MLR assays to evaluate the tolerogenic effect over CD4+ T cells. We observed that CD4+ T cells from HC and SLE donors cultured in the presence of monocyte‐derived tolDCs loaded with ApoCells from patients with SLE expressed reduced levels of CD25 and CD71 compared with those cultured in the presence of iDCs or mDCs. Likewise, CD4+ T cells co‐cultured with ApoCell‐loaded tolDCs showed decreased proliferation and secretion of IFN‐γ.
Although we presented encouraging data supporting the use of tolDCs as a potential therapy for SLE, further insight on ApoCell‐loaded tolDCs is needed to consolidate data to support their potential as a therapy for SLE, especially experiments using lupus mouse models. Despite this, we presented promising results that allow us to affirm that ApoCell‐loaded tolDCs generated from SLE monocytes with a stable immature phenotype are capable of exerting a tolerogenic effect, suggesting that they might be suitable for the generation of a specific immunotherapy for SLE. Nevertheless, further pre‐clinical studies are needed to address their competence to restore tolerance in autoimmunity, including studies in mouse models.
Disclosures
Patent applications for the use of ApoCell‐loaded tolDCs to treat SLE have been submitted in USA (62/008,319), Argentina (2015001001809) (Pending), WIPO (PCT, IB2015/054267) (Pending), and Chile (1539‐2015) (Pending).
Supporting information
Table S1. Characteristics of healthy controls recruited in the study, related to Fig. 1.
Table S2. Characteristics of healthy controls recruited in the study, related to Fig. 3.
Figure S1. UV‐B irradiation induces cell death in peripheral blood lymphocytes.
Figure S2. Systemic lupus erythematosus and healthy control mature DCs have high expression of co‐stimulatory molecules.
Figure S3. Rosiglitazone or dexamethasone alone do not prevent lipopolysaccharide‐induced maturation of healthy control or systemic lupus erythematosus dendritic cells.
Figure S4. Immunosuppressant drugs induce the expression of target genes.
Figure S5. Apoptotoc cell‐loaded tolerogenic dendritic cells modulate allogeneic activation of CD4+ T cells from a patient with systemic lupus erythematosus in a mixed lymphocyte reaction assay.
Acknowledgements
We would like to thank Dr Jaime Pereira and Carla Lorca for providing buffy coats from SLE patients and HC. We also thank Débora Contreras for coordinating clinical visits and sample collection of all individuals included in this study. We are also grateful for the contribution of SLE patients and HC who participated in this study. This work was supported by Corporación de Fomento de la Producción (CORFO) grants No. 12IDL2‐15146; 13CTI‐21526/P3 and FONDECYT grant No. 1150173.
References
- 1. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD et al Mortality in systemic lupus erythematosus. Arthritis Rheum 2006; 54:2550–7. [DOI] [PubMed] [Google Scholar]
- 2. Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex‐mediated neutrophil recruitment and tissue injury. Circulation 2009; 120:2012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Casciola‐Rosen L, Rosen A. Ultraviolet light‐induced keratinocyte apoptosis: a potential mechanism for the induction of skin lesions and autoantibody production in LE. Lupus 1997; 6:175–80. [DOI] [PubMed] [Google Scholar]
- 4. Mahajan A, Herrmann M, Munoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol 2016; 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doria A, Iaccarino L, Ghirardello A, Zampieri S, Arienti S, Sarzi‐Puttini P et al Long‐term prognosis and causes of death in systemic lupus erythematosus. Am J Med 2006; 119:700–6. [DOI] [PubMed] [Google Scholar]
- 6. Ugarte‐Gil MF, Alarcon GS. Systemic lupus erythematosus: a therapeutic challenge for the XXI century. Clin Rheumatol 2014; 33:441–50. [DOI] [PubMed] [Google Scholar]
- 7. Llanos C, Mackern‐Oberti JP, Vega F, Jacobelli SH, Kalergis AM. Tolerogenic dendritic cells as a therapy for treating lupus. Clin Immunol 2013; 148:237–45. [DOI] [PubMed] [Google Scholar]
