

Summary
Neuroinflammation is the hallmark of several infectious and neurodegenerative diseases. Synthetic glucocorticoids (GCs) are the first‐line immunosuppressive drugs used for controlling neuroinflammation. A delayed diffusion of GCs molecules and the high systemic doses required for brain‐specific targeting lead to severe undesirable effects, particularly when lifelong treatment is required. Therefore, there is an urgent need for improving this current therapeutic approach. The intranasal (i.n.) route is being employed increasingly for drug delivery to the brain via the olfactory system. In this study, the i.n. route is compared to the intravenous (i.v.) administration of GCs with respect to their effectiveness in controlling neuroinflammation induced experimentally by systemic lipopolysaccharide (LPS) injection. A statistically significant reduction in interleukin (IL)‐6 levels in the central nervous system (CNS) in the percentage of CD45+/CD11b+/lymphocyte antigen 6 complex locus G6D [Ly6G+ and in glial fibrillary acidic protein (GFAP) immunostaining was observed in mice from the i.n.‐dexamethasone (DX] group compared to control and i.v.‐DX‐treated animals. DX treatment did not modify the percentage of microglia and perivascular macrophages as determined by ionized calcium binding adaptor molecule 1 (Iba1) immunostaining of the cortex and hippocampus. The increased accumulation of DX in brain microvasculature in DX‐i.n.‐treated mice compared with controls and DX‐IV‐treated animals may underlie the higher effectiveness in controlling neuroinflammation. Altogether, these results indicate that IN‐DX administration may offer a more efficient alternative than systemic administration to control neuroinflammation in different neuropathologies.
Keywords: glucocorticoids, inflammation, intranasal route, LPS, neuroinflammation
Introduction
Neuroinflammation, defined as sustained microglia activation and the production of proinflammatory cytokines and reactive oxygen species, is a characteristic feature of different infectious and non‐infectious neuropathologies 1, 2. Increasing evidence supports the concept that a better control of neuroinflammation results in clear clinical benefits 3.
Corticosteroids play a pivotal role in the management of inflammation in a wide range of pathologies, such as allergies, asthma, autoimmune diseases and sepsis 4, 5. Synthetic glucocorticoids (GCs) with a much greater anti‐inflammatory activity than natural steroids have been developed. Dexamethasone (DX) and prednisone are the most extensively employed drugs in the clinic to control the acute neuroinflammation accompanying different infectious and non‐infectious neuropathologies 5, 6. High doses of both steroids have been used to reach the required concentration in the central nervous system CNS 7.
Unfortunately, severe side effects such as exacerbation or promotion of diabetes mellitus, gastro‐oesophageal reflux, osteoporosis, non‐traumatic osteonecrosis, Cushing syndrome, hypertension and behavioural changes after high‐dose, long‐term systemic steroid treatment limit their use, especially in chronic neuropathologies 5, 8, 9, 10, 11, 12. These undesirable adverse effects are difficult to avoid, as most steroid anti‐inflammatory and immunosuppressive activities are mediated by their binding to the ubiquitously expressed steroid receptors and result in affecting the functionality and survival of many different cell types, involved not only in innate and adaptive immunity but also in other crucial biological processes 13.
The intranasal (i.n.) route is a feasible alternative for drug delivery into the CNS 14, 15. Emerging evidence supports its potential to effectively deliver non‐steroidal drug and hormones into the brain, reaching higher central and lower systemic concentrations than the oral or parenteral routes 16, 17, 18, 19. To reach the brain, an intranasally delivered drug must be absorbed through the nasal mucosa into the blood and lymphatic vessels and penetrate across the endothelium of brain vessels and capillaries and the blood–brain barrier or reach the cerebral spinal fluid (CSF) through the choroid plexus 20. Once in the CSF, the drug may enter into the brain via the perivascular spaces. Small molecules can also enter the brain from the nose along the olfactory and trigeminal nerve structures without passing through the CSF, providing a rapid therapeutic effect 14, 21. These processes may last minutes or hours, depending on the size and the biochemical properties of the drug as well as its route of entry 22. The effectiveness in reaching the CNS may also differ between species, depending on the surface of the olfactory nerves. In humans, this surface is much smaller than in rodents, but the dense innervation by branches of the trigeminal nerves of the nasal cavity increases penetration efficacy 22, 23, 24. Currently, the i.n. administration of GC at very low doses is recommended as the first therapeutic choice to control human rhinitis, being found highly effective and causing minimal adverse effects 25.
