

Summary
Antibodies to neuronal antigens are associated with many neurological diseases including paraneoplastic neurological disorders, epilepsy, amyotrophic lateral sclerosis and multiple sclerosis. Immunization with neuronal antigens such as neurofilament light (NF‐L), a neuronal intermediate filament in axons, has been shown to induce neurological disease and spasticity in mice. Also, although antibodies to NF‐L are widely used as surrogate biomarkers of axonal injury in amyotrophic lateral sclerosis and multiple sclerosis, it remains to be elucidated if antibodies to NF‐L contribute to neurodegeneration and neurological disease. To address this, we examined the pathogenic role of antibodies directed to NF‐L in vitro using spinal cord co‐cultures and in vivo in experimental autoimmune encephalomyelitis (EAE) and optic neuritis animal models of multiple sclerosis. Here we show that peripheral injections of antibodies to NF‐L augmented clinical signs of neurological disease in acute EAE, increased retinal ganglion cell loss in experimental optic neuritis and induced neurological signs following intracerebral injection into control mice. The pathogenicity of antibodies to NF‐L was also observed in spinal cord co‐cultures where axonal loss was induced. Taken together, our results reveal that as well as acting as reliable biomarkers of neuronal damage, antibodies to NF‐L exacerbate neurological disease, suggesting that antibodies to NF‐L generated during disease may also be pathogenic and play a role in the progression of neurodegeneration.
Keywords: antibodies, autoimmunity, axonal damage, neurodegeneration, neurofilament light
Abbreviations
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- mAb
monoclonal antibody
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NF‐L
neurofilament light
- RGC
retinal ganglion cells
- RNFL
retinal nerve fibre layer
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) that has a strong neurodegenerative component.1 Several studies show that current therapies targeting the adaptive immune pathways are effective in people with MS who have active inflammatory lesions, but such approaches fail to show efficacy in progressive disease associated with irreversible axonal damage and neuronal loss. However, both B and T cells and immunoglobulin deposits are present in the CNS in primary and secondary progressive MS irrespective of the disease duration,2 indicating that immune responses may continue to play a role in progressive disease.
Although the aetiology of MS is still unclear, T cells and immunoglobulins directed towards myelin and axonal proteins as well as myelin‐associated antigens such as the heat‐shock protein hsp B5 are present in peripheral blood and the cerebrospinal fluid of people with MS,3, 4, 5, 6, 7 though some studies also show that these are also present in healthy controls.7, 8, 9 A clear role for immune responses in MS is evidenced by the efficacy of immunotherapy as a treatment strategy.10 Additional evidence comes from the similarities between experimental autoimmune encephalomyelitis (EAE) and MS. In EAE, autoimmune responses to myelin proteins and peptides11, 12, 13 as well as spinal cord homogenates14 induce chronic relapsing neurological disease and demyelination that is enhanced in the presence of demyelinating antibodies.15, 16
In the cerebrospinal fluid of people with MS, the presence of intrathecal oligoclonal antibody production has led to a search for the specificity of these antibodies based on the concept that they are involved in the pathogenesis of MS. Some of these antibodies are directed to the neuronal cytoskeleton protein neurofilament light (NF‐L) and are increased in the cerebrospinal fluid17 and serum18 in progressive MS. Likewise, CNS‐resident B cells have shown characteristics of an antigen‐driven response, including specificity to NF‐L.19 However, the question arises whether antibodies to CNS antigens such as NF‐L are pathogenic. Previously, we have shown that mice immunized with NF‐L develop neurological disease characterized by spasticity and limb paralysis associated with axonal degeneration and inflammation in the dorsal column and the grey matter.8, 20, 21 Furthermore, in these mice immunoglobulin is observed inside the axons of mice immunized with NF‐L, which develop neurological disease.8 This finding is in line with the observations that neuronal intracellular antigens are targets of specific humoral immune responses.22, 23, 24, 25, 26 However, in the NF‐L‐induced spasticity it was unclear whether the antibodies directed to NF‐L were themselves responsible for the pathogenicity in this model. In humans, it has recently been shown that IgG and IgM localize with neurofilaments in MS lesions.27 Nonetheless, the contribution of antibodies to NF‐L in MS remains unclear.
Here, we examined the pathogenic effect of antibodies directed to NF‐L in vitro using spinal cord co‐cultures and in vivo in mice. We show that a monoclonal antibody (mAb) to NF‐L induced axonopathy in rat dissociated spinal cord co‐cultures, promoted neurodegeneration in a mouse model of optic neuritis, exacerbated active EAE in mice and induced neurological signs after intracerebral injection in naive mice. Taken together, our results indicate a pathogenic role of antibodies to neuronal antigens and show that antibodies to NF‐L exacerbate neuronal damage in experimental disease. These findings indicate that NF‐L antibodies might also contribute to neurodegeneration in MS.
Materials and methods
Animals
Mice
For EAE studies Biozzi ABH (H‐2dq1) female and male mice between 8 and 10 weeks old were obtained from Harlan Ltd (UK) or bred at Queen Mary University of London (UK). For optical coherence tomography and retinal ganglion cell (RGC) analysis, 10‐week‐old female and male transgenic C57BL/6‐Tg(Tcra2D2,Tcrb2D2)1Kuch.Cg‐Tg(Thy‐1‐CFP)23Jrs/J mice expressing T‐cell receptors specific for myelin oligodendrocyte glycoprotein (MOG) and RGC expressing cyan fluorescent protein (CFP) were used (about 25% of the offspring).28 Animals were housed in a temperature‐controlled room (25°) and given access to food and water ad libitum.29 All procedures were performed following institutional ethical review in accordance with the United Kingdom Animals (Scientific Procedures) Act (1986), European Union Directive 2010/63/EU and the German Review Board for the Care of Animal Subjects of the district government (North Rhine‐Westphalia, Germany) under the recommendations of the Federation of European Laboratory Animal Science Associations. Further details relating to animal husbandry and operational experimental design relating to the ARRIVE guidelines reporting have been described previously.29
Rats
Time‐mated Sprague‐Dawley rats30, 31 purchased from Charles River (Germany) were housed in a temperature‐controlled room and given access to food and water ad libitum. All experimental procedures were reviewed and approved by the Ethical Committee for Animal Experiments of the VU Medical Centre, Amsterdam, the Netherlands (PA 13‐01).
