

Summary
Thalidomide (TM) has been reported to have anti‐cancer and anti‐inflammatory properties, and dexamethasone (DX) is known to reduce inflammation and inhibit production of inflammatory cytokines. Many studies have reported that combinatorial therapy with TM and DX is clinically used to treat multiple myeloma and lupus nephritis, but the mechanism responsible for its effects has not been elucidated. In this study, we determined that TM and DX co‐treatment had an enhanced immune‐modulatory effect on T cells through regulating the expression of co‐stimulatory molecules. Splenic naive T cells from C57BL/6 mice were sort‐purified and cultured for CD4+ T cell proliferation and regulatory T (Treg) cell conversion in the presence of TM and/or DX. Following incubation with the drugs, cells were collected and OX40, 4‐1BB, and glucocorticoid‐induced tumour necrosis factor receptor‐related protein (GITR) expression was quantified by flow cytometry. TM (1 or 10 μm) decreased CD4+ T cell proliferation in a dose‐dependent manner, whereas TM/DX (0·1 or 1 nm) co‐treatment further decreased proliferation. Treg cell populations were preserved following drug treatment. Furthermore, expression of co‐stimulatory molecules decreased upon TM/DX co‐treatment in effector T (Teff) cells and was preserved in Treg cells. Splenic CD4+ T cells isolated from TM‐ and DX‐treated mice exhibited the same patterns of Teff and Treg cell populations as observed in vitro. Considering the selective effect of TM on different T cell subsets, we suggest that TM may play an immunomodulatory role and that TM/DX combinatorial treatment could further enhance these immunomodulatory effects by regulating GITR, OX40, and 4‐1BB expression in CD4+ T cells.
Keywords: 4‐1BB, glucocorticoid‐induced tumour necrosis factor receptor‐related protein, OX40, T lymphocyte, thalidomide
Abbreviations
- APC
allophycocyanin
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- Cy
cyanine
- DX
dexamethasone
- FACS
fluorescence‐activated cell sorting
- FITC
fluorescein isothiocyanate
- FOXP3
forkhead box P3
- GC
glucocorticoid
- GITR
glucocorticoid‐induced tumour necrosis factor receptor‐related protein
- GITRL
GITR ligand
- IFN‐γ
interferon‐γ
- IL
interleukin
- IR
infrared
- MFI
median fluorescence intensity
- PE
phycoerythrin
- PerCP
peridinin chlorophyll protein
- Teff
effector T cell
- TM
thalidomide
- Tnaive
naive T cell
- TNF‐α
tumour necrosis factor‐α
- TNFSF
tumour necrosis factor superfamily
- TNFRSF
tumour necrosis factor receptor superfamily
- Treg
regulatory T cell
Introduction
Shortly after its introduction in the 1950s, thalidomide (TM) was banned due to its tragic teratogenic effect; however, as recent studies revealed its anti‐angiogenic and anti‐inflammatory effects, TM has regained attention in the fields of cancer and autoimmune diseases.1, 2 TM possesses an immunosuppressive function, which may be useful following transplantation,3, 4, 5, 6 and not only suppresses, but modulates the immune response by regulating key immune‐modulatory molecules, including nuclear factor‐κB and pro‐inflammatory cytokines such as tumour necrosis factor‐α (TNF‐α), interferon‐γ (IFN‐γ), interleukin (IL)‐6, IL‐10, IL‐12, and cyclooxygenase 2.2, 7, 8, 9, 10, 11 Our previous study further substantiated the immunomodulatory effects of TM, demonstrating that TM selectively suppresses effector T (Teff) cells among total CD4+ T cells.2, 12 We also found that the immunomodulatory effect of TM was potentiated with glucocorticoid (GC)‐combined treatment.9 Therefore, this study introduced GC as a complementary agent, as it may play an important role in extending the function of TM.
Glucocorticoid reduces inflammation and inhibits inflammatory cytokine production, so it is used clinically to treat immunological diseases and to maintain post‐transplantation homeostasis.13, 14, 15 However, the side effects of GC, which include the impairment of many anabolic processes, limit its therapeutic use. Therefore, it is used either in small doses or with a complementary agent. The latter method has proved successful in the treatment of cancer, such as multiple myeloma, when GC is combined with TM.16, 17, 18 TM and GC combinatorial treatment may also be a promising approach for post‐transplant immunomodulation. Although the mechanisms responsible for the success of TM and GC combinatorial treatment remain unclear, based on our previous study, we assumed that TM affects the action mechanisms of GC, as the effect of TM on the lupus nephritis mouse model was synergistically potentiated with the addition of GC.9 The action of GC is associated with the TNF and TNF receptor superfamily (TNFSF/TNFRSF). Therefore, analysis of the effect of TM and GC combinatorial treatment in relation to TNFRSF may lead to an understanding of their combinatorial activity.
Interactions of TNFSF and TNFRSF play critical roles in regulating both pro‐inflammatory and anti‐inflammatory responses in autoimmune diseases.19, 20 Their interactions transmit co‐stimulatory signals and regulate a broad range of immune activities by triggering, suppressing, or activating immune cell functions.2, 21, 22, 23
In this study, we determined the effect of TM and dexamethasone (DX), a synthetic GC, combinatorial treatment on the proliferation of CD4+ T cells and the conversion of naive T (Tnaive) cells to CD4+ forkhead box P3 (FOXP3)+ T cells. We also observed in vitro changes in the expression of TNFRSFs, primarily focusing on GC‐induced TNFR‐related protein (GITR), OX40, and 4‐1BB.
