

Summary
Seizures are due to excessive, synchronous neuronal firing in the brain and are characteristic of epilepsy, the fourth most prevalent neurological disease. We report handling‐induced and spontaneous seizures in mice deficient for CD39, a cell‐surface ATPase highly expressed on microglial cells. CD39−/− mice with handling‐induced seizures had normal input–output curves and paired‐pulse ratio measured from hippocampal slices and lacked microgliosis, astrogliosis or overt cell loss in the hippocampus and cortex. As expected, however, the cerebrospinal fluid of CD39−/− mice contained increased levels of ATP and decreased levels of adenosine. To determine if immune activation was involved in seizure progression, we challenged mice with lipopolysaccharide (LPS) and measured the effect on microglia activation and seizure severity. Systemic LPS challenge resulted in increased cortical staining of Iba1/CD68 and gene array data from purified microglia predicted increased expression of interleukin‐8, triggering receptor expressed on myeloid cells 1, p38, pattern recognition receptors, death receptor, nuclear factor‐κB , complement, acute phase, and interleukin‐6 signalling pathways in CD39−/− versus CD39+/+ mice. However, LPS treatment did not affect handling‐induced seizures. In addition, microglia‐specific CD39 deletion in adult mice was not sufficient to cause seizures, suggesting instead that altered expression of CD39 during development or on non‐microglial cells such as vascular endothelial cells may promote the seizure phenotype. In summary, we show a correlation between altered extracellular ATP/adenosine ratio and a previously unreported seizure phenotype in CD39−/− mice. This work provides groundwork for further elucidation of the underlying mechanisms of epilepsy.
Keywords: adenosine, ATP, CD39, epilepsy, microglia, seizure
Introduction
Epilepsy affects about 1% of the world population and is the fourth most common neurological disorder after tension headaches, migraines and Alzheimer's disease.1, 2 Epilepsy is a chronic disorder characterized by excessive, synchronous neuronal activity that leads to one or more unprovoked seizures. Only about 50% of newly diagnosed patients respond to the first anti‐epileptic drug prescribed and 30% of all patients are drug‐resistant.3, 4 Epilepsy is a heterogeneous and complex disease with several different phenotypes and over 400 associated genes.5
Although epilepsy is thought to be due to an imbalance of excitatory and inhibitory signalling in the brain,6 the mechanistic cause of epilepsy remains generally unknown and there is currently no cure. However, evidence suggests that extracellular nucleotides are involved. For example, altered levels of extracellular ATP7, 8 and adenosine9, 10, 11 as well as ectonucleotidase activity12, 13 are found in the brains of rat epilepsy models. In addition, levels of extracellular adenosine increased in the hippocampus of epilepsy patients during seizures.14
In the central nervous system (CNS), neurons,15 microglia16 and astrocytes17 are capable of releasing ATP in response to a variety of different stimuli via exocytosis or membrane channels.18, 19, 20 Once released to the extracellular space, ATP signals via P2X ligand‐gated ionotropic receptors and P2Y G protein‐coupled receptors21 that are widely expressed on neurons, microglia, astrocytes, oligodendrocytes and endothelial cells.21, 22, 23, 24
The effects of extracellular ATP depend on the context and the receptors they bind.21, 22, 23 The effects of ATP on neurons are largely driven by pre‐synaptic control of release of several neurotransmitters and has been extensively reviewed.25 A seminal paper by Rodrigues et al. showed that the effect of extracellular ATP on neurons is biphasic such that low concentrations inhibit glutamate release in a P2Y‐dependent fashion, whereas high concentrations induce glutamate release through a P2X‐dependent mechanism.26 In addition, P2 receptors have different spatial distributions as well as different affinities for various nucleotide ligands.27 Hence, the dynamics of ATP signalling and identification of receptors involved in a given context are difficult to assess but are of extreme interest and importance to our understanding of cellular communication within the CNS.
It is also difficult to study the effects of ATP on different P2 receptors due to its rapid catabolism to adenosine in 50 milliseconds.28 A series of enzymatic steps on the cell surface breakdown extracellular ATP to terminate receptor signalling.23 Breakdown of extracellular ATP is initiated by ectonucleoside triphosphate diphosphohydrolase 1, or CD39, which converts ATP and ADP into AMP. CD73 subsequently converts AMP into adenosine. In the CNS, CD39 is a prominent ATPase that is highly expressed on microglial and endothelial cells.29, 30, 31
Besides generation via breakdown of extracellular ATP, extracellular adenosine can be produced by cellular transport or release of cAMP.23 Extracellular adenosine signals via G‐coupled adenosine receptors, namely A1 and A2A in the brain.23 Similar to ATP, adenosine can fine‐tune synaptic transmission by affecting the secretion of many neurotransmitters in a process that is highly concentration‐ and context‐dependent.22 For example, adenosine also stimulates a biphasic release of glutamate; low concentrations inhibit it whereas high concentrations induce it.32 The inhibitory affects of adenosine on glutamate release have been shown to be mediated by A1 receptor activation,33 whereas the facilitatory effects are through A2A receptors.34, 35 Underlying this complexity is the co‐localization of A1 and A2A receptors in glutaminergic terminals36 and the formation of A1–A2A heterodimers.32
We report here for the first time a handling‐induced and idiopathic seizure phenotype of CD39−/− mice. We performed histopathology, cell frequency and gene expression analysis, and cell‐specific gene deletion to determine the cause of seizures in CD39−/− mice. Our results support previous findings that suggest a role for extracellular nucleotides in seizures and contribute to the field's understanding of the underlying mechanisms of epileptogenesis.
Materials and methods
Mice
C57BL/6 and Cx3Cr1creERT2 mice were obtained from Jackson Laboratories (Bar Harbor, ME) for breeding. CD39−/− mice, the generation of which has been described previously,37 were provided by Dr Simon Robson. CD39flox+/+ CX3CR1creErt2−/+ mice were generated and provided by the laboratory of Dr Francisco J. Quintana. All mice were housed with sterile food and water ad libitum. Mice were killed by CO2 inhalation. The Institutional Animal Care and Use Committee at Harvard Medical School approved all experimental procedures involving animals. Adult 100‐day‐old male mice were used unless otherwise indicated. Littermate controls were used for behavioural experiments. Intraperitoneal injection of 100 µg/day tamoxifen was given once a day for 5 days to induce Cre recombinase expression in CD39fl Cx3Cr1creERT2 mice.
Handling‐induced seizures
To induce seizures by handling, CD39−/− mice were tightly held behind the neck and turned upside down for 10 seconds. The mouse was then placed in the cage right side up and monitored until behaviour appeared normal. Each age group reported was composed of independent groups of mice that had never before been handled; the age groups do not represent one cohort that was monitored as it aged over time. Similarly, animals for cell‐based and molecular assays were confirmed to have handling‐induced seizures before being killed for the experiment.
The seizure progression observed in CD39−/− mice was slightly different from the traditional Racine scale described previously for rats and mice.38, 39 Therefore, we constructed a scale that described our observations that was loosely based on the Racine scale. This scale has not been published but describes the phenotype as we saw it. Score 0 represented normal behaviour during which the animal acted unaffected by handling and began exploring immediately when placed down in the cage. Score 1 was characterized by freezing and immobility while still having muscle tension and an upright posture. Score 2 was characterized by a duration of flaccidity during which the stomach and head were in contact with the floor of the cage. Minor or partial convulsions may also occur during a score 2. Score 3 was characterized by severe tonic–clonic convulsions (uncontrolled running or jumping or rigid and stretched extremities). Score 4 was a severe seizure resulting in death, of which there were none.
The prevalence of handling‐induced seizures was calculated by dividing the number of mice within each age group that had an observable handling‐induced seizure (score 1–4) by the total number of mice in the respective age group. The latency to seizure onset was determined by measuring the time from the point the mouse was placed in the cage right side up (after 10 seconds of handling) until the first indication of seizure activity was observed. The duration was defined as the amount of time from the onset of seizure activity until the last observable sign of seizure for each mouse.