The aim of this study was to compare the effectiveness of steroid delivery by the i.n. or intravenous (i.v.) route in controlling experimentally induced neuroinflammation by systemic injection of lipopolysaccharide (LPS) into mice.
Materials and methods
Mice
Male C57BL/6 mice, aged 7–8 weeks (approximately 25 g body weight), purchased originally from Harlan‐México (Cuidad de México, México) and further bred at our Institute, were employed. Mice were divided into groups of five to six animals, and kept in plexiglass boxes with food and water ad libitum before and during the experiments. The animal room was maintained at 22 ± 3°C with a 12 : 12 h light–dark cycle. Simultaneous experiments with five to six animals per group were performed to characterize the activation state of microglia and astrocytes by flow cytometry and immunofluorescence analysis.
All housing and experimental procedures were conducted under the guidelines established by the Committee on the Care and Use of Experimental Animals of the Instituto de Investigaciones Biomédicas (IBM) at the Universidad Nacional Autónoma de México (UNAM). Experimentation protocols were approved by the animal safety committee of the IBM, UNAM.
LPS‐induced inflammation
A model for systemic bacterial infection‐induced CNS inflammation, reported previously by Qin and collaborators, was employed 26. Mice received either 5 mg/kg of LPS from Escherichia coli serotype 0111:B4 (Sigma, St Louis, MO, USA) injected intraperitoneally (i.p.) or an equivalent volume of the vehicle (0·9% NaCl; endotoxin‐free isotonic saline solution; PiSA, Guadalajara, México) as a control.
Treatments
Three days after a single i.p. injection of LPS, one group of mice received one dose of either isotonic saline solution (ISS) or DX (0·25 mg/kg), i.v. or i.n. Mice were euthanized at different time‐points (3, 12, 24 or 72 h) after drug treatment (Fig. 1a) and the expression of ionized calcium binding adaptor molecule (Iba‐1) and glial fibrillary acidic protein (GFAP), which are expressed specifically in microglia and astrocytes, respectively, was performed by immunofluorescence analysis. The level of central cytokines [tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β and IL‐6] was measured 24 h after treatment. Based on results reported previously 27, other groups of mice were treated either with ISS or DX (0·25 mg/kg) as described above, 2 days after LPS injection, and euthanized 24 h later to characterize microglia activation and blood‐borne cell infiltration into the brain by flow cytometry analysis (Fig. 1b).
Figure 1.
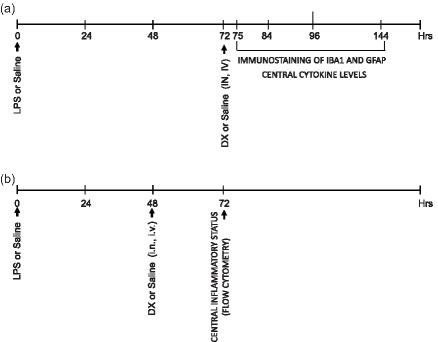
Experimental design. (a) Groups of five to six mice received lipopolysaccharide (LPS) or saline injected intraperitoneally (i.p.) (day 0). Three days later, mice were treated with saline or dexamethasone (DX), delivered intranasally (i.n.) or intravenously (i.v.). The expression of ionized calcium binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP) was studied 3, 12, 24 and 72 h later by immunostaining. Central cytokine levels were evaluated by enzyme‐linked immunosorbemt assay (ELISA) 24 h after DX treatment. (b) The percentage of central cell populations was measured 24 h after DX treatment applied 2 days after LPS injection.
A lethal intraperitoneal injection of 90–100 mg ketamine and 10 mg xylazine per kg was used to euthanize the mice.