Human subjects
People with relapsing–remitting MS were recruited at The Royal London Hospital, London, UK and La Fe Polytechnic and University Hospital in Valencia, Spain. People with secondary progressive MS were recruited from the National Hospital for Neurology and Neurosurgery, London, and the Royal Free Hospital, London, UK (see Supplementary material, Table S1). Sera were obtained with informed consent from the donors. All procedures were approved by the local ethical committees. Ethical approval was obtained from the North London REC 2 (10/H0724/36), Hospital Universitario La Fe Committee, University College London Hospitals Committee and National Research Ethics Committee (REC:06/Q0512/16, EudraCT: 2005‐005588‐27). All studies were carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
Antibodies
The mouse mAb to NF‐L used in the current study, NF‐L 10H9, has been described previously.4 The affinity‐purified anti‐human NF‐L rabbit polyclonal antibody was obtained from Abbexa (Cambridge, UK). Total human IgG preparations were purified from the serum of either people with MS who showed high reactivity to NF‐L protein in an ELISA (see Supplementary material, Table S1) or healthy controls. Pooled IgG fractions were diluted 1 : 10 in PBS and purified using a 1 ml HiTrap protein‐G HP column (GE Healthcare, Chalfont St Giles, UK). IgG bound to the column was eluted with 0·1 m citric acid (pH 3·0) and neutralized with 1 m Tris–HCl (pH 9·1). Samples were concentrated using Amicon® Ultra centrifugal filters (UFC501096, Millipore, Feltham, UK) and the buffer was exchanged for PBS. The IgG concentration was measured using a NanoDrop ND‐1000 spectrophotometer.
Western blotting
Western blotting was carried out to show the reactivity of NF‐L 10H9,4 and purified human IgG to spinal cord, brain homogenates and recombinant NF‐L. Serum from NF‐L immunized ABH mice32 was used as a positive control. Briefly, 0·49 mg/ml recombinant mouse NF‐L, or 10 μg mouse spinal cord or brain homogenate were separated using Mini‐Protean TGX Precast Gel (Bio‐Rad, Munich, Germany) and transferred onto Whatman nitrocellulose membranes. The nitrocellulose membranes were blocked with 5% fat‐free milk solution and 0·5% bovine serum albumin for 1 hr. Membranes were incubated with NF‐L 10H9 (10 μg/ml) or human IgG (50 μg/ml) overnight. After washing, goat anti‐mouse IgG‐horseradish peroxidase (Dako, Glostrup, Denmark) was added to the membranes and incubated for 1 hr at room temperature. Visualization was performed with the chromogen 3,3′‐diaminobenzidine (Dako) and enhanced chemiluminescence (Amersham, Bucks, UK).
Induction of EAE and administration of NF‐L antibodies
ABH mice were injected subcutaneously with 200 μg MOG35–55 peptide emulsified with incomplete Freund's adjuvant (Difco Laboratories, Oxford, UK) supplemented with 48 μg Mycobacterium tuberculosis and 6 μg Mycobacterium butyricum (Difco Laboratories) on days 0 and 7, as described previously.11, 29 Mice were also injected intraperitoneally with 200 ng of Bordetella pertussis toxin (Sigma, Poole, UK) immediately after the immunization and at 24 hr and 8 days after immunization. Mice were intraperitoneally injected with 0·5 mg NF‐L 10H9 or 0·5 mg human IgG from day 10 every other day until day 26. Mice were monitored daily and scored according to a neurological scale with 0 = normal, 1 = limp tail, 2 =impaired righting reflex, 3 = hind limb paresis and 4 = hind limb paralysis.8, 32
Induction of optic neuritis
Transgenic mice were used to study whether NF‐L 10H9 induced loss of RGC as described previously with anti‐myelin antibodies.28 Optic neuritis was induced in MOG‐specific T‐cell receptor transgenic mice with fluorescent RGC by injection with 150 ng B. pertussis toxin (Sigma) on day 0 and day 2.28 Subsequently, mice were randomly divided into two groups. One group (Group A) was injected with 0·5 mg fluorescently labelled NF‐L 10H9‐phycoerythrin on days 11, 12 and 13 and killed on day 15. The other group (Group B) was injected with 0·5 mg mAb unlabelled NF‐L 10H9 on days 14, 16 and 18 and killed at day 20. As a negative control, mice were injected with isotype control IgG1 (Sigma). Optical coherence tomography was used to measure changes in the retinal nerve fibre layer (RNFL) thickness comparing measurements before B. pertussis toxin injection (day 0) and after injection of antibodies to NF‐L (days 15 and 20).28 Retinal flat‐mounts were prepared for fluorescence microscopy and RGC density was calculated as described previously.28
Intracerebral administration of NF‐L antibodies
Intracerebral injection of NF‐L 10H9, mAb MOG Z1233 and human IgG was performed in female ABH mice anaesthetized by inhalation with isoflurane. Briefly, 30 μg of NF‐L 10H9, mAb MOG Z12 and 180 μg human IgG from people with MS or healthy controls was administered intracerebrally to the forebrain of mice.34
PC12 cell viability
Undifferentiated PC12 cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum (Invitrogen) and 10% horse serum (Sigma), 100 μg/ml streptomycin, 100 U/ml penicillin (Invitrogen) and incubated at 37° in a 5% CO2 humidified atmosphere. The cells were differentiated by plating at a density of 1 × 105 in 96‐well plates (Nunc, Thermofisher, UK) in Dulbecco's modified Eagle's medium with 0·1% horse serum supplemented with nerve growth factor (50 ng/ml). After 48 hr, the differentiated PC12 cells were treated for 24, 48 or 72 hr with 20 μg/ml of either rabbit IgG directed to human NF‐L (Abbexa, Cambridge, UK), rabbit IgG as control (Sigma) or medium only. The cell viability was determined using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. For analysis the treated cells were incubated with MTT (0·5 mg/ml) during the final 4 hr of incubation, the supernatants were discarded and 200 μl DMSO was added to solubilize the formazan crystals. The absorbance was determined at 590 nm using an ELISA reader (ELISA, Synergy HT microplate reader). The percentage of cell viability was calculated as the absorbance of experimental group/absorbance of control (media only).