- 8. Mackern‐Oberti JP, Llanos C, Vega F, Salazar‐Onfray F, Riedel CA, Bueno SM et al Role of dendritic cells in the initiation, progress and modulation of systemic autoimmune diseases. Autoimmun Rev 2015; 14:127–39. [DOI] [PubMed] [Google Scholar]
- 9. Mackern‐Oberti JP, Vega F, Llanos C, Bueno SM, Kalergis AM. Targeting dendritic cell function during systemic autoimmunity to restore tolerance. Int J Mol Sci 2014; 15:16381–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thompson AG, Thomas R. Induction of immune tolerance by dendritic cells: implications for preventative and therapeutic immunotherapy of autoimmune disease. Immunol Cell Biol 2002; 80:509–19. [DOI] [PubMed] [Google Scholar]
- 11. Torres‐Aguilar H, Aguilar‐Ruiz SR, Gonzalez‐Perez G, Munguia R, Bajana S, Meraz‐Rios MA et al Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen‐specific anergy and regulatory properties in memory CD4+ T cells. J Immunol 2010; 184:1765–75. [DOI] [PubMed] [Google Scholar]
- 12. Carreno LJ, Pacheco R, Gutierrez MA, Jacobelli S, Kalergis AM. Disease activity in systemic lupus erythematosus is associated with an altered expression of low‐affinity Fcγ receptors and costimulatory molecules on dendritic cells. Immunology 2009; 128:334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iruretagoyena MI, Wiesendanger M, Kalergis AM. The dendritic cell‐T cell synapse as a determinant of autoimmune pathogenesis. Curr Pharm Des 2006; 12:131–47. [DOI] [PubMed] [Google Scholar]
- 14. Fehr EM, Spoerl S, Heyder P, Herrmann M, Bekeredjian‐Ding I, Blank N et al Apoptotic‐cell‐derived membrane vesicles induce an alternative maturation of human dendritic cells which is disturbed in SLE. J Autoimmun 2013; 40:86–95. [DOI] [PubMed] [Google Scholar]
- 15. Casciola‐Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 1994; 179:1317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P et al Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum 2009; 60:1733–42. [DOI] [PubMed] [Google Scholar]
- 17. Cairns AP, Crockard AD, McConnell JR, Courtney PA, Bell AL. Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activity. Ann Rheum Dis 2001; 60:950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalergis AM, Iruretagoyena MI, Barrientos MJ, Gonzalez PA, Herrada AA, Leiva ED et al Modulation of nuclear factor‐κB activity can influence the susceptibility to systemic lupus erythematosus. Immunology 2009; 128:e306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreau A, Varey E, Bouchet‐Delbos L, Cuturi MC. Cell therapy using tolerogenic dendritic cells in transplantation. Transplant Res 2012; 1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikolic T, Roep BO. Regulatory multitasking of tolerogenic dendritic cells – lessons taken from vitamin D3‐treated tolerogenic dendritic cells. Front Immunol 2013; 4:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol 2013; 172:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen‐induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum 2010; 62:3656–65. [DOI] [PubMed] [Google Scholar]
- 23. Beriou G, Moreau A, Cuturi MC. Tolerogenic dendritic cells: applications for solid organ transplantation. Curr Opin Organ Transplant 2012; 17:42–7. [DOI] [PubMed] [Google Scholar]
- 24. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 2007; 7:610–21. [DOI] [PubMed] [Google Scholar]
- 25. Ferreira GB, Gysemans CA, Demengeot J, da Cunha JP, Vanherwegen AS, Overbergh L et al 1,25‐Dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J Immunol 2014; 192:4210–20. [DOI] [PubMed] [Google Scholar]
- 26. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis 2010; 69:2042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iruretagoyena MI, Sepulveda SE, Lezana JP, Hermoso M, Bronfman M, Gutierrez MA et al Inhibition of nuclear factor‐κB enhances the capacity of immature dendritic cells to induce antigen‐specific tolerance in experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther 2006; 318:59–67. [DOI] [PubMed] [Google Scholar]