Central inflammatory mediators
Mice were anaesthetized as described above and submandibular bleeding was performed before euthanasia. All mice were perfused by cardiac puncture with 250 ml of sterile NaCl 0·15 M to prevent the presence of peripheral molecules in central tissues. Brains were removed rapidly and one half was processed for protein extraction to determine TNF‐α, IL‐1β and IL‐6 concentrations. The other half of the brains were fixed for immunofluorescence analysis, as described below.
Protein extraction
Snap‐frozen brain hemispheres and spleens were homogenized in a lysis buffer [50 mM HEPES, 150 mM NaCl, 1% Nonidet‐p40, 0·5% sodium deoxycholate, 0·1% sodium dodecyl sulphate (SDS)] containing complete protease inhibitors (Roche, IN, USA). Samples were then centrifuged at 16 000 g for 15 min at 4°C and supernatants were collected for analysis. The total amount of proteins in the soluble extract was measured using the Lowry method 28.
Cytokine enzyme‐linked immunosorbent assay (ELISA)
Commercial kits were used to quantify the concentration of the proinflammatory cytokines IL‐1β, IL‐6 and TNF‐α in brain and spleen extracts (BioLegend, San Diego, CA, USA). Briefly, sandwich ELISAs were performed in 96‐well, flat‐bottomed MaxiSorp microtitre plates (Nunc, Roskilde, Denmark). Microplates were coated with the capture antibody for 18 h at 4°C. After washing with phosphate‐buffered saline (PBS)–Tween 20 (0·05%) and blocking for 60 min at room temperature with 2% PBS–bovine serum albumin (BSA), plates were incubated at room temperature for 2 h with standard or samples, washed three times, and incubated for 1 h with the detection antibody at room temperature. Bound detection antibodies were identified using 1 : 1000 diluted avidin–horseradish peroxidase (HRP) and 3,3',5,5'‐tetramethylbenzidine (TMB) as a substrate. Optical density was read before and after the reaction was stopped with H2SO4 2N at 450 and 630 nm, respectively. Results were expressed in pg/ml per mg of protein in the respective soluble extract.
Immunofluorescence (IFC) analysis
Each brain was fixed in 4% buffered paraformaldehyde at 4°C overnight. Free‐floating 30 µm‐thick mouse brain sections were processed as described previously 29. After antigen retrieval by incubating in citrate buffer (0·01 M citric acid, 0·05% Tween 20, pH 6.0) at 70°C for 50 min, samples were washed thoroughly several times with Tris‐buffered saline (TBS) and blocked with a solution of 2% immunoglobulin (Ig)G‐free albumin (Sigma, St Louis, MO, USA) in TBS for 20 min at room temperature. Brain sections were then incubated overnight at 4°C with either rabbit anti‐GFAP (Invitrogen, Carlsbad, CA, USA) or anti‐Iba‐1 (Wako Chemicals, Inc., Richmond, VA, USA), polyclonal antibody or rabbit anti‐dexamethasone (Abcam, Cambridge, UK) or rabbit anti‐CD31 (BioLegend, San Diego, CA, USA) in TBS–2% BSA to detect astrocytes, microglia, DX distribution and endothelial cells, respectively. After washing, sections were incubated for 1 h at room temperature with AlexaFluor 594 goat anti‐rabbit IgG (Molecular Probes, Eugene, OR, USA) diluted in TBS–2% BSA. Samples were mounted onto glass slides in Vectashield medium (Vector Laboratories, Burlingame, CA, USA) containing 4',6‐diamidino‐2‐phenylindole (DAPI) for nuclei imaging. Regions of the hippocampus [cornu ammonis (CA1 and CA2)] and the cortex (R1 and R2) of the images shown in Fig. 2 were selected and processed using Image J software (National Institute of Health, Bethesda, MD, USA). The same programme was employed to estimate the percentage of dexamethasone in the cortex (Fig. 6).
Figure 2.