Axonal density in myelinating spinal cord cultures
Myelinating co‐cultures were prepared as described previously.30, 31 Briefly, spinal cords were dissected from E15 rat pups and enzymatically dissociated. Cells were plated on a confluent monolayer of neurosphere‐derived astrocytes30 and allowed to develop to form a co‐culture containing axons, glia and the various cell types that make up the spinal cord for their detailed study. After 23–24 days, the co‐cultures were treated with the mAb to NF‐L. Spinal cord co‐cultures were incubated with 10 μg/ml NF‐L 10H9 or the isotype control (mouse IgG1, Sigma) for 0·5–48 hr at 37° in the presence of 5% CO2 in duplicate as described previously.35 Cultures were stained for phosphorylated axons/neurites with unlabelled anti‐mouse SMI310 IgG1 and goat‐anti mouse‐ Alexa Fluor‐594 F(ab')2 fragment was used as secondary antibody (1 : 1500; Abcam, Cambridge, UK). Cells were imaged on a Leica DMR fluorescence microscope using a Leica DFC4000 camera. Briefly, 20 images were analysed per condition using imageJ software (NIH systems, version 1.41o). For each time‐point, three independent myelinating co‐cultures were used and per condition the images were analysed (10 per coverslip, two coverslips per condition). The number of pixels of SMI310+ neurites was calculated and divided by the total number of pixels per field of view, resulting in the percentage of axonal density. Subsequently, for each time point, the percentage of axonal density from NF‐L 10H9‐treated co‐cultures was calculated in relation to the isotype control IgG1‐treated co‐cultures, resulting in the percentage of axonal loss.
Statistics
Data were analysed using graphpad Prism (5.01) and expressed as means ± standard error of the mean (SEM) unless otherwise specified. RNFL thinning and RGC density were analysed using Student's t‐test. Statistical analysis of clinical scores was assessed using the non‐parametric Mann–Whitney U‐test. Axonal density of rat spinal cord co‐cultures was quantified with imageJ and analysed with graphpad prism (5.01) using the Student's t‐test. P values of < 0·05 were considered statistically significant.
Results
Specificity of mAb to NF‐L
The specificity of NF‐L clone 10H9 was verified in Western blot analysis using mouse spinal cord, brain homogenate and recombinant NF‐L protein (Fig. 1). Similarly, human IgG purified from selected people with MS with high levels of NF‐L antibodies, showed reactivity mainly to a protein at the molecular weight of the linear NF‐L protein (68 000 MW) (see Supplementary material, Fig. S1).
Figure 1.
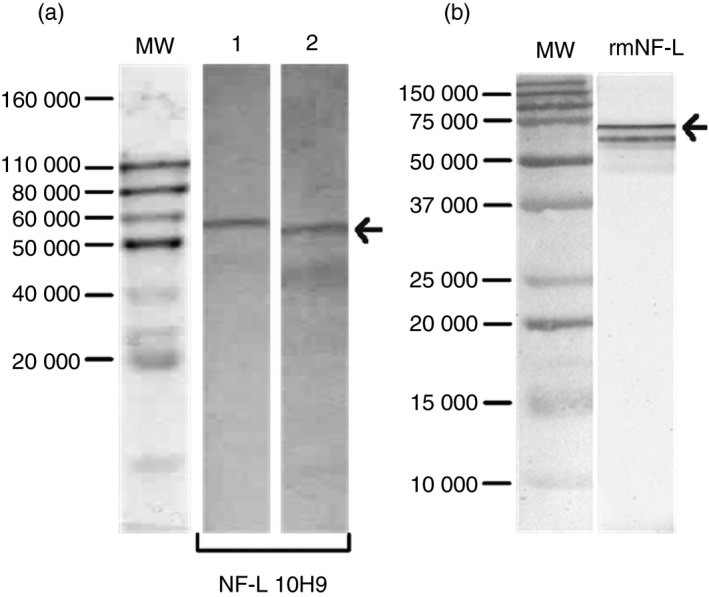
Specificity of the monoclonal antibody NF‐L 10H9. (a) Western blot analysis illustrates the specificity of NF‐L 10H9 to neurofilament light (NF‐L) protein (68 000 MW). Lane 1 shows the reactivity to spinal cord and lane 2 the reactivity to mouse brain homogenate. (b) Reactivity to the recombinant NF‐L protein. MW, protein standard.
Antibodies to NF‐L exacerbate MOG35–55‐induced EAE
To examine the in vivo pathogenic effect of NF‐L 10H9 antibody, mice were immunized with MOG35–55 (n = 11) and injected intraperitoneally with NF‐L 10H9 (n = 6) every other day from 10 days post‐immunization. The cumulative EAE score in mice immunized with MOG35–55 and injected with NF‐L 10H9 was significantly increased compared with control EAE mice (P = 0·01; Table 1). Likewise, injection of serum IgG from people with MS resulted in an early onset of clinical signs of disease compared with untreated mice (11·8 ± 1·0 days versus 14·1 ± 2·1 days; P < 0·05) (see Supplementary material,Fig. S2).