Materials and methods
Mice and reagents
Seven‐week‐old male C57BL/6 mice were purchased from Orient Bio Inc. (Seongnam, Korea) and maintained according to the ethical guidelines of our institution. The experimental protocol was approved by the Committee on Animal Investigation at Yonsei University. Anti‐mouse CD3, anti‐mouse CD28, fluorescein isothiocyanate (FITC)‐conjugated anti‐mouse CD44, peridinin chlorophyll protein (PerCP)‐cyanine (Cy)5.5‐conjugated anti‐mouse CD62 ligand (CD62L), FITC‐conjugated anti‐mouse FOXP3, phycoerythrin (PE)‐Cy7‐conjugated anti‐mouse OX40 (CD134), allophycocyanin (APC)‐conjugated anti‐mouse 4‐1BB (CD137), PE‐Cy7‐conjugated anti‐mouse IFN‐γ antibodies, and the Fixation/Permeabilization kit were purchased from eBioscience (San Diego, CA). APC‐Cy7‐conjugated anti‐mouse CD4, PerCP‐Cy5.5‐conjugated anti‐mouse GITR (CD357), PerCP‐Cy5.5‐conjugated anti‐mouse IL‐2, and APC‐conjugated anti‐mouse TNF‐α antibodies were purchased from BioLegend (San Diego, CA). Recombinant murine IL‐2 and transforming growth factor‐β 1 were purchased from PeproTech (London, UK). Carboxyfluorescein diacetate succinimidyl ester (CFSE; CellTrace™) and LIVE/DEAD™ Fixable near‐infrared (IR) Dead Cell Stain Kit were obtained from Invitrogen (Carlsbad, CA). The 70‐μm cell strainers and leucocyte Activation Cocktail, with BD GolgiPlug™ was purchased from BD Biosciences (Franklin Lakes, NJ). TM, DX, and red blood cell lysis buffer were purchased from Sigma‐Aldrich (St Louis, MO).
Tnaive cell isolation
Spleens taken from 8‐week‐old male C57BL/6 mice were homogenized through a 70‐μm cell strainer (BD Biosciences) and incubated in red blood cell lysis buffer for 5 min. To isolate Tnaive cells, expressing CD4+, CD44lo, and CD62Lhi, from the splenocytes, cells were stained with APC‐Cy7‐conjugated anti‐mouse CD4, FITC‐conjugated anti‐mouse CD44, and PerCP‐Cy5.5‐conjugated anti‐mouse CD62L antibodies, and sorted by fluorescence‐activated cell sorting (FACS) Aria III Cell Sorter; BD Biosciences).
Cell culture
Approximately 1·5 × 105 Tnaive cells were plated on 96‐well plates containing TM, DX, or TM/DX and incubated for 72 hr under standard culture conditions (37°, 5% CO2). To induce Teff cell proliferation, Tnaive cells were stimulated throughout the culture period with anti‐mouse CD3 (5 μg/ml) and anti‐mouse CD28 (2 μg/ml) antibodies, which were added to RPMI‐1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen), and 200 mm l‐glutamine and 50 mm β‐mercaptoethanol (Sigma‐Aldrich). Before plating, Tnaive cells were stained with CFSE (Invitrogen) to determine proliferation levels according to the manufacturer's instructions.
In order to convert Tnaive cells to regulatory T (Treg) cells, Tnaive cells were cultured with anti‐mouse CD3 (5 μg/ml), anti‐mouse CD28 (2 μg/ml) antibodies, IL‐2 (10 ng/ml), and transforming growth factor‐β 1 (50 ng/ml) within the same complete RPMI medium as in Teff cell proliferation.
Suppression assay
The function of Treg cells converted from Tnaive cells was determined in the presence or absence of DX or TM/DX. Treg cells were isolated by trypsinization and analysed by FACS to quantify conversion yields. Cells were then transferred to new wells containing sort‐purified Tnaive cells labelled with CFSE, and then incubated in media containing anti‐mouse CD3 and anti‐mouse CD28 under normal culture conditions for 72 hr. Treg and Tnaive cells were plated at Tnaive : Treg ratios of 1 : 1 (1 × 105 cells/well), a 2 : 1 (1 × 105 cells/well and 5 × 104 cells/well, respectively), and 4 : 1 (1 × 105 cells/well and 2·5 × 104 cells/well, respectively). Treg cell function was determined by measuring CFSE intensity, which indicated the division number during Teff cell proliferation.
Flow cytometry
Cultured or isolated T cells were incubated with the appropriately diluted antibodies for 40 min at 4°. To detect the changes in the expression of TNFRSF molecules, surface molecules were stained with PE‐Cy7‐conjugated anti‐mouse OX40, APC‐conjugated anti‐mouse 4‐1BB, and PerCP‐Cy5.5‐conjugated anti‐mouse GITR, and the cells were fixed and permeabilized using the Fixation/Permeabilization kit (eBioscience) according to the manufacturer's instructions. The cells cultured for Treg cell conversion were then stained intracellularly with FITC‐conjugated anti‐mouse FOXP3.
In order to examine the effect of drug toxicity on the survival of Teff and Treg cells, different sets of T cell samples that were plated with either TM, DX, or TM/DX in appropriate media for both Teff cell proliferation and Treg cell conversion were stained using LIVE/DEAD™ Fixable near‐IR Dead Cell Stain Kit (Invitrogen). To allow for clear detection of intracellular accumulations of cytokines in Teff cells, the cells were stimulated with leucocyte activation cocktail, with BD GolgiPlug™ (BD Biosciences). Dead cell staining and leucocyte activation were performed according to the manufacturer's instructions. The cells were then fixed and permeabilized, and intracellularly stained with PercP‐Cy5.5‐conjugated anti‐mouse IL‐2, APC‐conjugated anti‐mouse TNF‐α, and PE‐Cy7‐conjugated anti‐mouse IFN‐γ antibodies to determine the changes in Teff cells' ability to produce effector cytokines.
Flow cytometry was performed on an LSR II or FACS Verse flow cytometer (BD Biosciences). Data were analysed using flowjo v10.0.7 software (Tree Star, Inc., San Carlos, CA). Expression of FOXP3 on Treg cells and that of TNFSFs on differentiated CD4+ T cells were determined by relative median fluorescent intensity (MFI).