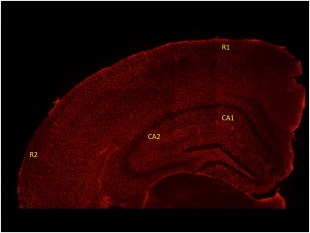
Overview of subregions, including cornu ammonis (CA1 and CA2). Two regions of the cortex were evaluated (R1 and R2). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 6.
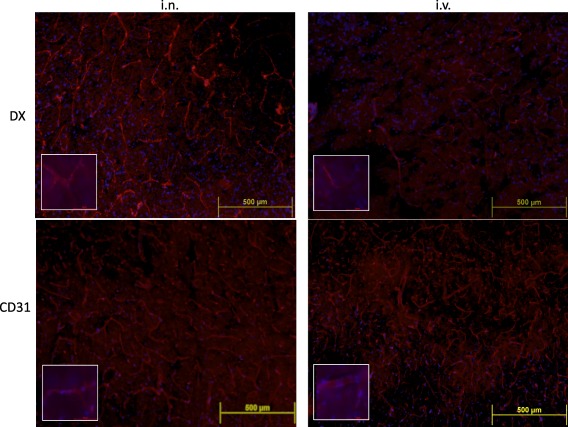
Representative immunofluorescence images of brain slices (four to five mice per group) obtained 24 h after intranasally (i.n.) or intravenously (i.v.) dexamethasone (DX) administration. Merged images of DX (red) or CD31 (red), and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue) staining are shown at ×10 (upper panel) and ×100 (bottom panel) magnification, respectively. Brain vessels in the cortex were distinguished more clearly in i.n. DX‐treated (4·82 pixels/µm2) than in brains of i.v. DX‐treated (1·97% pixels/µm2) mice. [Colour figure can be viewed at wileyonlinelibrary.com]
Brain cell isolation
Parallel experiments in other groups of mice were performed for flow cytometry analysis. Mice were anaesthetized with a mixture of ketamine and xylazine, as described above. Brain cells were obtained following a method reported previously, with minor modifications 30, 31. Briefly, mice were perfused by cardiac puncture with approximately 250 ml of sterile GKN (8 g/l NaCl, 0·4 g/L KCl, 3·56 g/l Na2HPO4·12H2O, 0·78 g/l NaH2PO4·2H2O, 2 g/L D‐(+)‐glucose, pH 7.4) to remove peripheral cells of the central tissues. Brains were collected in ice‐cold magnesium and calcium‐free saline solution (GKN) containing 0·02% (w/v) isotonic bovine serum albumin (GKN‐BSA), and dissociated mechanically using a 100‐μm mesh. The sediment was collected in a 50‐ml centrifuge tube, washed with GKN‐BSA and centrifuged at 400 g for 10 min at slow brake. The pellet was treated with 5 ml digestion buffer (4 g/l MgCl2, 2·55 g/l CaCl2, 3·73 g/l KCI and 8·95 g/l NaCl, pH 6.7) supplemented with 15 U of type‐II collagenase and 500 U of DNase I per brain for 1 h at 37°C, washed with GKN‐BSA and centrifuged at 400 g for 10 min at slow brake.
The cells were suspended in 4 ml of 37% isotonic Percoll and overlayed on top of 70% Percoll. Then, 4 ml of 30% Percoll were loaded slowly onto the top followed by 2 ml of GKN‐BSA. The tubes were centrifuged at 300 g for 40 min; 3 ml of the 70–37% interphase were collected carefully, washed with GKN and suspended in PBS (1X‐0·02% NaN3). Cells were counted, stained with specific antibodies and analysed by flow cytometry.
Flow cytometry
Isolated whole brain cells were treated with CD16/32 antibody to block Fc receptors and stained for surface markers, as reported previously 32. Cells were labelled with anti‐CD11b‐PE/anti‐CD45‐allophycocyanin (APC)/anti‐F4–80‐fluorescein isothiocyanate (FITC), anti‐CD11b‐phycoerythrin (PE)/anti‐CD45‐APC/anti‐lymphocyte antigen 6 complex locus G6D (Ly6G)‐FITC or isotype‐matched control antibodies (BioLegend) and analysed using Attune blue–violet flow cytometer and FlowJo software (Tree Star Inc., Ashland, OR, USA). For analysis, two cell populations were distinguished using antibodies against CD11b and CD45. As microglia, perivascular monocytes/macrophages, granulocytes, dendritic cells, natural killer (NK) cells, lymphocytes and neutrophils are CD11b‐positive, the difference in the level of CD45 expression was used to distinguish between microglia, which expresses low and intermediate levels, and perivascular macrophages and other infiltrated cells, which express high CD45 levels 31. Activation of microglia and macrophages was evaluated by the expression of F4–80 and the population of neutrophils by the LY6G expression.