Table 1.
Effect of neurofilament light (NF‐L) ‐specific antibodies in myelin oligodendrocyte glycoprotein 35–55 (MOG35–55)‐induced experimental autoimmune encephalomyelitis (EAE)
| Treatment | No. EAE | Mean group scorea | Mean EAE scoreb | Mean day of onsetc | AUCd** |
|---|---|---|---|---|---|
| MOG35–55 | 9/11 | 3·3 ± 1·6 | 4·0 ± 0·0 | 14·1 ± 2·1 | 46·4 ± 1·1 |
| MOG35–55 + NF‐L 10H9 | 6/6 | 4·0 ± 0·0 | 4·0 ± 0·0 | 12·3 ± 1·0 | 63·2 ± 1·4** |
Mean ± SEM of maximum clinical score of EAE from all mice in the group.
Mean ± SEM of maximum clinical score from mice exhibiting EAE within a group.
Mean ± SD of day of onset of clinical disease.
AUC ± SD Cumulative scores are expressed as area under the curve (AUC). Cumulative clinical scores were calculated as the sum of all the daily scores over the course of EAE, **P = 0·01.
RGC density is significantly affected by antibodies to NF‐L
As the majority of people with MS develop ocular deficits, we investigated whether the NF‐L 10H9 antibodies were pathogenic to RGCs. Damage to the RGC can occur as a downstream consequence of damage to the optic nerve during optic neuritis.36 Delivery of fluorescently labelled NF‐L 10H9‐phycoerythrin after the onset of optic neuritis caused a significant reduction in the mean RGC density at 20 days post‐induction (group B), compared with mice injected with the isotype IgG1 control (P < 0·05). This effect was not apparent at 15 days (Fig. 2a, group A). NF‐L 10H9 had no significant effect on the thickness of the RNFL in mice with optic neuritis (Fig. 2b).
Figure 2.
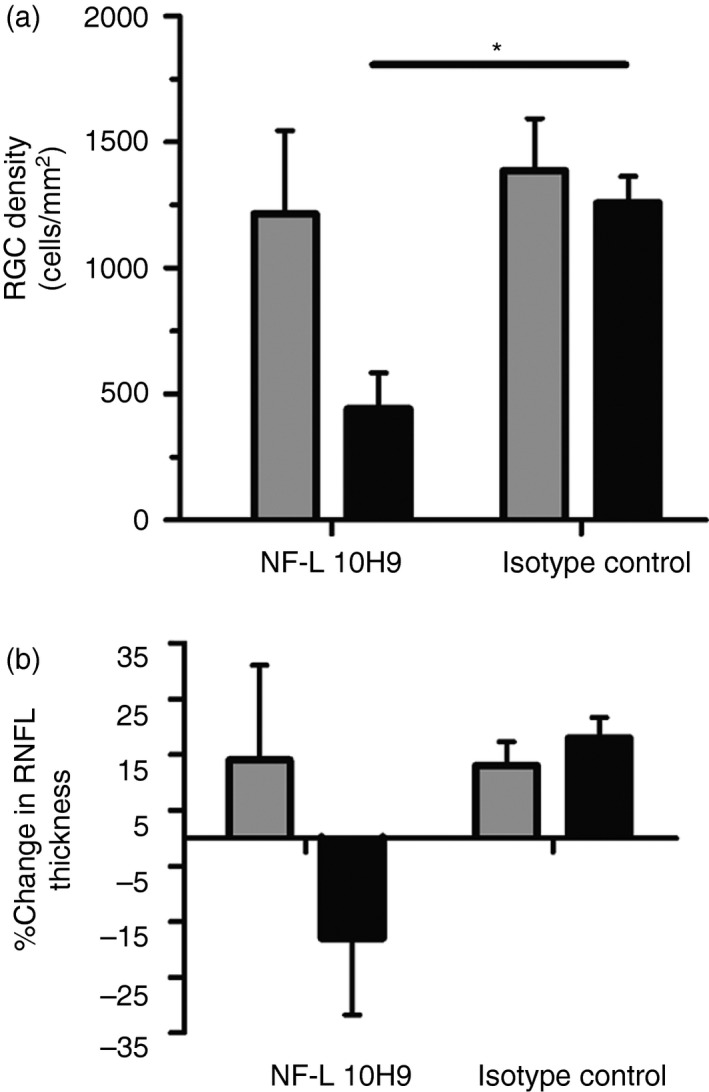
Retinal ganglion cell (RGC) density is significantly affected by NF‐L 10H9. (a) RGC density and (b) retinal nerve fibre layer (RNFL) thickness at 15 days (group A, grey bars, n = 3 mice per group, n = 2 for isotype control) or at 20 days (group B, black bars, n = 2 mice per group) after treatment with (un)labelled NF‐L 10H9 or isotype control IgG1. (a) Only mice in group B treated with unlabelled NF‐L 10H9 showed a statistically significant reduction in RGC density compared with mice treated with unlabelled isotype control IgG1 (Student's t‐test, P < 0·05). (b) No significant differences in percentage change in RNFL thickness were observed in NF‐L 10H9‐treated mice 15 or 20 days post‐induction. *P <0·05
Intracerebral injection of antibodies to NF‐L causes transient neurological signs
To further investigate the pathogenicity of antibodies to NF‐L, NF‐L 10H9 was injected intracerebrally into naive ABH mice. Significant neurological symptoms were apparent after recovery from the anaesthetic (see Supplementary material, Fig. S3a,b). This effect was also observed following injection of human IgG from people with MS (see Supplementary material, Fig. S3c). The neurological signs included stiffness of hind limbs, tail stiffness, paresis of the hind limbs and subsequent movement abnormalities. In contrast, mice injected with mouse serum, mAb MOG Z12 or saline solution did not develop signs of disease.