In vivo TM and DX treatment
To build upon our in vitro results, we examined changes in splenic T‐lymphocyte subsets in normal C57BL/6 mice treated with TM and/or DX. The experimental groups received chow containing either TM, DX, or TM/DX for 1 week. The control mice received regular chow for the same time period. TM and DX were administered at 10 or 1·5 mg/kg body weight per day, respectively, in all three groups. Doses were determined based on the results of our previous study.2, 9, 12
Statistical analysis
The significance of intergroup differences was determined using the Student's t‐test or one‐way analysis of variance. The analysis was conducted with sigma plot 2·0, and P values < 0·05 were considered statistically significant.
Results
Differentiation of CD4+ naive T cells
Treatment with TM (1 or 10 μm) suppressed Teff cell proliferation (not statistically significant), and TM/DX combinatorial treatments [TM (1 μm) and DX (0·1 nm), TM (10 μm) and DX (0·1 nm), TM (1 μm) and DX (1 nm), and TM (10 μm) and DX (1 nm)] further suppressed Teff cell proliferation in a dose‐dependent manner (Fig. 1). Although drug treatments were found to reduce the proliferation of Teff cells, the cells' ability to produce effector cytokines such as IFN‐γ, IL‐2, and TNF‐α was examined to reveal whether treatments compromised their functionality, as well. Secretion of IFN‐γ, IL‐2, and TNF‐α was also decreased upon drug treatments, although the change was less remarkable than that observed in the proliferation of Teff cells (see Supplementary material, Fig. S1).
Figure 1.
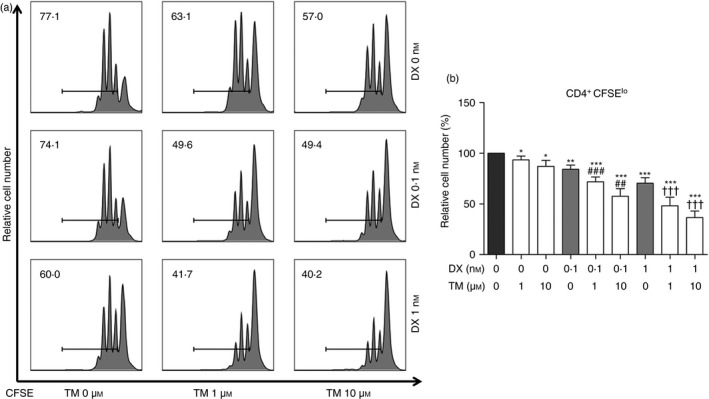
Proliferation of CD4+ T cells induced by thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment. Naive T (Tnaives) cells, which were labelled with CFSE, were incubated with anti‐CD3 and anti‐CD28 antibodies for 72 hr, and analysed by flow cytometry. TM/DX combinatorial treatment down‐regulated the proliferation of CD4+ T cells from Tnaive cells more than DX alone. (a) Histogram plots of CFSE intensities. (b) Ratios of CD4+ CFSElo T cells. The numbers on the histogram indicate percentages of proliferating cells among total T cells. The results shown are representative plots selected from five independent experiments (% of non‐treated controls, n = 5). *P < 0·05, **P < 0·01, and ***P < 0·001, compared with untreated controls; ## P < 0·01 and ### P < 0·001 compared with 0·1 nm DX treatment; and ††† P < 0·001 compared with 1 nm DX treatment.
The Treg cell population was slightly increased by TM‐alone treatments and significantly decreased by DX‐alone treatments. Interestingly, the impaired Treg cell conversion rate induced by DX was significantly improved by TM/DX treatments, but only as much as that in the control group (Fig. 2a and 2b). Because the function of Treg cells is dependent on their expression of FOXP3,24 the quality of each Treg cell was evaluated by comparing the MFI of FOXP3. The FOXP3 expression of Treg cells was impaired by DX, but unchanged on TM/DX combinatorial treatments (Fig. 2c). Both the conversion rate and the quality of Treg cells were preserved in TM/DX combinatorial treatments.
Figure 2.
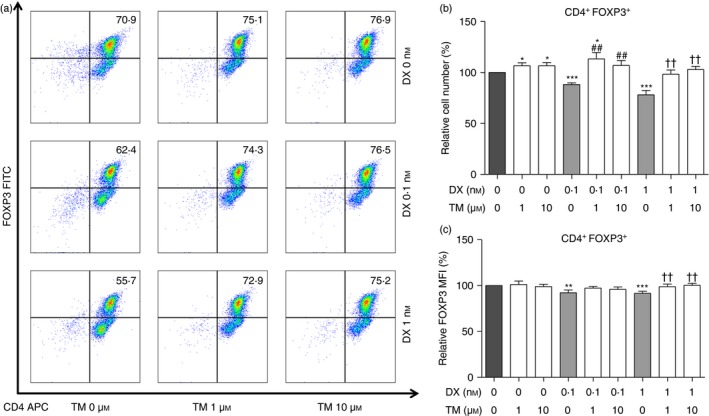
Differentiation of CD4+ T cells to CD4+ FOXP3+ T cells induced by thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment. Naive T cells were incubated with anti‐CD3 and anti‐CD28 antibodies, transforming growth factor‐β (TGF‐β), and interleukin‐2 (IL‐2) for 72 hr, and analysed by flow cytometry. (a) Dot plots showing regulatory T (Treg) cell analysis by flow cytometry. (b) Ratios of CD4+ FOXP3+ T cells. TM/DX combinatorial treatment increased the differentiation of CD4+ FOXP3+ T cells from Tnaive cells more so than DX‐only treatment. (c) Ratios of MFI of FOXP3 expression in CD4+ FOXP3+ T cells. (% of non‐treated controls, n = 5). *P < 0·05, **P < 0·01, and ***P < 0·001 compared with non‐treated controls; ## P < 0·01 compared with 0·1 nm DX treatment; and †† P < 0·01 compared with 1 nm DX treatment. [Colour figure can be viewed at wileyonlinelibrary.com]
Since Teff cell proliferation was reduced whereas Treg cell conversion was unaffected, it was critical to further evaluate the effect of drug toxicity on the survival of T cells. Results demonstrated that all T cells plated with each treatment showed no toxicity regardless of the degree of proliferation or conversion (see Supplementary material, Fig. S2).