- 28. Iruretagoyena MI, Tobar JA, Gonzalez PA, Sepulveda SE, Figueroa CA, Burgos RA et al Andrographolide interferes with T cell activation and reduces experimental autoimmune encephalomyelitis in the mouse. J Pharmacol Exp Ther 2005; 312:366–72. [DOI] [PubMed] [Google Scholar]
- 29. Carreno LJ, Riedel CA, Kalergis AM. Induction of tolerogenic dendritic cells by NF‐κB blockade and Fcγ receptor modulation. Methods Mol Biol 2011; 677:339–53. [DOI] [PubMed] [Google Scholar]
- 30. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011; 34:2026–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA et al Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis 2017; 76:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N et al Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype‐positive rheumatoid arthritis patients. Sci Transl Med 2015; 7:290ra87. [DOI] [PubMed] [Google Scholar]
- 33. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V et al Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 2010; 107:9813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gonzalez‐Rey E, Chorny A, Fernandez‐Martin A, Ganea D, Delgado M. Vasoactive intestinal peptide generates human tolerogenic dendritic cells that induce CD4 and CD8 regulatory T cells. Blood 2006; 107:3632–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalantari T, Karimi MH, Ciric B, Yan Y, Rostami A, Kamali‐Sarvestani E. Tolerogenic dendritic cells produced by lentiviral‐mediated CD40‐ and interleukin‐23p19‐specific shRNA can ameliorate experimental autoimmune encephalomyelitis by suppressing T helper type 17 cells. Clin Exp Immunol 2014; 176:180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Naranjo‐Gomez M, Raich‐Regue D, Onate C, Grau‐Lopez L, Ramo‐Tello C, Pujol‐Borrell R et al Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med 2011; 9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson AE, Sayers BL, Haniffa MA, Swan DJ, Diboll J, Wang XN et al Differential regulation of naive and memory CD4+ T cells by alternatively activated dendritic cells. J Leukoc Biol 2008; 84:124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li R, Zheng X, Popov I, Zhang X, Wang H, Suzuki M et al Gene silencing of IL‐12 in dendritic cells inhibits autoimmune arthritis. J Transl Med 2012; 10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eloranta ML, Lovgren T, Finke D, Mathsson L, Ronnelid J, Kastner B et al Regulation of the interferon‐α production induced by RNA‐containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheum 2009; 60:2418–27. [DOI] [PubMed] [Google Scholar]
- 40. Herrada AA, Llanos C, Mackern‐Oberti JP, Carreno LJ, Henriquez C, Gomez RS et al Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology 2012; 136:414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steinbach F, Henke F, Krause B, Thiele B, Burmester GR, Hiepe F. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann Rheum Dis 2000; 59:283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S et al Anti‐dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med 2016; 14:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Voigtlander C, Rossner S, Cierpka E, Theiner G, Wiethe C, Menges M et al Dendritic cells matured with TNF can be further activated in vitro and after subcutaneous injection in vivo which converts their tolerogenicity into immunogenicity. J Immunother 2006; 29:407–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of healthy controls recruited in the study, related to Fig. 1.
Table S2. Characteristics of healthy controls recruited in the study, related to Fig. 3.
Figure S1. UV‐B irradiation induces cell death in peripheral blood lymphocytes.
Figure S2. Systemic lupus erythematosus and healthy control mature DCs have high expression of co‐stimulatory molecules.
Figure S3. Rosiglitazone or dexamethasone alone do not prevent lipopolysaccharide‐induced maturation of healthy control or systemic lupus erythematosus dendritic cells.
Figure S4. Immunosuppressant drugs induce the expression of target genes.
Figure S5. Apoptotoc cell‐loaded tolerogenic dendritic cells modulate allogeneic activation of CD4+ T cells from a patient with systemic lupus erythematosus in a mixed lymphocyte reaction assay.