Antibodies to NF‐L decrease neuronal viability
The pathogenic effect of antibodies to NF‐L was examined using the PC12 cell line. No effect of the control rabbit immunoglobulin was observed over the study. In contrast a neurotoxic effect of NF‐L antibody was observed within 24 hr that decreased the cell viability compared with control rabbit IgG (P < 0·01) and medium only (P < 0·001) (Fig. 3). This was mirrored at 48 and 72 hr. In contrast, the control IgG antibody had no significant effect on cell viability.
Figure 3.
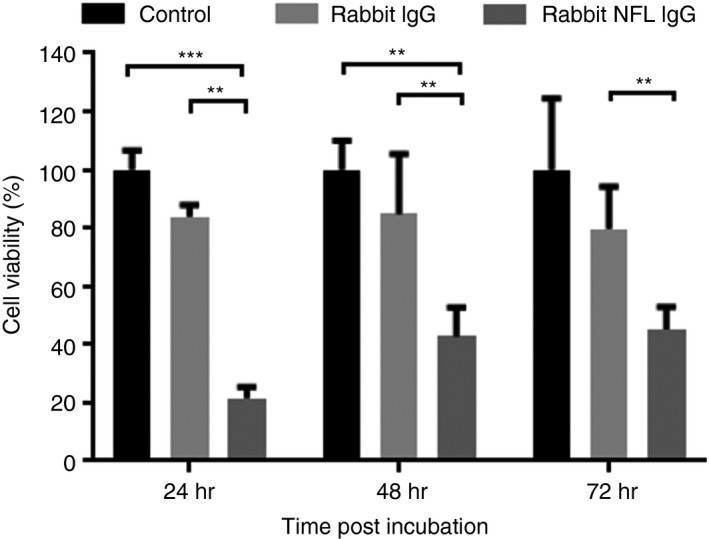
Effect of an anti‐human neurofilament light (NF‐L) antibody on neuronal viability. Differentiated PC12 cells were treated for 24, 48 and 72 hr with 20 μg/ml rabbit anti‐human NF‐L antibody or rabbit IgG as a control of the assay. Cells were incubated with MTT (0·5 mg/ml) for the last 4 hr of incubation, then supernatants were discarded and 200 μl DMSO was added to solubilize the formazan crystals. Absorbance was determined at 590 nm using an ELISA reader. ***P < 0·001, **P < 0·01.
Pathogenic effect of mAb to NF‐L in spinal cord cultures
To further examine the in vivo effect of NF‐L antibodies the antibody 10H9 was applied to rat spinal cord co‐cultures and the density of phosphorylated axons and neurites was used as a measure of axonal damage as described previously.31, 35, 37 The monoclonal NF‐L 10H9 induced axonal loss in the culture system relative to the isotype control IgG1 (Fig. 4a; n = 3 or n = 4; P < 0·05). After 1 hr of incubation with NF‐L 10H9, axonal loss was significantly increased compared with isotype control‐treated cultures (P < 0·001; Fig. 4a). Representative immunofluorescent images of cultures incubated with isotype control IgG1 (Fig. 4b) and NF‐L 10H9 (Fig. 4c) reveal significant loss of the axonal network.
Figure 4.
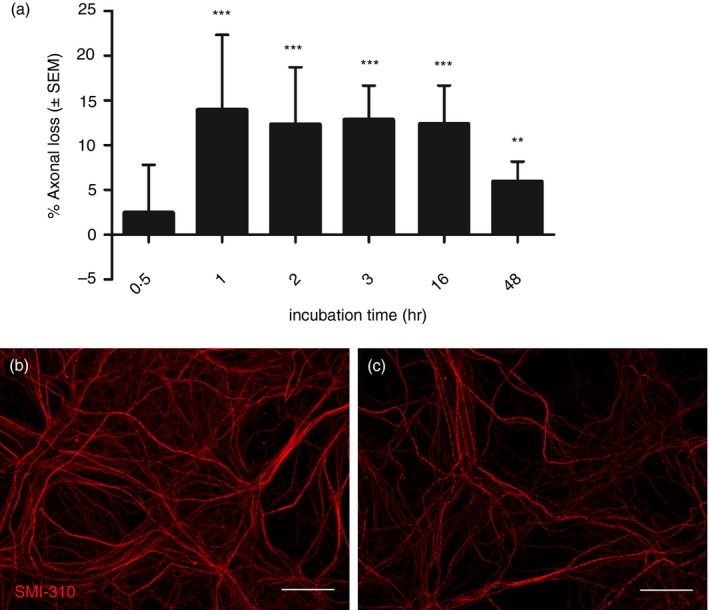
Monoclonal antibody (mAb) to neurofilament light (NF‐L) reduces axonal density in rat spinal cord co‐cultures. (a) Co‐cultures were treated with NF‐L 10H9 or isotype control for 0·5, 1, 2, 3, 16 or 48 hr with 10 μg/ml mAb. Subsequently, cultures were stained for SMI310 and axonal density was determined using imageJ and represented as percentage axonal loss relative to the isotype control IgG1. Axonal loss was significant after 1, 2, 3, 16 or 48 hr of treatment with NF‐L 10H9 (n = 4 for 0·5 and 48 hr, n = 3 for 1–16 hr, ***P < 0·001, **P < 0·01, *P < 0·05, Student's t‐test). Representative immunofluorescent images of cultures treated with (b) 10 μg/ml isotype control IgG1 or (c) NF‐L 10H9 for 16 hr and stained with SMI310 (scale bars = 35 μm). [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Antibodies to NF‐L have been previously evaluated as surrogate biomarkers of neuronal damage. We show, for the first time, that antibodies to neurofilaments contribute to neuronal damage. Our data indicate that antibodies to NF‐L might be involved in disease progression in neurodegenerative diseases such as MS and other neurodegenerative diseases in which antibodies to NF‐L have been reported.17, 18, 38 Hence, therapeutic approaches targeting humoral immunity to neurons and axons could be beneficial in a variety of neurological disorders. There is some evidence of the efficacy of this approach already. In MS therapeutic strategies, targeting B cells reduces disease severity39, 40 although such efficacy might not be due to a direct effect of reducing autoantibodies. In general, subgroups of people with MS respond well to plasma exchange,41, 42, 43 indicating that pathogenic antibodies contribute to disease.