Furthermore, the suppressive function of Treg cells on Teff cells was slightly impaired by TM or DX treatment; however, this effect was significantly restored after TM/DX combinatorial treatment (Fig. 3).
Figure 3.
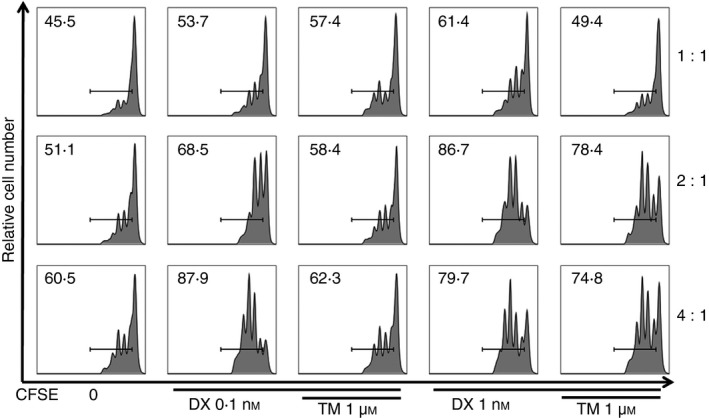
Histogram plots of CFSE intensity indicating suppressive function of regulatory T (Treg) cells on effector T (Teff) cells. Treg cells stimulated by thalidomide (TM)/dexamethasone (DX) showed more potent suppressive effects than those given DX‐only treatment. The numbers on the histogram indicate percentages of proliferating cells among total T cells. The results shown are representative plots selected from five independent experiments.
Expression of OX40, 4‐1BB, and GITR
During Teff cell proliferation, DX treatments (0·1 nm and 1 nm) reduced surface expression of OX40 and 4‐1BB in a dose‐dependent manner, and TM/DX combinatorial treatments [TM (1 μm) and DX (0·1 nm), TM (10 μm) and DX (0·1 nm), TM (1 μm) and DX (1 nm), and TM (10 μm) and DX (1 nm)] further significantly decreased OX40 and 4‐1BB expression in a dose‐dependent manner. Neither DX‐only treatment (0·1 nm and 1 nm) nor TM/DX combinatorial treatment (10 μm and 1 nm, respectively) significantly influenced the GITR expression on Teff cells (Fig. 4).
Figure 4.
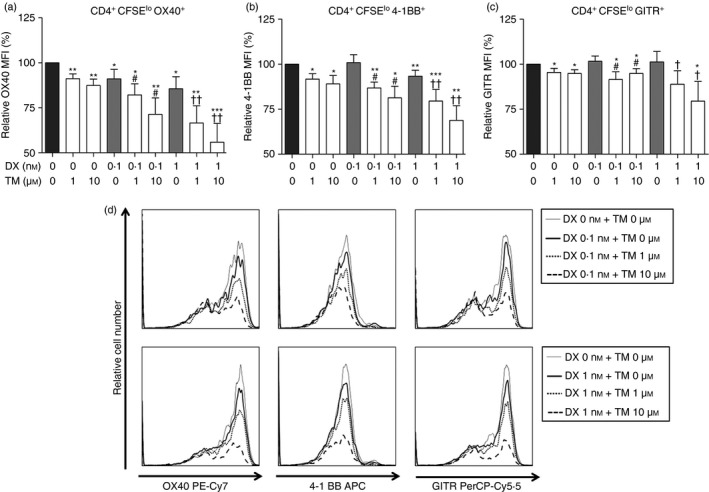
Tumour necrosis factor receptor superfamily expression on effector T (Teff) cells induced by thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment. (a) OX40 (CD134; TNFSF4), (b) 4‐1BB (CD137; TNFSF9), (c) GITR (CD357; TNFSF18). (d) Histogram plots showing changes in expression of each molecule upon drug treatment. These changes were analysed on gated CD4+ CFSElo T cells, which were viewed as Teff cells by FACS analysis using relative MFI (% of non‐treated controls, n = 5). *P < 0·05, **P < 0·01, and ***P < 0·001 compared with untreated controls; # P < 0·05 compared with 0·1 nm DX treatment; and † P < 0·05 and †† P < 0·01 compared with 1 nm DX treatment.
In converted CD4+ FOXP3+ T cells, TM/DX combinatorial treatments down‐regulated OX40, 4‐1BB, and GITR expression only slightly compared with the control groups, and overall, the changes were not significant and remained relatively constant (Fig. 5).
Figure 5.
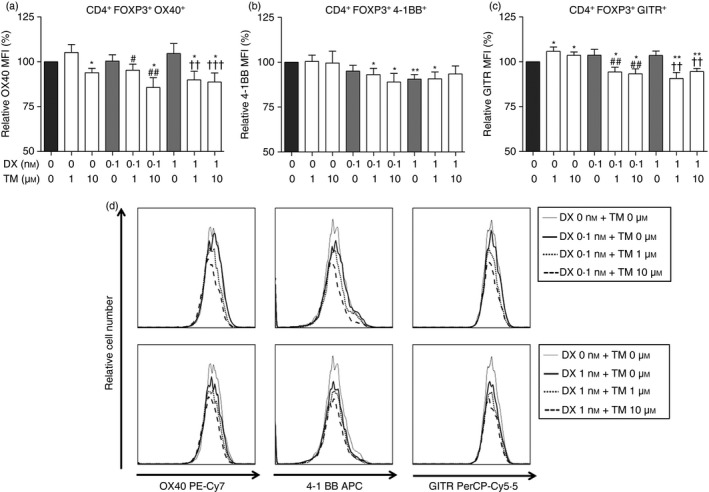
Tumour necrosis factor receptor superfamily expression on differentiated CD4+ FOXP3+ T cells (Treg cells) induced by thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment. (a) OX40 (CD134; TNFSF4), (b) 4‐1BB (CD137; TNFSF9), (c) GITR (CD357; TNFSF18). (d) Histogram plots showing changes in expression levels of each molecule upon drug treatment. Changes in protein expression were analysed by FACS using relative MFI (% of non‐treated controls, n = 5). *P < 0·05 and **P < 0·01 compared with untreated controls; # P < 0·05 and ## P < 0·01 compared with 0·1 nm DX treatment; and †† P < 0·01 and ††† P < 0·001 compared with 1 nm DX treatment.