Behavioural testing
All behavioural tests were performed in the Harvard NeuroBehavioral Laboratory Core within the Harvard NeuroDiscovery Center. Before behavioural testing, mice (3–5 months of age) were ear tagged and handled for 1–2 min daily for at least 2 days to habituate subjects to the experimenter and to monitor health status. Before each experiment, mice were placed in the testing room 15 min before the test to acclimate to the environment. The apparatus (test chambers or mazes) were cleaned with 70% ethanol between individual subjects. Behavioural tests were performed at about 10.00 h. The same cohort of 10 male mice per group was used for open field, elevated plus maze, contextual fear conditioning, rotarod and social interaction test.
Open field
The open field apparatus consisted of a 27·9 × 27·9 cm, clear Plexiglas arena equipped with 16‐beam infrared arrays (Med Associates; St Albans, VT). Mice were placed in the centre of the arena and allowed to freely explore for 60 min. Movements were tracked and recorded automatically using Med Associates software. The open field test assessed general locomotor activity, exploratory behaviour, adaptability and anxiety.
Elevated plus maze
The elevated plus‐maze apparatus consisted of two open and two closed arms extended out from a central platform. Each arm of the maze was 30 cm long and 5 cm wide. The maze surface was 85 cm above the floor. Mice were placed near the centre platform of the maze, facing the centre, and allowed to explore the apparatus for 5 min. A computer‐assisted video‐tracking system (topscan software; CleverSys Inc., Reston, VA) was used to record the number of open and closed arm entries as well as the total time spent in open and closed arms. The elevated plus‐maze test was used to assess anxiety.
Contextual fear conditioning
The testing apparatus was a standard‐sized operant chamber (Med Associates) with Plexiglas sidewalls and stainless steel end‐walls as described previously.40 The floor consisted of steel bars 4·8 mm in diameter and spaced 1·6 cm apart in which a scrambled electric shock could be delivered. A fan was also attached to the chamber to provide masking noise. topscan software (Cleversys Inc.) computer‐assisted video tracking system was used to record and assess freezing behaviour defined as lack of body movement. Testing occurred over two sessions separated by 24 hr. To begin Session 1, a subject was placed in an illuminated chamber and allowed to explore for 2 min. At the end of this 2‐min period, a 2‐second, 0·5‐mA shock was delivered via the grid floor. A second, identical shock followed 2 min later. One minute recording without shock took place at the end of this session. Session 2 consisted of a 3‐min period during which the subject was allowed to freely explore the chamber in the absence of any foot shock. The contextual fear conditioning test was used to assess contextual memory and shock‐induced fear response.
Rotarod
Before the rotarod test, mice completed a training session to become accustomed to the rotarod apparatus (Ugo Basile Apparatus; Stoelting, Wood Dale, IL). For the rotarod training, the mice complete a 5‐min session at a constant speed of 4 rpm. If the mouse fell off during this session, it was placed back on the rotarod until the 5 min were completed. After the training session, mice were placed back in their cage to rest for about 10 min before starting the rotarod test. For the test, the rotarod speed increased from 4 rpm up to 40 rpm over a period of 3 min. The duration of time spent on the apparatus was recorded for each mouse. Rotarod was used to assess the mobility of mice.
Social interaction
The apparatus for Crawley's sociability and preference for social novelty test was used. The apparatus comprised a rectangular, three‐chamber box. Each chamber was 19 × 45 cm and the dividing walls were made from clear Plexiglas. The middle section had entryways to each end chamber, allowing free access to the subject mouse. In the corners of each side chamber, we placed wire cup‐like containers. The Topscan (Cleversys Inc.) video‐tracking system was used to record and track location of the mouse during test.
First, the subject mouse completed a habituation trial during which it was allowed to freely roam the apparatus for 10 min. Next, for the sociability trial, a naive/unfamiliar target mouse was placed into one of the cup‐like containers in one of the end chambers. The subject mouse was then placed back into the apparatus.
The amount of time spent near the empty cup versus the cup containing the mouse was calculated for each subject mouse. The average amount of time near the empty versus target cup was then calculated for each group. The social interaction test was used to assess social behaviour of the mice.
Field excitatory postsynaptic potential recordings
Mouse brains were collected and placed in chilled (4°) low‐Ca2+, low‐Na+ slicing solution consisting of (in mm): 234 sucrose, 11 glucose, 24 NaHCO2, 2·5 KCl, 1·25 NaH2PO4, 10 MgSO4 and 0·5 CaCl2, equilibrated with a mixture of 95% O2 : 5% CO2. The brain was glued to the slicing stage of a Vibratome 3000 sectioning system and 400‐µm slices were cut in a coronal orientation. The slices were then incubated in 32° oxygenated artificial cerebrospinal fluid (CSF) (in mm): 126 NaCl, 2·5 KCl, 1·25 NaH2PO4, 1 MgSO4, 2 CaCl2, 10 glucose, 26 NaHCO2) for 1 hr. Samples were subsequently allowed to cool to room temperature.
For field recordings, slices were placed in an interface chamber maintained at 34°, superfused with oxygenated artificial CSF at 2 ml/min. The Schaffer collateral was stimulated with a tungsten concentric bipolar electrode. Stimulation intensity was adjusted such that a field excitatory postsynaptic potential > 0·5 mV was elicited with a 100‐microsecond pulse, but not a 50‐microsecond pulse (World Precision Instruments, Sarasota, FL). Glass micropipettes (resistance ≅ 1 MΩ) were filled with artificial CSF and placed in CA1. Electrophysiological data were recorded with an Axon Multiclamp 700A amplifier and Digidata 1322A digitizer (sampling rate = 20 kHz) with labchart software (ADInstruments, Colorado Springs, CO). Input–output curves were generated by stimulating the Schaffer collateral every 30 seconds with increasing duration (50–600 microseconds). Paired‐pulse recordings were generated by giving a pair of 200‐microsecond stimulations separated by 50 milliseconds. All data were analysed in clampfit (Molecular Devices, Sunnyvale, CA).
Histology
Mice were transcardially perfused with 10–20 ml Hanks’ balanced salt solution (HBSS,; Invitrogen, Carlsbad, CA). Brains were removed and placed in 10% formalin diluted in PBS. Samples were embedded in paraffin, cut into 5‐µm slices and stained with haemotoxylin & eosin (H&E). An Aperio ScanScope System was used to image the stained slides.
Cerebrospinal fluid measurements
The method for CSF collection was adapted from a previously described protocol.41 Briefly, the mice were deeply anaesthetized and placed in an inverted fashion with the head and body forming a 90‐degree angle. The cisterna magna was exposed and a small puncture was made in the arachnoid membrane using a micro‐needle. Cerebrospinal fluid was collected from the puncture hole with a polypropylene narrow bore pipette. The mice were killed immediately afterwards. CSF samples were immediately placed on dry ice and stored at −80° until thawed for assay within 24 hr of collection. The ATP assay (Invitrogen; cat# A22066) and adenosine assay (BioVision, Milpitas, CA; cat# K327) were conducted according to the manufacturer's protocols.
Flow cytometry
Mononuclear cells were isolated from the brains of adult mice as previously described.30 Briefly, mice were transcardially perfused with cold HBSS and brains were dissected. Single‐cell suspensions were prepared using a Teflon‐stick homogenizer and centrifuged over a 37% to 70% discontinuous Percoll gradient (GE Healthcare, Chalfont St Giles, UK). Mononuclear cells were isolated from the interface and collected in blocking buffer containing 0·2% bovine serum albumin (Sigma‐Aldrich, St Louis, MO) in HBSS.