Although people with MS experience various symptoms, visual problems commonly precede other neurological symptoms. Optical coherence tomography can be used to assess damage to the retina. Here, we used a new optic neuritis model28 to investigate whether mAb to NF‐L have a pathogenic effect on RNFL thickness and RGC density consequent to damage of axons within the optic nerve. Similar to effects noted with injection of mAb to MOG,28 injection of NF‐L 10H9 into animals with optic neuritis significantly reduced RGC density 20 days post‐injection. The significance of antibodies to NF‐L in people with MS who develop optic neuritis has not been accounted before and deserves further investigation.
Here we also show early onset of disease and exacerbation of MOG35–55‐induced EAE, following injection of IgG purified from sera of multiple people with MS with high levels of NF‐L antibodies (see Supplementary material, Fig. S1), and a significant increase in the cumulative score induced following injection with the mAb NF‐L. This suggests that antibodies to neuronal antigens, such as those to NF‐L, may elicit an additional reaction causing neuronal damage. The differential effect on the EAE course observed after injection of the mAb NF‐L or human IgG could be due to differences in specificity and therefore pathogenicity of these antibodies in people with MS compared with the mouse mAb. This is in line with previous studies by Pedotti et al.16 showing that IgG purified from plasma from a patient with relapsing–remitting MS exacerbated proteolipid protein‐induced EAE in mice. Absorption and affinity studies are required to identify the contribution of the NF‐L antibodies as well as other antibodies present in the purified human IgGs. Nevertheless, we have previously shown that autoimmune T‐cell responses to neuronal antigens augment experimental autoimmune disease32 whereas other reports show that antibodies to neurofascin augments neuronal damage3 underscoring the potential importance of autoimmunity to neurons in humans.
To overcome the influence of the blood–brain barrier in the penetration of antibodies into the CNS, mAb were injected intracerebrally in mice. These antibodies induced the development of MS‐like symptoms, including ataxia, spasticity and paralysis. Some studies have shown that antibodies to an intracellular antigen have the ability to enter neurons, causing depletion of adenosine triphosphate and increase of caspase levels that could lead to apoptosis and cell death.44 This mechanism together with an elevation in intracellular Ca2+ mediated by anti‐α‐enolase antibodies has also been described in autoimmune retinopathy.45 In addition, activation of FcγRI by IgG–immune complex has been shown to cause an increase in intracellular calcium and enhance excitability in sensory neurons.46 Patch‐clamping might be used to investigate whether mAb to NF‐L impact on neuronal signalling. Our data also show that both NF‐L‐specific and isotype antibodies can be internalized by neurons (data not shown). Once internalized, antibodies to NF‐L might disturb axonal transport processes by binding to their target. Here we show that an anti‐human NF‐L antibody decreased remarkably the viability of differentiated neurons as measured in an MTT assay that reflects the amount of mitochondrial activity. We also show that mAb to NF‐L reduced axonal density in rat spinal cord co‐cultures, which might be the consequence of failure of proper axonal transportation. The ability of antibodies directed to intracellular neuronal targets to disrupt axonal transport has been shown before.47, 48
Further research needs to be performed to determine the mechanism by which antibodies to NF‐L cause cytotoxicity. It is also possible that antibody–antigen complexes of neurofilament proteins and antibodies crosslink with Fc‐receptors on immune cells, which can trigger phagocytosis, antibody‐dependent cell‐mediated cytotoxicity and cytokine release49 thereby contributing to progression in neuroinflammatory conditions.
Our previous studies show that antibodies to NF‐L are diminished in response to disease‐modifying therapies in MS50 and are also correlated with fast progression of amyotrophic lateral sclerosis,38 which further suggest a pathogenic role of these antibodies. In this study, we expand some of our previous observations of the presence of immunoglobulin deposits within axons in lesions of mice exhibiting clinical disease.8 Our results show that antibodies to intracellular antigens contribute to axonal damage and are not just surrogate markers of disease. This may open novel potential therapeutic targets for antibodies interfering with these responses.
Authors’ contribution
FP, BJS and SA contributed to the conception and design of the study. FP, BJS, SB, MK, LB, JS and SCB performed the experiments. IB, JR and SG provided the samples from MS patients. FP, BJS, SB, MK, LB, IB, JS, JR, SG, PV, SCB, DB and SA contributed to the interpretation of the results, writing and revision of the manuscript.
Disclosures
The authors declare that there are no conflicts of interest.
Supporting information
Figure S1. Specificity of IgG from multiple sclerosis patient serum.
Figure S2. Exacerbation of myelin oligodendrocyte glycoprotein 35–55 (MOG35–55)‐induced experimental autoimmune encephalomyelitis (EAE) by human IgG.
Figure S3. Intracerebral injection of monoclonal antibody to neurofilament light (NF‐L) causes transient neurological signs.
Table S1. Clinical data of multiple sclerosis patients.
Acknowledgements
We thank Wouter Gerritsen for his exceptional technical skills and Kamilla Nurtaeva and Adib Zendedel for excellent technical assistance with stereotactic injections. PC12 cells were kindly provided by Dr Alexandra Chittka. We also thank Katie Lidster for technical assistance on optic neuritis studies. This work was supported by Stichting MS Research, the Netherlands (grant number 07–627), the DANA Foundation and by the MS Society of the United Kingdom (grant number NSCG‐1F7R).