Statistical analysis
Data are presented as mean ± standard deviation and statistical analyses were performed with GraphPad InStat (GraphPad Software, San Diego, CA, USA). Differences between groups were evaluated using non‐parametric tests (Kruskal–Wallis followed by Mann–Whitney U‐test). A difference was considered statistically significant at P‐value less than 0·05.
Results
Cytokine levels
As shown in Table 1, i.n. administration of DX reduced significantly the level of IL‐6 in the brain extracts 1 day after treatment (Fig. 1a), while no effect was observed in i.v.‐treated mice. TNF‐α levels were not detectable in any of the groups included. No effect in the reduction of IL‐1β was observed between i.n. and i.v. DX‐treated mice.
Table 1.
Cytokine (pg/mg protein) levels in soluble extract from brain of untreated and treated mice 4 days after lipopolysaccharide (LPS) injection
| TNF‐α | IL‐6 | IL‐1β | |
|---|---|---|---|
| Non‐treated | n.d. | 0·72 ± 1·9a | 12·16 ± 2·5a |
| LPS | n.d. | 24·99 ± 8·7b | 15·36 ± 1·4b,c |
| LPS + ISS i.v. | n.d. | 19·44 ± 5·1b,c | 15·03 ± 1·1b,c |
| LPS + ISS i.n. | n.d. | 24·70 ± 7·9b | 18·70 ± 6·8b |
| LPS + DX i.v. | n.d. | 20·73 ± 12·6b,c | 13·48 ± 3·1a,b,c |
| LPS + DX i.n. | n.d. | 14·97 ± 5·1c | 13·77 ± 2·9a,c |
Mean ± standard deviation (s.d.) of brain cytokine levels in groups of five to six C57BL/6 male mice each; n.d. = not detectable; TNF = tumour necrosis factor; IL = interleukin; ISS = isotonic saline solution. Three days after intraperitoneal (i.p.) LPS injection, mice received saline or dexamethasone (DX) (i.v.) or intranasally (i.n.). Levels of central cytokines were measured 24 h later. The effects of the different treatment in each cytokine were compared. Data labelled with the same letter are not significantly different from each other, whereas those with different letters are significantly different. Values were considered statistically significant at P < 0·05 using the Kruskal–Wallis test followed by Mann–Whitney U‐test.
Intranasal delivery of DX effectively reduced the percentage of GFAP+ but not ionized calcium binding adaptor molecule 1 (IBA1)+ brain cells 24 h after administration
Mice were randomized into five groups (five to six mice per group). Based on a previously reported kinetic study 27, 3 days after LPS treatment mice received saline or DX administered i.n. or i.v., and the effect on microglia and astrocytes was evaluated 3, 12, 24 and 72 h thereafter.
Four days after intraperitoneal injection, LPS increased intensively the expression of GFAP in astrocytes (Fig. 3) and Iba1 in microglia (Supporting information, Fig. S1).
Figure 3.