For staining, cells were pre‐blocked with blocking antibody CD16/CD32 FcBlock (clone 2.4G2, 0·5 mg/ml; BD Biosciences, San Jose, CA) and stained on ice for 30 min. Staining antibodies included phycoerythrin (PE)‐Cy7 anti‐CD11b (M1/70; eBioscience, San Diego, CA), FITC anti‐CD45 (30‐F11, eBioscience), PE anti‐Ly6C (HK1.4; BioLegend, San Diego, CA), 7AAD (BD Biosciences), AlexaFluor 700 anti‐CD19 (eBio1D3, eBioscience), Peridinin chlorophyll proteinP anti‐CD4 (RM4‐5, BioLegend), FITC anti‐CD8 (53‐6.7, BD Pharmingen, San Diego, CA), PE‐Cy7 anti‐CD11c (N418, eBioscience) and allophycocyanin anti‐Ly6G (1 A8, BD Pharmingen). All antibodies for co‐stimulatory molecules were PE‐conjugated antibodies from BD Biosciences. These were anti‐CD40 (3/23), anti‐CD80 (16‐10A1), anti‐CD86 (GL1) and anti‐MHCII (AF6‐120.1).
FACS analysis was performed on an LSR machine (BD Biosciences), and data were analysed with flowjo Software (TreeStar, Ashland, OR). Cell sorting was performed using a Becton Dickinson FACS ARIA cell sorter.
Fluorescence microscopy
Mice were transcardially perfused with 10–20 ml HBSS. Brains were removed and placed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA; #15714) diluted in PBS (Invitrogen) for 24 hr at 4°. Tissue was washed three times with PBS and then placed in 30% sucrose (in PBS) for 48 hr at 4°.
Next, the brains were incubated in a plastic mould for 1 hr at room temperature in a 2 : 1 mixture of 30% sucrose to OCT. Moulds were frozen on dry ice and 14‐μm sections were made by cryosectioning and were attached to glass slides. Unused tissue and slides were stored at −80°.
For staining with primary antibody, slides were thawed at room temperature for 30 min. The borders of the slides were outlined with ImmEdge Hydrophobic Barrier PAP Pen (Burlingame, CA). Slides were washed twice with 1× PBS for 10 min. Tissue was blocked with blocking buffer containing 1× Tris‐buffered saline, 5% bovine serum albumin, 100 mm l‐lysine, 0·04% azide, 0·2% Triton‐X‐100, pH 7·4 for 1 hr at room temperature. Slides were incubated with primary antibody diluted in 4 : 1 antibody buffer : block overnight at 4°. Antibody buffer contained 1× Tris‐buffered saline, 1% bovine serum albumin, 100 mm l‐lysine, 0·04% azide, pH 7·4.
The next day, tissues were washed in 1× PBS for 10 min, four times. Secondary antibody (1 : 200) in 4 : 1 antibody buffer : block was added to slides and incubated for 2 hr at room temperature. Slides were washed in PBS for 15 min three times and mounted with coverslip using Prolong® Gold anti‐fade reagent with DAPI (Life Technologies, Grand Island, NY; P36931). Slides were imaged using Zeiss 710 confocal microscope and analysed using fiji software.
Primary antibodies used were anti‐mouse CD68 (AbD Sortec BioRad, Hercules, CA; #MCA1957), Iba1 (Wako, Richmond, VA; #019‐19741), NeuN (clone A60, Millipore, Billerica, MA; MAB377), Iba1 (Wako) and GFAP (BD Pharmingen; 556330). Secondary antibodies were donkey anti‐rabbit IgG AlexaFluor® 594 (Life Technologies; A‐21207) and donkey anti‐rat IgG (H+L) secondary antibody AlexaFluor® 488 conjugate (A‐21208; Life Technologies).
Microglia activation assay
Mice were injected with PBS or 3 mg/kg lipopolysaccharide (LPS; Sigma). Seizure severity was measured 24 hr post injection. After 48 hr, brain sections were mounted on glass slides, washed and stained with anti‐mouse CD68 and anti‐mouse Iba1 as described above. Slices were imaged using a Zeiss 710 confocal microscope and analysed using fiji software. Microglia activation score was calculated as previously described.42 Briefly, every cell in the field of view was assigned a morphology and CD68 score by a blinded investigator. Iba1 morphology score ranged from 0 to 3; score 3 was given to cells with round, compact cell body and score 0 was given to larger ramified cells. CD68 expression score was also on a 0–3 scale; cells with high, aggregated CD68 expression received a score of 3 whereas cells with no CD68 received a score of 0. The individual morphology and CD68 scores where then added together to obtain a total activation score for each cell in the field of view; score (0+0) reflected a quiescent cell whereas a maximum score of 6 (3+3) represented a highly activated cell. The average activation score in each group (n = 3) was then calculated.
NanoString
After sorting, microglial cells were stored in lysis buffer from a mirVana miRNA isolation kit (Ambion) in −80° until further processed. Total RNA was extracted using a mirVana™ miRNA isolation kit (Ambion) following the manufacturer's protocol.
NanoString ncounter with the immunology code set was used to perform mRNA transcription profiling according to the manufacturer's suggested protocol. nsolver analysis software was used to analyse the expression data. Background subtraction was performed using geometric mean (+2*SD) of the negative control probes. Data were normalized to the geometric mean of seven housekeeping genes that had variation < 62%. These housekeeping genes were Tubb5, Eef1g, Gapdh, Ppia, Gusb, Oaz1, Rpl19. Low‐expressing genes that had expression values equal to or below the negative controls (5·31) were removed from analysis. Student's t‐tests (two‐tailed, unpaired) were performed between CD39+/+ versus CD39−/− mice treated with PBS or LPS. Genes that had P‐value > 0·05 were removed from further analysis. heatmap was created in nsolver using agglomerative cluster analysis type, Euclidean distance for metric type, and cluster for gene data usage.
Ingenuity pathway analysis
The NanoString data set was analysed by Ingenuity Pathway Analysis (Ingenuity Systems, Qiagen, Hilden, Germany; http://www.ingenuity.com). Differentially expressed genes (with corresponding fold changes and P‐values) were incorporated in canonical pathways and bio‐functions.
Statistical analysis
graphpad prism 6·0 was used for statistical analysis. Unpaired, two‐tailed Student's t‐test or one‐way analysis of variance, followed by Tukey multiple comparisons test, were used. α was set to 0·05. Data are presented as averages ± standard error of the mean.
Results
Adult CD39−/− mice exhibit handling‐induced and spontaneous seizures
When performing routine animal husbandry, we noticed that aged, breeding CD39−/− mice exhibited seizure‐like behaviour after handling (see Supplementary material, [Link], [Link], [Link], [Link]). We consistently observed handling‐induced seizures in our CD39−/− mouse colony over a period of 4 years and more than 10 generations. On rare occasions, mice had unprovoked seizure events in the absence of handling. We monitored seizure behaviour in mice of different ages to determine if seizure severity was correlated with age. Although we observed similar seizure phenotype in male and female mice, further characterization and experiments were performed on male mice to exclude the effect of oestrous cycle and hormone fluctuation on behaviour. No handling‐induced seizures were observed in mice < 20 days of age, possibly because it was difficult to handle the smaller, younger mice in the same manner as the older mice.
We developed a seizure severity scale that described the evolution of seizure severity that we observed, which was loosely based on the Racine scale used to describe chemically induced seizures in rats38 and mice.39 Based on our scoring system, severity of handling‐induced seizures in CD39−/− mice significantly increased as mice aged from 20 to 100 days [0·4 ± 0·2 (20 days) n = 5 versus 1·2 ± 0·3 (75 days) n = 16 versus 2·2 ± 0·3 (100 days) n = 18] (Fig. 1a). The prevalence of seizure events also increased from 40% at 20 days old to 94·5% at 100 days old (Fig. 1b).
Figure 1.
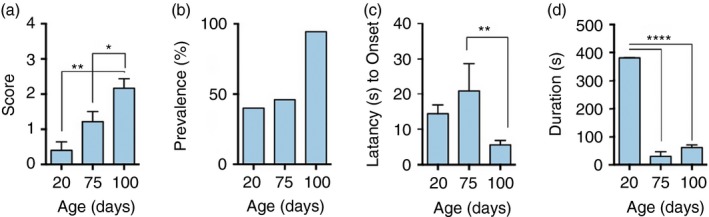
Spontaneous and handling‐induced seizures in CD39−/− mice. Handling‐induced seizure (a) score, (b) prevalence, (c) latency to onset and (d) duration in CD39−/− male mice of different ages. For 20‐day‐old n = 5, 75‐day‐old n = 16 and 100‐day‐old n = 18. *P ≤ 0·05; **P < 0·005; ****P ≤ 0·0001.