Senior author: Prof. Sandra Amor
Email: s.amor@vumc.nl
References
- 1. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008; 31:247–69. [DOI] [PubMed] [Google Scholar]
- 2. Frischer JM, Bramow S, Dal‐Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009; 132:1175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Derfuss T, Parikh K, Velhin S, Braun M, Mathey E, Krumbholz M et al Contactin‐2/TAG‐1‐directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci U S A 2009; 106:8302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huizinga R, van der Star BJ, Kipp M, Jong R, Gerritsen W, Clarner T et al Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia 2012; 60:422–31. [DOI] [PubMed] [Google Scholar]
- 5. Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR et al Neurofascin as a novel target for autoantibody‐mediated axonal injury. J Exp Med 2007; 204:2363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson EJ, Kaufmann P, Shortman RC, Rudge P, McDonald WI. Oligoclonal immunoglobulins and plasma cells in spinal fluid of patients with multiple sclerosis. Br Med J 1979; 1:16–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Noort JM, Van Sechel AC, Bajramovic JJ, El Ouagmiri M, Polman CH, Lassmann H et al The small heat‐shock protein α B‐crystallin as candidate autoantigen in multiple sclerosis. Nature 1995; 375:798–801. [DOI] [PubMed] [Google Scholar]
- 8. Huizinga R, Heijmans N, Schubert P, Gschmeissner S, ‘t Hart BA, Herrmann H et al Immunization with neurofilament light protein induces spastic paresis and axonal degeneration in Biozzi ABH mice. J Neuropathol Exp Neurol 2007; 66:295–304. [DOI] [PubMed] [Google Scholar]
- 9. Van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L et al αb‐crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 2010; 69:694–703. [DOI] [PubMed] [Google Scholar]
- 10. Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372:1502–17. [DOI] [PubMed] [Google Scholar]
- 11. Amor S, Baker D, Groome N, Turk JL. Identification of a major encephalitogenic epitope of proteolipid protein (residues 56–70) for the induction of experimental allergic encephalomyelitis in Biozzi AB/H and nonobese diabetic mice. J Immunol 1993; 150:5666–72. [PubMed] [Google Scholar]
- 12. Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV et al Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol 1994; 153:4349–56. [PubMed] [Google Scholar]
- 13. Amor S, O'Neill JK, Morris MM, Smith RM, Wraith DC, Groome N et al Encephalitogenic epitopes of myelin basic protein, proteolipid protein, myelin oligodendrocyte glycoprotein for experimental allergic encephalomyelitis induction in Biozzi ABH (H‐2Ag7) mice share an amino acid motif. J Immunol 1996; 156:3000–8. [PubMed] [Google Scholar]
- 14. Baker D, O'Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol 1990; 28:261–70. [DOI] [PubMed] [Google Scholar]
- 15. Morris‐Downes MM, Smith PA, Rundle JL, Piddlesden SJ, Baker D, Pham‐Dinh D et al Pathological and regulatory effects of anti‐myelin antibodies in experimental allergic encephalomyelitis in mice. J Neuroimmunol 2002; 125:114–24. [DOI] [PubMed] [Google Scholar]
- 16. Pedotti R, Musio S, Scabeni S, Farina C, Poliani PL, Colombo E et al Exacerbation of experimental autoimmune encephalomyelitis by passive transfer of IgG antibodies from a multiple sclerosis patient responsive to immunoadsorption. J Neuroimmunol 2013; 262:19–26. [DOI] [PubMed] [Google Scholar]
- 17. Silber E, Semra YK, Gregson NA, Sharief MK. Patients with progressive multiple sclerosis have elevated antibodies to neurofilament subunit. Neurology 2002; 58:1372–81. [DOI] [PubMed] [Google Scholar]
- 18. Ehling R, Lutterotti A, Wanschitz J, Khalil M, Gneiss C, Deisenhammer F et al Increased frequencies of serum antibodies to neurofilament light in patients with primary chronic progressive multiple sclerosis. Mult Scler 2004; 10:601–6. [DOI] [PubMed] [Google Scholar]
- 19. Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O'Connor KC. Investigating the antigen specificity of multiple sclerosis central nervous system‐derived immunoglobulins. Front Immunol 2015; 6:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huizinga R, Gerritsen W, Heijmans N, Amor S. Axonal loss and gray matter pathology as a direct result of autoimmunity to neurofilaments. Neurobiol Dis 2008; 32:461–70. [DOI] [PubMed] [Google Scholar]
- 21. Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben‐Nun A et al Myelin‐specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med 2009; 15:626–32. [DOI] [PubMed] [Google Scholar]
- 22. Stein TD, Fedynyshyn JP, Kalil RE. Circulating autoantibodies recognize and bind dying neurons following injury to the brain. J Neuropathol Exp Neurol 2002; 61:1100–8. [DOI] [PubMed] [Google Scholar]
- 23. Tampellini D, Magrane J, Takahashi RH, Li F, Lin MT, Almeida CG et al Internalized antibodies to the Aβ domain of APP reduce neuronal Aβ and protect against synaptic alterations. J Biol Chem 2007; 282:18895–906. [DOI] [PubMed] [Google Scholar]
- 24. Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T et al Stiff person syndrome‐associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 2010; 133:3166–80. [DOI] [PubMed] [Google Scholar]
- 25. Fewou SN, Rupp A, Nickolay LE, Carrick K, Greenshields KN, Pediani J et al Anti‐ganglioside antibody internalization attenuates motor nerve terminal injury in a mouse model of acute motor axonal neuropathy. J Clin Invest 2012; 122:1037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michalak Z, Lebrun A, Di Miceli M, Rousset MC, Crespel A, Coubes P et al IgG leakage may contribute to neuronal dysfunction in drug‐refractory epilepsies with blood–brain barrier disruption. J Neuropathol Exp Neurol 2012; 71:826–38. [DOI] [PubMed] [Google Scholar]