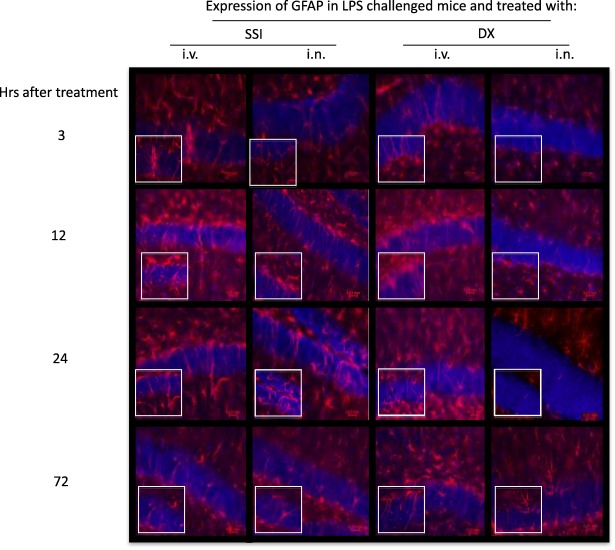
Effect of dexamethasone (DX) on glial fibrillary acidic protein (GFAP) expression in lipopolysaccharide (LPS)‐treated mice. Representative immunofluorescence of 30 μm coronal sections of mouse brain of the different groups stained with anti‐GFAP antibodies (red) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue nuclei). The pictures derive from LPS‐injected mice, treated intranasally (i.n.) or intravenously (i.v.) with saline or DX 3, 12, 24 and 72 h later. The highest number of astrocytes with morphological characteristics of activated cells was observed 24 h after DX treatment. Bottom images represent a fivefold magnification of the region outlined in the box in the corresponding upper image. [Colour figure can be viewed at wileyonlinelibrary.com]
A reduction in GFAP expression was observed 24 h after IN DX administration (Fig. 4). Iba1 expression remained the same at the different times after DX administered i.n. or i.v. (Supporting information, Fig. S1).
Figure 4.

Immunohistological staining and quantification of glial fibrillary acidic protein (GFAP) 24 h after dexamethasone (DX) administration. (a) Representative immunofluorescence of 30 μm coronal sections stained with anti‐GFAP antibodies (red) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue nuclei) in two cornu ammonis (CA1 and CA2) regions and the cortex (R1 and R2). The pictures derive from naive, lipopolysaccharide (LPS)‐treated mice without additional treatment, and animals treated with saline or DX. (b) Table shows the mean ± standard deviation of the percentage of pixels/µm2. The effects of the different treatment in GFAP expression in each region were compared. Data labelled with the same letter are not significantly different from each other, whereas those with different letters are significantly different. [Colour figure can be viewed at wileyonlinelibrary.com]
Thus, a further evaluation of GFAP expression in the CA1 and CA2 and in the cortex (Fig. 2) was performed 24 h after DX administration.
The quantification of GFAP expression 24 h after DX treatment was performed in the cortex and dentate gyrus, as shown in Fig. 4. The mean staining intensity in an average of two sections per mouse was analysed to calculate the area for each photomicrograph and data were plotted. As shown in Fig. 4, a significant reduction in GFAP expression was observed in all the areas analysed in DX‐treated mice. A decrease in GFAP expression was observed in the i.n. treatment compared to the i.v. treatment in all regions, but the difference was statistically significant only in CA2.
Intranasal administration of DX effectively reduced the percentage of CD45high/CD11b/ly‐6G cells 24 h after administration
Taking into account the results described above, the effect of administration of i.n. or i.v.‐DX on microglia was assessed by flow cytometry 24 h after administration (Fig. 1). No effect was observed in the percentage of CD45high/CD11b+ microglial cells in mice treated with the different treatments or in the percentage of activated CD45high/CD11b/F4–80 microglial cells (Fig. 5).
Figure 5.