Latency to seizure onset, defined as the time from the end of handling until the first signs of observable seizures, significantly decreased as mice aged [21 ± 7·8 seconds (75 days) n = 16 versus 5·6 ± 1·3 seconds (100 days) n = 18] (Fig. 1c). Interestingly, seizure duration also significantly decreased as mice aged [381 ± 1 seconds (20 days) n = 5 versus 29·7 ± 16·8 seconds (75 days) n = 16 versus 61·2 ± 9·6 seconds (100 days) n = 18] (Fig. 1d). CD39+/+ littermate controls did not have any observable spontaneous or handling‐induced seizures (data not shown). All subsequent experiments were performed using 100‐day‐old male mice at the peak of the measured seizure severity. CD39+/+ wild‐type littermates were used as controls.
To determine if CD39−/− had any other behavioural phenotype, we performed several neurobehavioural tests on CD39+/+ and CD39−/− mice including open field, elevated plus maze, fear conditioning, rotarod and social interaction tests (Fig. 2 and data not shown). The open field test was used to assess locomotor activity, exploratory behaviour, anxiety and adaptability. The elevated plus maze test was used to assess anxiety. The fear‐conditioning test was used to assess fear response and contextual memory. The rotarod test measured motility and the social interaction test measured sociability. The only difference in neurobehaviour detected by the tests conducted was in the elevated plus maze test. CD39−/− spent significantly less time in the closed arm compared with CD39+/+ mice [286 ± 3·2 seconds (wild‐type n = 10) versus 365·3 ± 7·0 seconds (knockout n = 10); P < 0·05] (Fig. 2f), indicating that CD39−/− mice are less anxious than their wild‐type counterparts. There was no difference in the weight of CD39+/+ and CD39−/− mice (data not shown).
Figure 2.
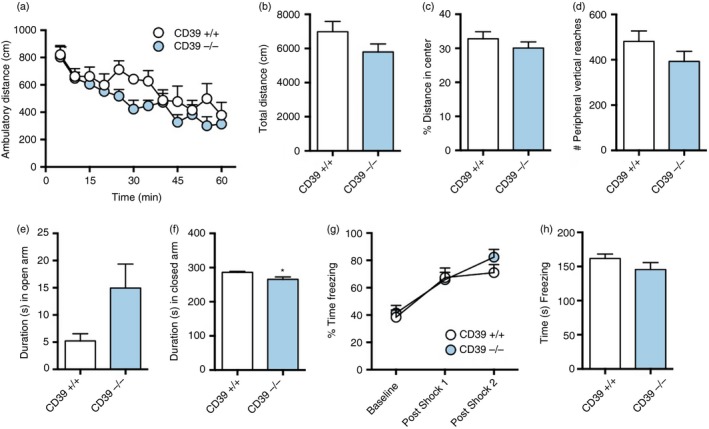
CD39−/− mice have normal exploratory behaviour, anxiety levels, shock response and recall. The 100‐day‐old male littermate mice were exposed to a variety of neurobehavioural tests. Results from open field test of (a) ambulatory distance over time, (b) total ambulatory distance, (c) per cent distance travelled in centre zone, and (d) number of vertical reaches in the peripheral zone for CD39+/+ and CD39−/− mice. Duration in (e) open arm and (f) closed arm in elevated plus maze for CD39+/+ and CD39−/− mice. (g) Per cent time freezing during fear conditioning for CD39+/+ and CD39−/− mice. (h) Context test 1 day after fear conditioning. The same cohort of 10 male mice/group were used for all behavioural tests performed in the order presented. *P ≤ 0·05.
Imbalance of extracellular ATP and adenosine in the CSF of CD39−/− mice
We next investigated whether seizures in CD39−/− mice correlated with altered neuronal response, brain histology, gliosis or immune cell recruitment. Neuronal function was assessed in acute hippocampal slices by measuring the response to electrical stimuli. The Shaffer collateral pathway was stimulated using a concentric bipolar stimulation electrode. The duration of stimulation was varied from 50 to 600 microseconds and the resulting field excitatory postsynaptic potentials in the CA1 stratum radiatum were measured. There was no significant difference in the threshold stimulation intensity or the peak normalized input–output relationship from CD39+/+ and CD39−/− slices (Fig. 3a). The two groups also exhibited similar paired pulse ratios when paired stimuli were delivered to the Schaffer collateral pathway with a 50‐millisecond interval (Fig. 3b). These data suggest that neurons in the hippocampus of CD39−/− mice do not have altered axonal excitability or pre‐synaptic function.
Figure 3.
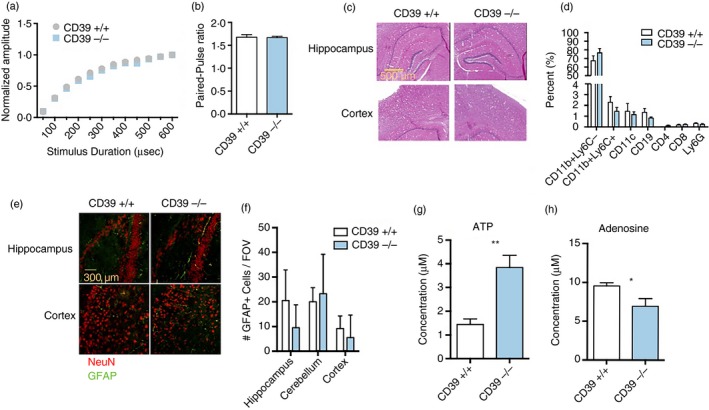
Imbalance of extracellular ATP and adenosine in the cerebrospinal fluid (CSF) of CD39−/− mice. (a) Normalized field excitatory postsynaptic potential (fEPSPs) recoded in stratum radiatum following stimulation of the Shaffer collateral (SC) pathway. Stimulation duration was varied from 50 to 600 microseconds (µs). (b) Paired‐pulse ratio computed from fEPSPs evoked by giving 200‐µs stimulation with a 50‐millisecond interval. (c) Haematoxylin & eosin staining of hippocampus and motor cortex. (d) Frequencies of immune cell populations in the brain of naive mice. CD39+/+ (n = 6) and CD39−/− (n = 8) male mice were used. (e) NeuN (red) and GFAP (green) staining of hippocampus and cortex. (f) Quantification of GFAP + cells in hippocampus, cerebellum and cortex from confocal images. ATP (g) and adenosine (h) measurements in the CSF of CD39+/+ and CD39−/− mice. For histology experiments, n = 3 mice per group were used. For electrophysiology experiments, n = 7 for the CD39+/+ group and n = 6 for the CD39−/− group. For CSF measurements, n = 6 mice/group. *P < 0·05; **P < 0·005.
Cell death and gliosis are common hallmarks of temporal lobe epilepsy in mice43, 44 and humans.45, 46, 47 Therefore, we performed H&E staining, FACS analysis and fluorescence imaging of CD39+/+ and CD39−/− brains to assess brain structure, cellular composition and gliosis, respectively. Brains of CD39−/− mice were indistinguishable from CD39+/+ brains by H&E staining (Fig. 3c) and did not contain abnormal frequencies of major immune cell populations (Fig. 3d). NeuN and GFAP staining was also normal in CD39−/− mice and there was no difference in number of GFAP+ astrocytes in the hippocampus or cortex of these mice (Fig. 3e,f).
We hypothesized that CD39−/− mice would have increased extracellular ATP and reduced adenosine in the CNS due to inherently decreased CD39‐mediated breakdown of ATP. To test this hypothesis, we measured ATP and adenosine in the CSF of CD39−/− and CD39+/+ mice. As expected, CD39−/− had about 2·5‐fold more ATP in the CSF compared with CD39+/+ mice [1·4 ± 0·2 µm (wild‐type) versus 3·8 ± 0·5 µm (knockout); n = 7 mice/group; P < 0·005] (Fig. 3g). In addition, adenosine was significantly decreased in the CSF of CD39−/− versus CD39+/+ mice [9·6 ± 0·4 µm (wild‐type n = 6) versus 6·9 ± 1·0 µm (knockout n = 7); P < 0·05] (Fig. 3h). These data, in addition to the hippocampal slice recordings, suggest that altered extra‐synaptic, but not synaptic, ATP : adenosine ratio may play a role in seizure progression of CD39−/− mice.