T cell subset changes in vivo
The body weight of the mice in the TM group increased by more than 12% compared with the DX and TM/DX groups, and the body weight of mice in all test groups increased compared with the controls (Fig. 6a, P < 0·01). T lymphocytes were isolated and stained with anti‐CD4 and anti‐CD44 antibodies to label Teff cells or with anti‐CD4 and anti‐FOXP3 antibodies to label Treg cells. TM/DX treatment down‐regulated the population of Teff cells (CD4+ CD44hi T cells), whereas TM‐only treatment had no effect (Fig. 6b and 6d). To determine how the drug treatments influenced solely CD4+ CD44hi T cells, which represent memory characterized cells, the population of CD4+ CD44lo T cells was also analysed (see Supplementary material, Fig. S3). Figure S3a, which shows the ratio of CD4+ CD44hi to CD4+ CD44lo T cells, demonstrates that CD4+ CD44hi T cell population was significantly less than that of CD4+ CD44lo on TM/DX treatment compared with the control. TM/DX combinatorial treatment appears to preserve the population of Treg cells (CD4+ FOXP3+ T cells), whereas neither TM alone nor DX alone had any effect on Treg cell populations (Fig. 6c and 6d).
Figure 6.
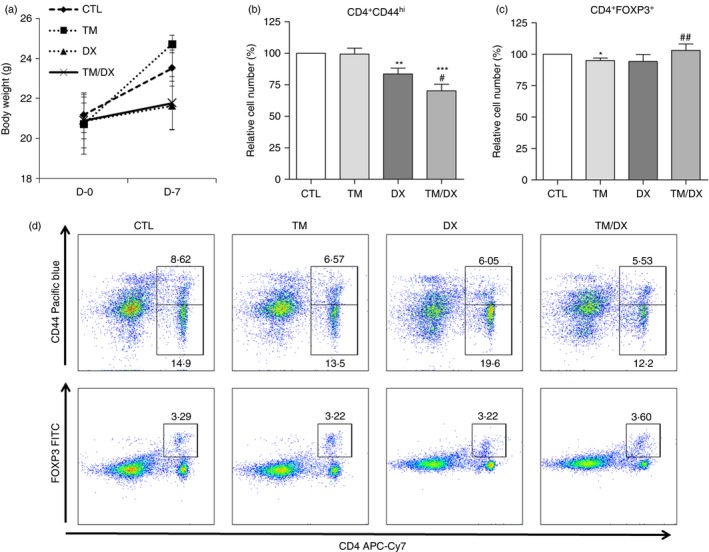
In vivo testing of the effect of thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment in C57BL/6 mice. Mice were provided with chow containing TM (10 mg/kg), DX (1·5 mg/kg), or TM and DX (10 and 1·5 mg/kg, respectively) daily for 7 days. (a) Body weight. Animals in the TM group were heavier than those in the DX or TM/DX combinatorial treatment group (CO), and even higher than those in the untreated control group (CTL) (n = 6, P < 0·01). (b) Ratios of CD4+ CD44hi and (c) CD4+ FOXP3+ T cells to total splenocytes. CD4+ CD44hi T cells (effector T cells) were more down‐regulated by TM/DX combinatorial treatment than by TM or DX alone. CD4+ FOXP3+ T cells (regulatory T cells) were preserved by TM/DX combinatorial treatment. (d) Dot plots of the control, TM alone, DX alone, and TM/DX combinatorial treatment groups analysed by flow cytometry. The plots show APC‐Cy7‐conjugated anti‐CD4 antibody and Pacific blue‐conjugated anti‐CD44 antibody, or APC‐Cy7‐conjugated anti‐CD4 antibody and FITC‐conjugated anti‐FOXP3 antibody. The results shown are representative plots selected from six independent experiments (% of non‐treated controls, n = 6). *P < 0·05, **P < 0·01, and ***P < 0·001 compared with untreated controls; # P < 0·05 and ## P < 0·01 compared with DX‐treated groups. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Following its withdrawal from the market as a sedative, TM has been found to be an effective anti‐inflammatory drug for patients with leprosy and many other autoimmune diseases, such as multiple myeloma, systemic lupus erythematosus and rheumatoid arthritis.1, 2, 9, 11, 25 In our previous study, we demonstrated the immune‐modulatory role of TM, but isolated use of TM was not sufficiently potent to effectively suppress the proliferation of T cells.2 Based on the encouraging results obtained with TM plus prednisolone treatment in a lupus nephritis mouse model,9 we hypothesized that TM and GC combinatorial treatment may generate enhanced effects on regulating each T cell subset. We chose DX as a GC, and the dose of 0·1 and 1 nm was based on previous studies.26, 27
We previously reported that isolated TM treatment could alter CD4+ T cell subset populations by down‐regulating Teff cells while preserving Treg cells.2 In the current study, this result was reproduced in TM‐only treated groups, but it was further enhanced in TM/DX combinatorial groups, both in vitro and in vivo. As seen in the in vitro data, Teff cell proliferation following 1 and 10 μm TM treatment showed only subtle decreases, but following TM/DX combinatorial treatment, Teff cell proliferation was significantly inhibited. The inhibitory effect of TM/DX combinatorial treatment on Teff cell subsets increased in a dose‐dependent manner (Fig. 1).