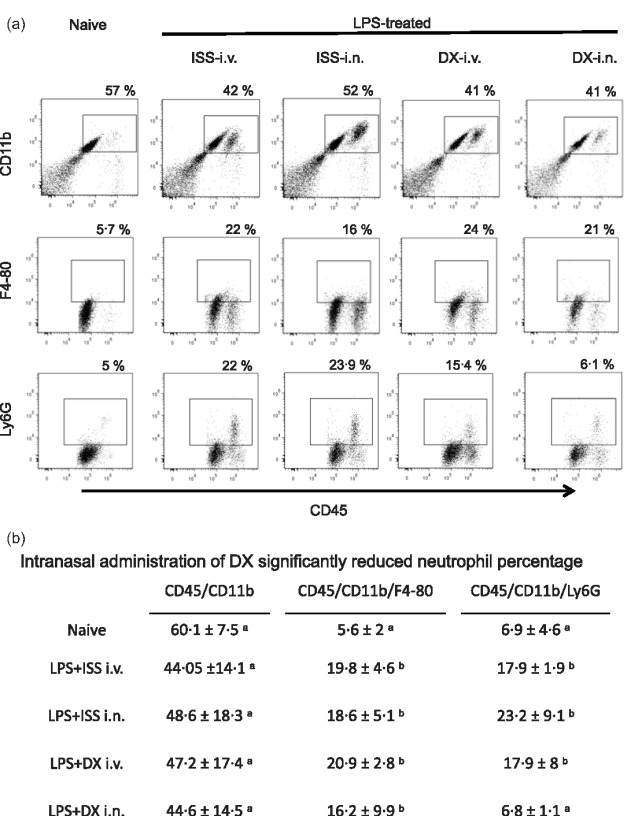
(a) Representative dot‐plots of isolated brain cells from naive and lipopolysaccharide (LPS)‐treated mice that received saline or dexamethasone (DX), stained with anti‐CD11b, CD45 and F4/80 antibodies, and analysed by flow cytometry. (b) Mean ± standard deviation of the percentage of cells before and 48 h after LPS‐treated mice without additional treatment and 24 h after saline or DX treatment. The effects of the different treatment in each cell phenotype were compared. Data labelled with the same letter are not significantly different from each other, whereas those with different letters are significantly different.
After LPS treatment, there was neutrophil infiltration into the brain, and i.n.‐DX was able to reduce the presence of neutrophils in the brain statistically significantly (Ly6G+/CD45+/CD11b+ cells).
Higher levels of DX are observed in the cerebrovasculature of mice after in administration compared with the i.v. route
Fig. 6 shows the distribution of DX in the cortex of i.n. and i.v.‐DX treated mice. As can be appreciated, a more intense and extensive distribution of DX was observed in the brains of i.n.‐treated mice. A similar staining was also observed when slices were treated with anti‐CD31 antibodies to stain endothelial cells.
Discussion
This study demonstrates for the first time the effectiveness of the i.n. route for GC administration to control peripheral infection‐induced neuroinflammation; i.n.‐DX administration to LPS‐treated mice controlled astrocytes activation and proliferation effectively, accompanied by a decreased IL‐6 expression and a reduced infiltration of neutrophils (CD11b/CD45+/Ly6G+) into the brain. When DX was administered at the same dose by the i.v. route, the protective effects were less pronounced.
It is important to mention that DX as well as other drugs administered by the i.n. route may bypass the blood–brain barrier and reach brain regions such as the olfactory bulb, hippocampus and hypothalamus, as was demonstrated previously for other neurosteroids 16. This non‐invasive route is more efficient in reaching cerebral blood vessels compared with systemic administration (Fig. 6).
It has been demonstrated previously that peripheral LPS injection activates microglia and astrocytes, and microglia with intensified Iba‐1 immunostaining density and astrocytes with enhanced GFAP expression have been detected in the cerebral cortex and hippocampus 33, 34. In this study, we confirmed that microglia/macrophages and astrocytes with activated morphology and intensified Iba‐1 and GFAP, respectively, are observed after LPS injection. Importantly, a statistically significant decrease of GFAP levels detected by immunofluorescence in the hippocampus and in the cortex of i.n.‐DX‐treated mice was observed, suggesting decreased astrocyte activation. This decrease was more pronounced in the CA2 region after intranasal administration of DX compared with the i.v. route. Astrocytes are essential for controlling the environment in the brain and regulate neural plasticity providing trophic and metabolic support 34. Although physiological activation of astrocytes is an important protective mechanism in the brain, uncontrolled activation of these cells has detrimental effects, leading to the release of inflammatory mediators and neurodegeneration 35, 36. The results reported in this study suggest that i.n.‐DX may be of interest in controlling the excessive astrocyte activation that accompanies different neuropathological conditions.