Lipopolysaccharide‐induced immune activation does not affect seizure severity in CD39−/− mice
CD39 is considered an immunosuppressive molecule because of its role in the generation of adenosine48 and because the immune system is activated in human epilepsy patients49 and mouse models of epilepsy.50, 51 Therefore, we hypothesized that CD39−/− microglia were more activated than CD39+/+ microglia and that immune activation was involved in seizures of CD39−/− mice. To test this hypothesis, we injected mice with PBS or LPS, a component of bacterial cell wall, and examined the effect on microglia activation and seizure severity.
Brains from PBS‐ or LPS‐treated mice were stained for Iba1 and CD68, markers of macrophage and lysosomal activation, respectively (Fig. 4a). Microglia activation scores were calculated by adding the Iba1 morphological scores and CD68 expression scores for each cell in the field of view as previously described.42 There were no differences in microglia activation scores in PBS‐injected mice (Fig. 4b,c). After LPS injection, however, the microglia activation score in the cortex of CD39−/− mice was significantly greater than in CD39+/+ mice [2·9 ± 0·3 (wild‐type) versus 3·6 ± 0·07 (knockout); n = 3 mice/group; P < 0·05] (Fig. 4b). Microglia activation score in the hippocampus of LPS‐treated CD39−/− mice was higher than in CD39+/+ mice, though not statistically significant [3·2 ± 0·2 (wild‐type) versus 3·6 ± 0·2 (knockout); n = 3 mice/group] (Fig. 4c).
Figure 4.
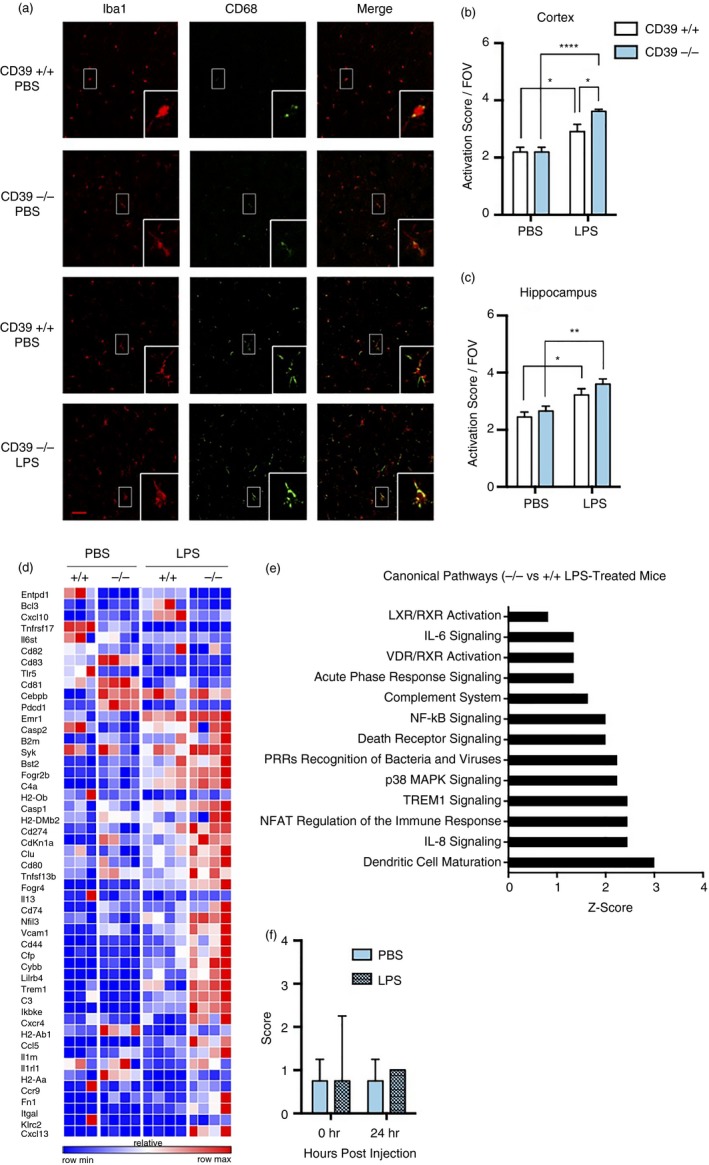
Increased activation of microglia in lipopolysaccharide (LPS) ‐treated CD39−/− versus CD39+/+ mice does not impact seizure severity. (a) Representative confocal images of brain 42 hr after injection with PBS or 3 mg/kg LPS. Images are representative of two independent experiments with four mice per group total. Scale bar in merged images measures 50 µm and insets are magnified images from representative boxed cells. Microglial activation score representing sum of Iba1 morphological and CD68 expression score for individual microglia cells in (b) cortex and (c) hippocampus. (d) Heat map of significantly affected genes measured by NanoString in CD11b+ CD45mid cells isolated from mice treated with PBS or LPS. (e) Ingenuity pathway analysis predicted several canonical pathways to be increased in microglia isolated from brains of CD39−/− compared with CD39+/+ mice. (f) Seizure scores of PBS‐treated and LPS‐treated CD39−/− mice at time of injection and 24 hr later. *P < 0·05; **P < 0·005; ****P < 0·001.
Consistent with the CD68 and Iba1 activation scores, there were no differences in gene expression of CD11b+ CD45lo Ly6C− microglia isolated from PBS‐treated mice. Only after LPS treatment did microglia in CD39−/− mice become more activated than their CD39+/+ counterparts (Fig. 4d). Ingenuity pathway analysis predicted several inflammation‐related canonical pathways to be increased in CD39−/− versus CD39+/+ microglia isolated from LPS‐treated mice including interleukin‐8, triggering receptor expressed on myeloid cells 1, p38, pattern recognition receptors, death receptor, nuclear factor‐κB, complement, acute phase and interleukin‐6 signalling pathways (Fig. 4e). Although microglia were more activated in LPS‐treated mice, LPS treatment had no effect on overall seizure severity (Fig. 4f). Taken together, these results suggest that microglia in CD39−/− mice are more sensitive to LPS activation compared with CD39+/+ microglia, but that microglia activation does not affect handling‐induced seizure severity in CD39−/− mice.
Microglia‐specific deletion of CD39 is not sufficient to induce seizures
We next hypothesized that mice with microglia‐specific CD39 deletion would phenocopy mice with global CD39 deletion on all cell types since CD39 is predominantly expressed on microglia in the brain.29, 30 To test this hypothesis, we conditionally deleted CD39 on microglial cells and measured seizure severity in adult mice. CD39flox+/+ CX3CR1creErt2−/+ and CD39flox+/+ CX3CR1creErt2−/− littermate controls were injected with tamoxifen to induce Cre‐mediated deletion of CD39 in cells expressing CX3CR1, all myeloid cells including tissue macrophages (Fig. 5a). FACS analysis confirmed that tamoxifen injection resulted in deletion of CD39 on brain‐resident CD11b+ microglia cells in CD39flox+/+ CX3CR1creErt2−/+ mice 2 weeks post injection (Fig. 5b). In the spleen on the other hand, CD39flox+/+ CX3CR1creErt2−/+ and CD39flox+/+ CX3CR1creErt2−/− tamoxifen‐treated mice had equal levels of CD39, suggesting that CD39 deletion in CD39flox+/+ CX3CR1creErt2−/+ mice was specific for microglia and not peripheral monocytes/macrophages that can be newly and frequently generated from the bone marrow in adult mice. Surprisingly, CD39flox+/+ CX3CR1creErt2−/+ tamoxifen‐treated mice did not have handling‐induced seizures (Fig. 5c). Of note, deletion of CD39 on microglia during early developmental stages, instead of in fully grown adult mice, may be sufficient for seizure generation. These data also suggest the importance of CD39 deletion in non‐microglia cells, such as endothelial cells, in driving seizures in CD39−/− mice.