- 27. Sadaba MC, Tzartos J, Paino C, Garcia‐Villanueva M, Alvarez‐Cermeno JC, Villar LM et al Axonal and oligodendrocyte‐localized IgM and IgG deposits in MS lesions. J Neuroimmunol 2012; 247:86–94. [DOI] [PubMed] [Google Scholar]
- 28. Lidster K, Jackson SJ, Ahmed Z, Munro P, Coffey P, Giovannoni G et al Neuroprotection in a novel mouse model of multiple sclerosis. PLoS ONE 2013; 8:e79188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al‐Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S et al Practical guide to the mouse induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH. Mult Scler Relat Disord 2012; 1:29–38. [DOI] [PubMed] [Google Scholar]
- 30. Sorensen A, Moffat K, Thomson C, Barnett SC. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro . Glia 2008; 56:750–63. [DOI] [PubMed] [Google Scholar]
- 31. Boomkamp SD, Riehle MO, Wood J, Olson MF, Barnett SC. The development of a rat in vitro model of spinal cord injury demonstrating the additive effects of Rho and ROCK inhibitors on neurite outgrowth and myelination. Glia 2012; 60:441–56. [DOI] [PubMed] [Google Scholar]
- 32. Puentes F, van der Star BJ, Victor M, Kipp M, Beyer C, Peferoen‐Baert R et al Characterization of immune response to neurofilament light in experimental autoimmune encephalomyelitis. J Neuroinflammation 2013; 10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van der Goes A, Kortekaas M, Hoekstra K, Dijkstra CD, Amor S. The role of anti‐myelin (auto)‐antibodies in the phagocytosis of myelin by macrophages. J Neuroimmunol 1999; 101:61–7. [DOI] [PubMed] [Google Scholar]
- 34. Baker D, O'Neill JK, Davison AN, Turk JL. Control of immune‐mediated disease of the central nervous system requires the use of a neuroactive agent: elucidation by the action of mitoxantrone. Clin Exp Immunol 1992; 90:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J et al Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain 2012; 135:1819–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lidster K, Baker D. Optical coherence tomography detection of neurodegeneration in multiple sclerosis. CNS Neurol Disord Drug Targets 2012; 11:518–27. [DOI] [PubMed] [Google Scholar]
- 37. Thomson CE, Hunter AM, Griffiths IR, Edgar JM, McCulloch MC. Murine spinal cord explants: a model for evaluating axonal growth and myelination in vitro . J Neurosci Res 2006; 84:1703–15. [DOI] [PubMed] [Google Scholar]
- 38. Puentes F, Topping J, Kuhle J, van der Star BJ, Douiri A, Giovannoni G et al Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2014; 85:274–8. [DOI] [PubMed] [Google Scholar]
- 39. Gredler V, Mader S, Schanda K, Hegen H, Di Pauli F, Kuenz B et al Clinical and immunological follow‐up of B‐cell depleting therapy in CNS demyelinating diseases. J Neurol Sci 2013; 328:77–82. [DOI] [PubMed] [Google Scholar]
- 40. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al B‐cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med 2008; 358:676–88. [DOI] [PubMed] [Google Scholar]
- 41. Keegan M, Konig F, McClelland R, Bruck W, Morales Y, Bitsch A et al Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005; 366:579–82. [DOI] [PubMed] [Google Scholar]
- 42. Weinshenker BG, O'Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW et al A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999; 46:878–86. [DOI] [PubMed] [Google Scholar]
- 43. Kaynar L, Altuntas F, Aydogdu I, Turgut B, Kocyigit I, Hacioglu SK et al Therapeutic plasma exchange in patients with neurologic diseases: retrospective multicenter study. Transfus Apher Sci 2008; 38:109–15. [DOI] [PubMed] [Google Scholar]
- 44. Douglas J, Gardner L, Levin M. Antibodies to an intracellular antigen penetrate neuronal cells and cause deleterious effects. J Clin Cell Immunol 2013; 4:134. [Google Scholar]
- 45. Magrys A, Anekonda T, Ren G, Adamus G. The role of anti‐α‐enolase autoantibodies in pathogenicity of autoimmune‐mediated retinopathy. J Clin Immunol 2007; 27:181–92. [DOI] [PubMed] [Google Scholar]
- 46. Qu L, Zhang P, LaMotte RH, Ma C. Neuronal Fc‐γ receptor I mediated excitatory effects of IgG immune complex on rat dorsal root ganglion neurons. Brain Behav Immun 2011; 25:1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnston KM, Brady ST, van der Kooy D, Connolly JA. A unique tubulin antibody which disrupts particle movement in squid axoplasm. Cell Motil Cytoskeleton 1987; 7:110–5. [DOI] [PubMed] [Google Scholar]
- 48. Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci U S A 1990; 87:1061–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34–47. [DOI] [PubMed] [Google Scholar]
- 50. Amor S, van der Star BJ, Bosca I, Raffel J, Gnanapavan S, Watchorn J et al Neurofilament light antibodies in serum reflect response to natalizumab treatment in multiple sclerosis. Mult Scler 2014; 20:1355–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Specificity of IgG from multiple sclerosis patient serum.
Figure S2. Exacerbation of myelin oligodendrocyte glycoprotein 35–55 (MOG35–55)‐induced experimental autoimmune encephalomyelitis (EAE) by human IgG.
Figure S3. Intracerebral injection of monoclonal antibody to neurofilament light (NF‐L) causes transient neurological signs.
Table S1. Clinical data of multiple sclerosis patients.