Meanwhile, Treg cell conversion rates in the TM/DX combinatorial treatment groups were similar to those in the control and the TM‐only groups (Fig. 2a and 2b). When compared with the control group, MFI of FOXP3 expression on Treg cells did not show any significant changes by TM/DX (Fig. 2c), indicating that the quality of each Treg cell was also preserved. Our in vivo data showed similar results of reduced Teff cell proliferation. In our experimental setting, it was not clear whether the Treg cell population observed in vivo were the pre‐existing Treg cells or the Treg cells converted due to drug treatment, but there is a slight increase in Treg cell conversion upon TM/DX combinatorial treatment (Fig. 6b and 6c).
Upon analysis of the suppression assay data, we found that the suppressive function of Treg cells was impaired by DX treatments, but when coupled with TM treatments, the suppressive function of Treg cells was restored. As seen in Fig. 3, at a Teff : Treg ratio of 4 : 1, with TM/DX combinatorial treatment (1 μm and 0·1 nm, respectively), the suppressive function of Treg cells recovered nearly to the level of the control groups. This finding may indicate that both the function and population of Treg cells was preserved by the TM/DX combinatorial treatments.
Teff cells are key participants in executing immune functions; however, their pathogen clearance and anti‐tumour abilities can cause tissue damage and elicit unwanted self‐reactivity.28 Treg cells actively suppress this activation of immune responses mediated by Teff cells,29 thereby making it vital for any immune‐modulatory drug to influence each T cell subset independently from each other. Many immunosuppressant drugs, such as calcineurin inhibitors, reduce T cell populations regardless of their subsets,30, 31 but the TM/DX combinatorial treatment showed clear modulating effects and can be potentially used as a viable immunomodulatory therapy.
The TNF receptors are primarily involved in apoptosis and inflammation, and interactions of TNFRSF and TNFSF deliver co‐stimulatory signals to regulate the function of many immune cells.19, 20 In our previous study, we suggest that the selected co‐stimulatory molecules, OX40, 4‐1BB, and GITR, may be involved in the TM‐induced regulation of proliferation and conversion of Treg cells from Tnaive cells.12 It has already been proposed by several studies that TM increases the therapeutic efficacy for treatment of multiple myeloma and lupus nephritis when combined with GCs;16, 17, 18, 27 so, we hypothesized that DX may enhance the action of TM toward the co‐stimulatory molecules. Indeed, in this study, we demonstrated that co‐stimulatory molecule expression changed upon treatment with TM and/or DX.
OX40 (CD134, TNFRSF4) is a well‐known T cell activation marker. OX40 and its ligand, OX40L (CD252), are expressed on many lymphoid cells, including activated CD4+ T cells. OX40 conducts co‐stimulatory signals to CD4+ T cells not only to promote their division and survival but to augment Teff cell expansion.32, 33 While Teff cell populations are increased by OX40 and OX40L ligation, the differentiation and activity of Treg cells are suppressed. Therefore, the interactions of OX40 and OX40L play a critical role in the development of autoimmune and inflammatory diseases.21, 23, 32, 34, 35
Our in vitro data demonstrated that OX40 expression on Teff cells decreased in TM/DX combinatorial groups in a dose‐dependent manner (Fig. 4a), whereas expression on Treg cells did not (Fig. 5a). In the high‐dose TM/DX (10 μm and 1 nm, respectively) group, OX40 expression on Teff cells decreased to approximately 60% of the control, indicating that the Teff cell‐expanding function of OX40 was diminished upon TM/DX combinatorial treatment. Even under high‐dose conditions, however, OX40 expression on Treg cells only decreased to approximately 90% of the control, which may indicate that TM/DX combinatorial treatment has no significant effect on Treg cell activity. These results also correspond with the data represented in Figs. 1 and 2; the Teff cell population decreased whereas the Treg cell population was preserved, and the TM/DX combinatorial treatment influenced each T cell subset differently.
Similar to OX40, one of the primary functions of 4‐1BB (CD137, TNFRSF9) is to activate Teff cells to proliferate. Its function on Treg cells, however, is opposite to that of OX40 in that 4‐1BB up‐regulates Treg cell differentiation and activity.36, 37, 38
Our results demonstrated that expression of 4‐1BB decreased as the drug dose increased. Whereas each TM‐only group (1 and 10 μm) exhibited minimal down‐regulation of 4‐1BB, the group treated with the highest dosages of TM and DX (10 μm and 1 nm, respectively) exhibited significantly greater decreases of up to 30% compared with the control group (Fig. 4b). This finding indicates that the TM/DX combinatorial treatment is more effective in inhibiting 4‐1BB's function of Teff cell expansion and activation compared with the control and TM‐ or DX‐only isolated treatments. However, 4‐1BB expression on Treg cells did not change significantly (Fig. 5b). Considering that Treg cell differentiation is controlled by 4‐1BB and 4‐1BB ligand (CD137L) interaction, the relatively preserved level of 4‐1BB on Treg cells may reflect little change in the Treg cell population. These results also support our data described in Figs. 1 and 2, that TM/DX combinatorial treatment affected each T cell subset differently.
As with OX40 and 4‐1BB, interactions of GITR (CD357, TNFRSF19) with its binding partner, GITR ligand (GITRL), induced Teff cell proliferation and rendered Teff cells less susceptible to Treg cells. Interestingly, GITR/GITRL ligation not only activates Teff cells but inhibits the suppressive activity of Treg cells, while still promoting Treg cell proliferation.22, 39, 40, 41, 42, 43 The effects of GITR on T cells remains controversial, but it is clear that GITR plays an important role in modulating immune responses.
There was no significant change in GITR expression on Teff cells in all groups compared with the control, except in the TM/DX combinatorial treatment groups (Fig. 4c). GITR expression decreased to approximately 80% of the control in the TM/DX group (10 μm and 1 nm, respectively), indicating that a reduction in GITR molecules may result in reduced Teff cell proliferation. Similar to OX40 and 4‐1BB, GITR expression in Treg cells did not significantly change, which indicates that the Treg cell population is preserved (Fig. 5c).