Figure 5.
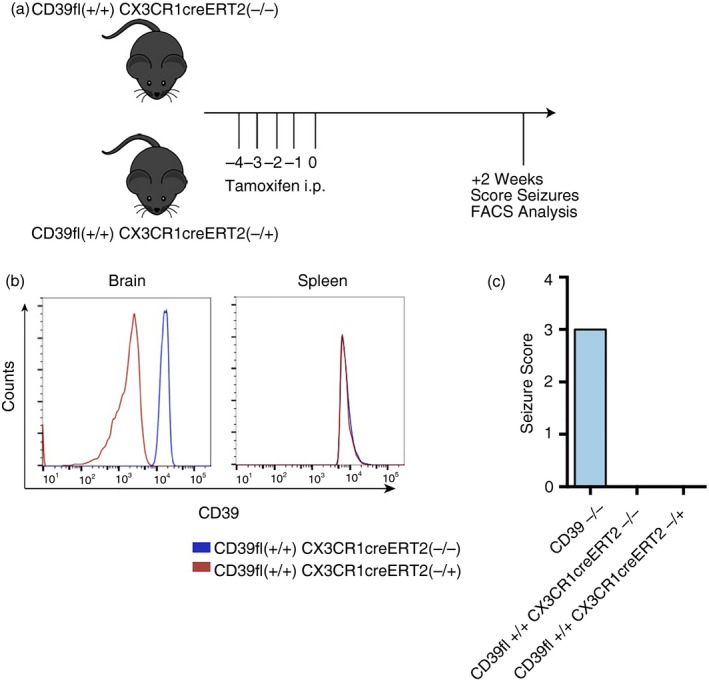
Microglia‐specific deletion of CD39 does not induce seizures. (a) Experimental timeline highlighting tamoxifen treatment and subsequent analysis. (b) CD39 expression of CD11b+ CD45midLy6C− cells in brain and spleen of tamoxifen‐treated CD39flox+/+ CX3CR1creErt2−/+ and CD39flox+/+ CX3CR1creErt2−/+ mice. (c) Seizure scores of CD39−/− and tamoxifen‐treated CD39flox+/+ CX3CR1creErt2−/+ and CD39flox+/+ CX3CR1creErt2−/+ mice. n = 5 mice per group.
Discussion
We describe a novel, idiopathic, epileptic phenotype in CD39−/− mice. CD39 has not been directly implicated in epilepsy, although human CD39 co‐localizes with a gene involved in partial human epilepsy with audiogenic symptoms.52, 53 In addition, mutations in CD39 cause spastic paraplegia 64, an autosomal recessive neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs.54
CD39−/− mice used in this study were originally developed by Enjyoji et al.37 in 1999 and reported to have prolonged bleeding times and altered platelet function. An independent group generated CD39−/− mice by a different approach.55 These mice also displayed no obvious phenotypic abnormalities and exhibited a pro‐thombotic phenotype only after stroke induction.55 We hypothesize that seizure phenotype was not previously reported due to group differences in the (i) ages of mice used, (ii) handling techniques of different investigators, (iii) interest and focus of laboratories or (iv) microbiome, which has been shown to affect behaviour in mice.56, 57
Of note, there have been other instances in which investigators have observed seizures in a transgenic mouse line long after its original publication and characterization. For example, Palop et al. detected intermittent non‐convulsive EEG seizures in a hAPP mouse model of Alzheimer's disease58 and Papaleo et al. described how brain‐derived neurotrophic factor over‐expressing (BDNF‐transgenic) mice had seizures when placed in a clean, new cage.59 In addition to seizures, the hAPP60 and BDNF‐transgenic59 mice exhibited other neurobehavioural abnormalities such as impaired learning and memory. Interestingly, CD39−/− mice spent less time in the closed arm and greater time in the open arm, although not significant, of the elevated plus maze test, indicating that they have reduced levels of anxiety compared with CD39+/+ mice. Future assessment with different tests may reveal other unreported behavioural defects.
The observational handling‐induced seizures occurred in about 100% of 100‐day‐old CD39−/− mice and ranged from absence (score 1) to tonic–clonic (score 3). The range of seizure types observed may be suggestive of the mechanism involved. For example, absence seizures indicate the involvement of the thalamo–cortical circuit.61 On the other hand, the range of seizure types may be due to inconsistencies in the investigator's tightness of grip or the height, duration, or angle at which the mouse was held. Nonetheless, handling‐induced seizures were specific for the type of handling performed. CD39−/− mice did not have seizures during any of the neurobehavioural tests performed or in response to vestibular (tail suspension) or auditory (70–80 dB tone 15–30 seconds) stimuli, all of which may be considered stressful. In addition, seizure severity diminished when animals were repeatedly handled. This pattern indicates a lack of kindling despite increased handling‐induced seizure severity in aged mice. In addition to likely sickness behaviour, habituation may also explain why seizure severity did not increase 24 hr after LPS treatment. Future studies using simultaneous video, EEG and EMG recordings will help to characterize seizures in CD39−/− mice.
CD39−/− mice did not have any gross brain pathology related to cell loss, astrogliosis or microgliosis. The absence of sclerosis and inflammation in CD39−/− mice may be due to the acute and idiopathic nature of their seizures. In fact, neuronal damage correlates with frequency and duration of seizures;62 brief seizures result in necrosis and minimal apoptosis63 and patients with idiopathic epilepsy have subtle brain abnormalities.64 Future studies could investigate potentially microscopic differences in microglia–neuron interactions, astrocytic phyllopodia, vasculature or number of excitatory/inhibitory synapses.
The CSF of CD39−/− mice contained increased levels of ATP and decreased levels of adenosine compared with CD39+/+ mice. The range of concentrations of ATP (about 1·4–3·8 µm) and adenosine (about 6·9–9·6 µm) were much higher than reported in the literature. Xu et al. reported mouse CSF ATP levels to be about 0·2 nm in basal conditions and about 0·8 nm after radiation.65 For adenosine, concentrations in the hippocampus measured by dialysis can reach about 135 nm in rats with bicuculline‐induced seizures9 and 1·5 µm during seizures in humans.14 Differences in techniques or anaesthetics used between studies may contribute to the inconsistencies observed.
Future studies should be performed to investigate whether altered ATP/adenosine levels in the CSF are cause or consequence of seizures in CD39−/− mice by taking measurements in mice of different ages (newborn to 100 days), with different seizure severity scores (0–3) and in mice with or without microglia‐specific CD39 deletion. In addition, purine receptor targeting will also help to determine the contribution of altered extracellular nucleotide signalling in seizures of CD39−/− mice.
Endogenous adenosine is released within hippocampal cultures where it has inhibitory effects.66 Adenosine exerts most of its effects in the synapse67 raising the question of whether altered extracellular adenosine levels are biologically significant in CD39−/− mice. As mentioned previously, this question may be addressed by examining the effect of adenosine receptor targeting on handling‐induced seizures in CD39−/− mice. It will also be important to determine if there are any differences in synaptic glutamate levels as both ATP26 and adenosine32 affect glutamate release in a biphasic manner and glutamate is an important excitatory neurotransmitter that may play a role in seizures in CD39−/− mice.
We investigated the role of microglial cells in seizures in CD39−/− mice as these cells contribute significantly to the levels of CD39 in the brain.29, 30, 31 It was surprising that there were no differences in gene and protein expression of CD39−/− versus CD39+/+ microglia in PBS‐treated (relatively naive) mice despite an altered extracellular ATP/adenosine ratio in the microenvironment. This is consistent with the finding that microglia have no effect on epileptogenesis in hippocampal cultures.68 However, our results are inconsistent with reports showing that ATP and adenosine affect microglia activation69 and migration.70 Matyash et al. also recently showed that CD39−/− microglia had attenuated ramification,71 a phenotype that has been correlated with microglia quiescence. Interestingly, microglia‐derived ATP and adenosine mediate synaptic plasticity and transmission72 and microglia‐dependent modulation of neuronal activity has been shown to depend on neuronal hemi‐channels,73 channels important for ATP release. Therefore, our findings do not rule out the possibility that CD39−/− mice may have altered neuron–microglia communication that could contribute to the observed seizure phenotype. These studies further highlight the need to examine synaptic transmission and neuronal wiring in CD39−/− mice.
Although microglia were not altered in PBS‐injected mice, CD39−/− microglia appeared more activated than CD39+/+ when mice were injected with LPS, suggesting that CD39−/− microglia have attenuated compensatory mechanisms. In contrast to a report by Sayyah et al.,74 LPS treatment of CD39−/− mice did not alter seizure severity. In addition to the different model systems, possible habituation to handling or induced‐sickness behaviour, our study used a higher dose of LPS and increased incubation time compared with the study by Sayyah et al. It was also surprising that conditional and specific deletion of CD39 on microglia cells in adult mice was not sufficient to induce seizures. This suggests that expression of CD39 on microglia during development or on other cell types, such as vascular endothelial cells, may contribute to the phenotype. Of note, purine signalling affects blood–brain barrier permeability,75, 76 which increases in epilepsy.77 Consistent with a recent review,22 our report stresses the need for future studies to address the many unanswered questions related to the role of purinergic signalling in the brain.
Disclosures
The authors have no conflicts of interest to disclose.
Supporting information
Movie S1. Handling‐induced seizure of score 3.
Movie S2. Handling‐induced seizure of score 2.
Movie S3. Handling induced seizure of score 2 without convulsions.
Movie S4. Wild‐type mouse showing no disturbances upon handling.
Acknowledgements
We thank Oleg Butovsky and Gopal Murugaiyan for their help with techniques and initial planning and brainstorming of this project, Vanessa Beynon for the suggestion to measure ATP and adenosine from CSF, Deneen Kozoriz for FACS sorting, Maria Mazzola for assistance with cytokine array, Victoria Beja‐Glasser for assistance and training with fluorescence microscopy. We thank Kevin Staley and Yero Saponjian for their help with hippocampal cultures that were not presented here. We greatly appreciate the general oversight and direction from Vijay Kuchroo, Beth Stevens and Robert Brown.
References
- 1. Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol 2011; 7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moshe SL, Perucca E, Ryvlin P, Tomson T. Epilepsy: new advances. Lancet 2015; 385:884–98. [DOI] [PubMed] [Google Scholar]
- 3. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia 2001; 42:1255–60. [DOI] [PubMed] [Google Scholar]
- 4. Brodie MJ, Perucca E, Ryvlin P, Ben‐Menachem E, Meencke HJ. Comparison of levetiracetam and controlled‐release carbamazepine in newly diagnosed epilepsy. Neurology 2007; 68:402–8. [DOI] [PubMed] [Google Scholar]
- 5. Ran X, Li J, Shao Q, Chen H, Lin Z, Sun ZS et al EpilepsyGene: a genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res 2015; 43:D893–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Staley K. Molecular mechanisms of epilepsy. Nat Neurosci 2015; 18:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dona F, Conceicao IM, Ulrich H, Ribeiro EB, Freitas TA, Nencioni AL et al Variations of ATP and its metabolites in the hippocampus of rats subjected to pilocarpine‐induced temporal lobe epilepsy. Purinergic Signal 2016; 12:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lietsche J, Imran I, Klein J. Extracellular levels of ATP and acetylcholine during lithium‐pilocarpine induced status epilepticus in rats. Neurosci Lett 2016; 611:69–73. [DOI] [PubMed] [Google Scholar]
- 9. Berman RF, Fredholm BB, Aden U, O'Connor WT. Evidence for increased dorsal hippocampal adenosine release and metabolism during pharmacologically induced seizures in rats. Brain Res 2000; 872:44–53. [DOI] [PubMed] [Google Scholar]
- 10. Dona F, Ulrich H, Persike DS, Conceicao IM, Blini JP, Cavalheiro EA et al Alteration of purinergic P2X4 and P2X7 receptor expression in rats with temporal‐lobe epilepsy induced by pilocarpine. Epilepsy Res 2009; 83:157–67. [DOI] [PubMed] [Google Scholar]
- 11. Rebola N, Coelho JE, Costenla AR, Lopes LV, Parada A, Oliveira CR et al Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci 2003; 18:820–8. [DOI] [PubMed] [Google Scholar]
- 12. Bonan CD, Amaral OB, Rockenbach IC, Walz R, Battastini AM, Izquierdo I et al Altered ATP hydrolysis induced by pentylenetetrazol kindling in rat brain synaptosomes. Neurochem Res 2000; 25:775–9. [DOI] [PubMed] [Google Scholar]
- 13. Bonan CD, Walz R, Pereira GS, Worm PV, Battastini AM, Cavalheiro EA et al Changes in synaptosomal ectonucleotidase activities in two rat models of temporal lobe epilepsy. Epilepsy Res 2000; 39:229–38. [DOI] [PubMed] [Google Scholar]
- 14. During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 1992; 32:618–24. [DOI] [PubMed] [Google Scholar]
- 15. Fields RD. Nonsynaptic and nonvesicular ATP release from neurons and relevance to neuron‐glia signaling. Semin Cell Dev Biol 2011; 22:214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imura Y, Morizawa Y, Komatsu R, Shibata K, Shinozaki Y, Kasai H et al Microglia release ATP by exocytosis. Glia 2013; 61:1320–30. [DOI] [PubMed] [Google Scholar]
- 17. Koizumi S. Synchronization of Ca2+ oscillations: involvement of ATP release in astrocytes. FEBS J 2010; 277:286–92. [DOI] [PubMed] [Google Scholar]
- 18. Burnstock G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 2016; 104:4–17. [DOI] [PubMed] [Google Scholar]
- 19. Lazarowski ER, Sesma JI, Seminario‐Vidal L, Kreda SM. Molecular mechanisms of purine and pyrimidine nucleotide release. Adv Pharmacol 2011; 61:221–61. [DOI] [PubMed] [Google Scholar]
- 20. Bodin P, Burnstock G. Purinergic signalling: ATP release. Neurochem Res 2001; 26:959–69. [DOI] [PubMed] [Google Scholar]
- 21. Rodrigues RJ, Tome AR, Cunha RA. ATP as a multi‐target danger signal in the brain. Front Neurosci 2015; 9:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cunha RA. How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem 2016; 139:1019–55. [DOI] [PubMed] [Google Scholar]
- 23. Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol 2005; 63:191–270. [DOI] [PubMed] [Google Scholar]
- 24. Engel T, Alves M, Sheedy C, Henshall DC. ATPergic signalling during seizures and epilepsy. Neuropharmacology 2015; 104:140–53. [DOI] [PubMed] [Google Scholar]
- 25. Cunha RA, Ribeiro JA. ATP as a presynaptic modulator. Life Sci 2000; 68:119–37. [DOI] [PubMed] [Google Scholar]
- 26. Rodrigues RJ, Almeida T, Richardson PJ, Oliveira CR, Cunha RA. Dual presynaptic control by ATP of glutamate release via facilitatory P2X1, P2X2/3, and P2X3 and inhibitory P2Y1, P2Y2, and/or P2Y4 receptors in the rat hippocampus. J Neurosci 2005; 25:6286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol 2014; 99:16–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunwiddie TV, Diao L, Proctor WR. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci 1997; 17:7673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Braun N, Sevigny J, Robson SC, Enjyoji K, Guckelberger O, Hammer K et al Assignment of ecto‐nucleoside triphosphate diphosphohydrolase‐1/cd39 expression to microglia and vasculature of the brain. Eur J Neuorsci 2000; 12:4357–66. [PubMed] [Google Scholar]
- 30. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G et al Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G et al Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Investig 2012; 122:3063–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ciruela F, Casado V, Rodrigues RJ, Lujan R, Burgueno J, Canals M et al Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1‐A2A receptor heteromers. J Neurosci 2006; 26:2080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barrie AP, Nicholls DG. Adenosine A1 receptor inhibition of glutamate exocytosis and protein kinase C‐mediated decoupling. J Neurochem 1993; 60:1081–6. [DOI] [PubMed] [Google Scholar]
- 34. d'Alcantara P, Ledent C, Swillens S, Schiffmann SN. Inactivation of adenosine A2A receptor impairs long term potentiation in the accumbens nucleus without altering basal synaptic transmission. Neuroscience 2001; 107:455–64. [DOI] [PubMed] [Google Scholar]
- 35. Popoli P, Betto P, Reggio R, Ricciarello G. Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 1995; 287:215–7. [DOI] [PubMed] [Google Scholar]