Taken together, our results indicate that changes in surface expression of OX40, 4‐1BB, and GITR lead to a decrease in Teff cells and preservation of the Treg cell population. Though the outcomes after TM‐ or DX‐only treatments are somewhat different for each molecule, it is notable that all of the selected members of TNFRSF demonstrate the same influence on the Teff and Treg cell populations upon TM/DX combinatorial treatment.
We hope to address the limitations of this study in future experiments by co‐culturing T cells with other lymphocytes, such as dendritic cells, to fully understand the ligation levels of co‐stimulatory molecules. In vivo experiments with transplant or autoimmune disease animal models may also be required to strengthen our findings.
In conclusion, the immunomodulatory characteristics of TM are further enhanced by DX, and the TM/DX combinatorial treatment influences Teff and Treg cell populations differently, suppressing the Teff cells while preserving the Treg cells. This enhanced effect may be related to the expression of co‐stimulatory molecules altered by TM and DX. TM/DX combinatorial treatment as an immunomodulatory therapy may possess a promising capacity to maintain post‐transplant homeostasis and a tolerogenic state for autoimmune diseases. Further study will be needed to determine the underlying molecular links between the TM‐ and DX‐enhanced effects in T cells.
Author contributions
JGL, BSK, and YSK conceived and designed the experiments. JYK and EJK performed the experiments. BSK, EJK, and JGL analysed the data. DJJ, MSK, KHH, and YSK contributed reagents, materials, and/or analysis tools. BSK, EJK, JGL, JYK, SHS, and YSK wrote the paper.
Disclosures
The authors declare no conflicts of interest.
Supporting information
Figure S1. Changes in cytokines production of effector T cells upon thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment.
Figure S2. Live/Dead analysis results of CD4+ T cells from effector T cell proliferation assay and regulatory T cell conversion assay.
Figure S3. Subset‐specific changes in CD4+ T cell population in vivo.
Acknowledgements
This work was supported by a research grant from Yonsei University Health System IACF (#2013‐31‐0453).
Contributor Information
Beom Seok Kim, Email: docbsk@yuhs.ac.
Yu Seun Kim, Email: yukim@yuhs.ac.
References
- 1. Bersani‐Amado LE, Dantas JA, Damiao MJ, Rocha BA, Besson JC, Bastos RL et al Involvement of cytokines in the modulation and progression of renal fibrosis induced by unilateral ureteral obstruction in C57BL/6 mice: effects of thalidomide and dexamethasone. Fundam Clin Pharmacol 2016; 30:35–46. [DOI] [PubMed] [Google Scholar]
- 2. Kim BS, Kim JY, Lee JG, Cho Y, Huh KH, Kim MS et al Immune modulatory effect of thalidomide on T cells. Transplant Proc 2015; 47:787–90. [DOI] [PubMed] [Google Scholar]
- 3. Chen C, Kuehn C, Bretzel RG, Linn T. Anti‐inflammatory thalidomide improves islet grafts survival and functions in a xenogenic environment. PLoS ONE 2009; 4:e6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Carvalho JBV, Petroianu A, Alberti LR. Efficacy of thalidomide as immunossupressor in heterotopic heart transplantation. Chirurgia 2008; 103:429–34. [PubMed] [Google Scholar]
- 5. Carvalho JB, Petroianu A, Travolo E, de Oliveira BH, Duarte AB, Alberti LR. Effects of immunosuppression induced by thalidomide and cyclosporine in heterotopic heart transplantation in rabbits. Transplant Proc 2007; 39:1640–1. [DOI] [PubMed] [Google Scholar]
- 6. Chaves DNB, Alberti LR, Petroianu A. Assessment of immunossupresion induced by thalidomide, cyclosporine and diclofenac on skin allograft survival in rabbits. Rev Assoc Med Bras 2008; 54:42–7. [DOI] [PubMed] [Google Scholar]
- 7. Majumdar S, Lamothe B, Aggarwal BB. Thalidomide suppresses NF‐κB activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J Immunol 2002; 168:2644–51. [DOI] [PubMed] [Google Scholar]
- 8. Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet 2004; 363:1802–11. [DOI] [PubMed] [Google Scholar]
- 9. Lee SW, Park YB, Yang J, Park KH, Lee SK, Choi KH et al Attenuation of nephritis in lupus‐prone mice by thalidomide. Rheumatology (Oxford) 2012; 51:2131–40. [DOI] [PubMed] [Google Scholar]
- 10. Tseng CM, Hsiao YH, Su VY, Su KC, Wu YC, Chang KT et al The suppression effects of thalidomide on human lung fibroblasts: cell proliferation, vascular endothelial growth factor release, and collagen production. Lung 2013; 191:361–8. [DOI] [PubMed] [Google Scholar]
- 11. Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC. Thalidomide and lenalidomide: mechanism‐based potential drug combinations. Leuk Lymphoma 2008; 49:1238–45. [DOI] [PubMed] [Google Scholar]
- 12. Kim BS, Kim JY, Kim EJ, Lee JG, Joo DJ, Huh KH et al Role of thalidomide on the expression of OX40, 4‐1BB, and GITR in T cell subsets. Transplant Proc 2016; 48:1270–4. [DOI] [PubMed] [Google Scholar]
- 13. Prado C, Gomez J, Lopez P, de Paz B, Gutierrez C, Suarez A. Dexamethasone upregulates FOXP3 expression without increasing regulatory activity. Immunobiology 2011; 216:386–92. [DOI] [PubMed] [Google Scholar]
- 14. Wang XQ, Zhou X, Zhou Y, Rong L, Gao L, Xu W. Low‐dose dexamethasone alleviates lipopolysaccharide‐induced acute lung injury in rats and upregulates pulmonary glucocorticoid receptors. Respirology 2008; 13:772–80. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki S, Koyama K, Darnel A, Ishibashi H, Kobayashi S, Kubo H et al Dexamethasone upregulates 11β‐hydroxysteroid dehydrogenase type 2 in BEAS‐2B cells. Am J Respir Crit Care Med 2003; 167:1244–9. [DOI] [PubMed] [Google Scholar]