- 36. Rebola N, Rodrigues RJ, Lopes LV, Richardson PJ, Oliveira CR, Cunha RA. Adenosine A1 and A2A receptors are co‐expressed in pyramidal neurons and co‐localized in glutamatergic nerve terminals of the rat hippocampus. Neuroscience 2005; 133:79–83. [DOI] [PubMed] [Google Scholar]
- 37. Enjyoji K, Sevigny J, Lin Y, Frenette PS, Christie PD, Esch JS 2nd et al Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat Med 1999; 5:1010–7. [DOI] [PubMed] [Google Scholar]
- 38. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972; 32:281–94. [DOI] [PubMed] [Google Scholar]
- 39. Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 1997; 94:4103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzuki R, Ferris HA, Chee MJ, Maratos‐Flier E, Kahn CR. Reduction of the cholesterol sensor SCAP in the brains of mice causes impaired synaptic transmission and altered cognitive function. PLoS Biol 2013; 11:e1001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu L, Duff K. A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp 2008; 21:960. doi: 10.3791/960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016; 352:712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH et al Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol 2003; 182:21–34. [DOI] [PubMed] [Google Scholar]
- 44. Borges K, McDermott D, Irier H, Smith Y, Dingledine R. Degeneration and proliferation of astrocytes in the mouse dentate gyrus after pilocarpine‐induced status epilepticus. Exp Neurol 2006; 201:416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blumcke I, Vinters HV, Armstrong D, Aronica E, Thom M, Spreafico R. Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord 2009; 11:181–93. [DOI] [PubMed] [Google Scholar]
- 46. Søvik O, Schubbert S, Houge G, Steine SJ, Norgard G, Engelsen B et al De novo HRAS and KRAS mutations in two siblings with short stature and neuro‐cardio‐facio‐cutaneous features. BMJ Case Rep 2009; 2009:bcr07.2008.0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wirenfeldt M, Clare R, Tung S, Bottini A, Mathern GW, Vinters HV. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen's encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol Dis 2009; 34:432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A et al Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007; 204:1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Vries EE, van den Munckhof B, Braun KP, van Royen‐Kerkhof A, de Jager W, Jansen FE. Inflammatory mediators in human epilepsy: a systematic review and meta‐analysis. Neurosci Biobehav Rev 2016; 63:177–90. [DOI] [PubMed] [Google Scholar]
- 50. Okuneva O, Korber I, Li Z, Tian L, Joensuu T, Kopra O et al Abnormal microglial activation in the Cstb–/– mouse, a model for progressive myoclonus epilepsy, EPM1. Glia 2015; 63:400–11. [DOI] [PubMed] [Google Scholar]
- 51. Henshall DC, Engel T. P2X purinoceptors as a link between hyperexcitability and neuroinflammation in status epilepticus. Epilepsy Behav 2015; 49:8–12. [DOI] [PubMed] [Google Scholar]
- 52. Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker‐Cummings C et al Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet 1995; 10:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maliszewski CR, Delespesse GJ, Schoenborn MA, Armitage RJ, Fanslow WC, Nakajima T et al The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. J Immunol 1994; 153:3574–83. [PubMed] [Google Scholar]
- 54. Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD et al Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014; 343:506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R et al Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest 2002; 109:1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J et al The intestinal microbiota affect central levels of brain‐derived neurotropic factor and behavior in mice. Gastroenterology 2011; 141(599–609):e1–3. [DOI] [PubMed] [Google Scholar]
- 57. Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 2012; 10:735–42. [DOI] [PubMed] [Google Scholar]
- 58. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien‐Ly N et al Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 2007; 55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Papaleo F, Silverman JL, Aney J, Tian Q, Barkan CL, Chadman KK et al Working memory deficits, increased anxiety‐like traits, and seizure susceptibility in BDNF overexpressing mice. Learn Mem 2011; 18:534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J et al Neuronal depletion of calcium‐dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease‐related cognitive deficits. Proc Natl Acad Sci USA 2003; 100:9572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kostopoulos GK. Involvement of the thalamocortical system in epileptic loss of consciousness. Epilepsia 2001; 42(Suppl. 3):13–9. [DOI] [PubMed] [Google Scholar]
- 62. Dam AM. Epilepsy and neuron loss in the hippocampus. Epilepsia 1980; 21:617–29. [DOI] [PubMed] [Google Scholar]
- 63. Bengzon J, Mohapel P, Ekdahl CT, Lindvall O. Neuronal apoptosis after brief and prolonged seizures. Prog Brain Res 2002; 135:111–9. [DOI] [PubMed] [Google Scholar]
- 64. Braga AM, Fujisao EK, Verdade RC, Paschoalato RP, Paschoalato RP, Yamashita S et al Investigation of the cingulate cortex in idiopathic generalized epilepsy. Epilepsia 2015; 56:1803–11. [DOI] [PubMed] [Google Scholar]
- 65. Xu P, Xu Y, Hu B, Wang J, Pan R, Murugan M et al Extracellular ATP enhances radiation‐induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav Immun 2015; 50:87–100. [DOI] [PubMed] [Google Scholar]
- 66. Dunwiddie TV. Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia 1980; 21:541–8. [DOI] [PubMed] [Google Scholar]
- 67. Thompson SM, Haas HL, Gahwiler BH. Comparison of the actions of adenosine at pre‐ and postsynaptic receptors in the rat hippocampus in vitro . J Physiol 1992; 451:347–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park KI, Dzhala V, Saponjian Y, Staley KJ. What elements of the inflammatory system are necessary for epileptogenesis in vitro? eNeuro 2015; 2:ENEURO.0027–14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. George J, Goncalves FQ, Cristovao G, Rodrigues L, Meyer Fernandes JR, Goncalves T et al Different danger signals differently impact on microglial proliferation through alterations of ATP release and extracellular metabolism. Glia 2015; 63:1636–45. [DOI] [PubMed] [Google Scholar]
- 70. Farber K, Markworth S, Pannasch U, Nolte C, Prinz V, Kronenberg G et al The ectonucleotidase CD39/ENTPDase1 modulates purinergic‐mediated microglial migration. Glia 2008; 56:331–41. [DOI] [PubMed] [Google Scholar]
- 71. Matyash M, Zabiegalov O, Wendt S, Matyash V, Kettenmann H. The adenosine generating enzymes CD39/CD73 control microglial processes ramification in the mouse brain. PLoS ONE 2017; 12:e0175012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. George J, Cunha RA, Mulle C, Amedee T. Microglia‐derived purines modulate mossy fibre synaptic transmission and plasticity through P2X4 and A1 receptors. Eur J Neurosci 2016; 43:1366–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo . Dev Cell 2012; 23:1189–202. [DOI] [PubMed] [Google Scholar]
- 74. Sayyah M, Javad‐Pour M, Ghazi‐Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience 2003; 122:1073–80. [DOI] [PubMed] [Google Scholar]
- 75. Bynoe MS, Viret C, Yan A, Kim DG. Adenosine receptor signaling: a key to opening the blood–brain door. Fluids Barriers CNS 2015; 12:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang F, Zhao K, Zhang X, Zhang J, Xu B. ATP induces disruption of tight junction proteins via IL‐1β‐dependent MMP‐9 activation of human blood–brain barrier in vitro . Neural Plast 2016; 2016:8928530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oby E, Janigro D. The blood–brain barrier and epilepsy. Epilepsia 2006; 47:1761–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Handling‐induced seizure of score 3.
Movie S2. Handling‐induced seizure of score 2.
Movie S3. Handling induced seizure of score 2 without convulsions.
Movie S4. Wild‐type mouse showing no disturbances upon handling.