- 16. Amador‐Medina LF, Lacayo‐Lenero D, Crespo‐Solis E, Aguayo A, Martinez‐Banos D. Thalidomide and dexamethasone induction therapy until best response in recently diagnosed patients with multiple myeloma: results from a pilot study. Rev Invest Clin 2015; 67:304–12. [PubMed] [Google Scholar]
- 17. Takashima S, Miyamoto T, Kadowaki M, Ito Y, Aoki T, Takase K et al Combination of bortezomib, thalidomide, and dexamethasone (VTD) as a consolidation therapy after autologous stem cell transplantation for symptomatic multiple myeloma in Japanese patients. Int J Hematol 2014; 100:159–64. [DOI] [PubMed] [Google Scholar]
- 18. Zamagni E, Petrucci A, Tosi P, Tacchetti P, Perrone G, Brioli A et al Long‐term results of thalidomide and dexamethasone (thal‐dex) as therapy of first relapse in multiple myeloma. Ann Hematol 2012; 91:419–26. [DOI] [PubMed] [Google Scholar]
- 19. Croft M. The role of TNF superfamily members in T‐cell function and diseases. Nat Rev Immunol 2009; 9:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol 2012; 33:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Armitage RJ. Tumor necrosis factor receptor superfamily members and their ligands. Curr Opin Immunol 1994; 6:407–13. [DOI] [PubMed] [Google Scholar]
- 22. Esparza EM, Lindsten T, Stockhausen JM, Arch RH. Tumor necrosis factor receptor (TNFR)‐associated factor 5 is a critical intermediate of costimulatory signaling pathways triggered by glucocorticoid‐induced TNFR in T cells. J Biol Chem 2006; 281:8559–64. [DOI] [PubMed] [Google Scholar]
- 23. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 2005; 23:23–68. [DOI] [PubMed] [Google Scholar]
- 24. Wan YY, Flavell RA. Regulatory T‐cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007; 445:766–70. [DOI] [PubMed] [Google Scholar]
- 25. Direskeneli H, Ergun T, Yavuz S, Hamuryudan V, Eksioglu‐Demiralp E. Thalidomide has both anti‐inflammatory and regulatory effects in Behcet's disease. Clin Rheumatol 2008; 27:373–5. [DOI] [PubMed] [Google Scholar]
- 26. Cole TJ. Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol Annu Rev 2006; 12:269–300. [DOI] [PubMed] [Google Scholar]
- 27. Lowenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T‐cell immunosuppression. Trends Mol Med 2007; 13:158–63. [DOI] [PubMed] [Google Scholar]
- 28. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133:775–87. [DOI] [PubMed] [Google Scholar]
- 29. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441:235–8. [DOI] [PubMed] [Google Scholar]
- 30. Tsuda K, Yamanaka K, Kitagawa H, Akeda T, Naka M, Niwa K et al Calcineurin inhibitors suppress cytokine production from memory T cells and differentiation of naive T cells into cytokine‐producing mature T cells. PLoS ONE 2012; 7:e31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gillett NA, Chan C. Applications of immunohistochemistry in the evaluation of immunosuppressive agents. Hum Exp Toxicol 2000; 19:251–4. [DOI] [PubMed] [Google Scholar]
- 32. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T‐cell biology and immune disease. Immunol Rev 2009; 229:173–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M et al OX40 ligand shuts down IL‐10‐producing regulatory T cells. Proc Natl Acad Sci USA 2006; 103:13138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Demirci G, Li XC. Novel roles of OX40 in the allograft response. Curr Opin Organ Transplant 2008; 13:26–30. [DOI] [PubMed] [Google Scholar]
- 35. So T, Lee SW, Croft M. Immune regulation and control of regulatory T cells by OX40 and 4‐1BB. Cytokine Growth Factor Rev 2008; 19:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vinay DS, Kwon BS. Role of 4‐1BB in immune responses. Semin Immunol 1998; 10:481–9. [DOI] [PubMed] [Google Scholar]
- 37. Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS et al 4‐1BB‐dependent inhibition of immunosuppression by activated CD4+ CD25+ T cells. J Leukoc Biol 2004; 75:785–91. [DOI] [PubMed] [Google Scholar]
- 38. Arch RH, Thompson CB. 4‐1BB and Ox40 are members of a tumor necrosis factor (TNF)‐nerve growth factor receptor subfamily that bind TNF receptor‐associated factors and activate nuclear factor κB. Mol Cell Biol 1998; 18:558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nocentini G, Ronchetti S, Cuzzocrea S, Riccardi C. GITR/GITRL: more than an effector T cell co‐stimulatory system. Eur J Immunol 2007; 37:1165–9. [DOI] [PubMed] [Google Scholar]
- 40. Nocentini G, Ronchetti S, Petrillo MG, Riccardi C. Pharmacological modulation of GITRL/GITR system: therapeutic perspectives. Br J Pharmacol 2012; 165:2089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol 2012; 24:217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shevach EM, Stephens GL. The GITR–GITRL interaction: co‐stimulation or contrasuppression of regulatory activity? Nat Rev Immunol 2006; 6:613–8. [DOI] [PubMed] [Google Scholar]
- 43. Chen X, Du Y, Lin X, Qian Y, Zhou T, Huang Z. CD4+ CD25+ regulatory T cells in tumor immunity. Int Immunopharmacol 2016; 34:244–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Changes in cytokines production of effector T cells upon thalidomide (TM), dexamethasone (DX), or TM/DX combinatorial treatment.
Figure S2. Live/Dead analysis results of CD4+ T cells from effector T cell proliferation assay and regulatory T cell conversion assay.
Figure S3. Subset‐specific changes in CD4+ T cell population in vivo.