Surprisingly, we did not observe decrease of microglia activation after DX treatment as assessed by flow cytometry and immunofluorescence staining. It is possible that the dose of DX employed was not sufficient to reduce the expression of Iba‐1 and F4/80, both markers used commonly to evaluate microglia activation. It is worth mentioning that the morphological profile of microglia and Iba‐1 immunoreactivity did not correlate with the corresponding inflammatory mRNA profile after peripheral LPS administration, as reported recently, and further research is needed to clarify this aspect 36.
It is interesting to emphasize the effectiveness of IN‐DX in reducing neutrophil infiltration into the brain. Previous studies demonstrated the involvement of neutrophils in brain damage and inflammation, and the development of novel therapeutic strategies interfering with pathological neutrophil trafficking is warranted 37.
Finally, we observed a reduction in IL‐6 levels in the brain after i.n.‐DX treatment. It has been demonstrated previously that IL‐6 is not simply a biomarker of brain injury or neuroinflammation but, instead, is a mediator of pathophysiology and an important driving factor for the neuroinflammatory response 38.
Altogether, we observed multi‐target effects of i.n.‐DX in a mouse model of peripheral LPS‐induced neuroinflammation. This murine model resembles the sustained inflammatory milieu that occurs in human sepsis and in other neuroinflammatory conditions. Thus, the effectiveness of this treatment in benefit of the outcome of these pathologies and the quality of life will be studied in the next future.
Independently of the mechanisms involved in central delivery after i.n. administration, there is an increasing interest in this non‐invasive route to deliver drugs and macromolecules into the CNS 39, 40. Enhanced uptake of drugs by the nasal epithelium has been explored by including cell‐penetrating peptides (CPP) in i.n. formulations. This route has also been effective to administer hormones and non‐steroidal anti‐inflammatory drugs, such as ibuprofen, but to our knowledge it has not been employed for the delivery of GC into the central nervous system.
In conclusion, the results obtained in this study support the effectiveness of GC i.n. administration as a new therapeutic alternative of potential value in many different neuropathologies in which neuroinflammation is related to the disease. Importantly, i.n. administration may overcome the non‐desirable effects mediated by the high systemic doses that must be employed to reach GC therapeutic levels to control CNS inflammation.
Author contributions
E. S., G. F. and G. M. conceived and designed the experiments. G. M., M. B., A. E., A. F., G. D. and G. A. performed the experiments. E. S., H. B., A. F. and G. G. analysed the data. G. G. and D. M. contributed reagents, materials and analysis tools. E. S., G. F., G. G., d. R. A. and H. B. wrote the paper.
Disclosure
Authors declare that they have no competing interests.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Immunohistological staining and quantification of the microglial marker ionized calcium binding adaptor molecule 1 (Iba1). Representative immunofluorescence of 30 μm coronal sections stained with Iba1, microglia (red) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue nuclei). The pictures derive from mice that received lipopolysaccharide (LPS) and 3, 12, 24 and 72 h after treatment with saline or dexamethasone (DX), either intranasally (i.n.) or intravenously (i.v.). The highest number of microglia with morphological characteristics of activated cells was observed in mice 24 h after DX administration. Bottom images represent a fivefold magnification of the region outlined in the box in the corresponding upper image.
Acknowledgements
The authors thank Jacquelynne Cervantes, Martha E. Carrasco and Marisela Hernández for their technical support and Guillermo Olvera for experimental support and Daniel Garzón for his assistance with animal care. Juan Francisco Rodriguez edited the English version of the manuscript. This work was supported by CONACyT (SALUD‐2013–01‐201448 to ES) and DGAPA‐UNAM (IN201116 to G. G.) México. This study was also supported by the Institutional program ‘Programa de Investigación para el Desarrollo y la Optimización de Vacunas, Inmunomoduladores y Métodos Diagnósticos del IIB’ (PROVACADI).
References
- 1. Corrigan F, Mander KA, Leonard AV, Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation 2016; 13:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schwartz M, Deczkowska A. Neurological disease as a failure of brain‐immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol 2016; 37:668–79. [DOI] [PubMed] [Google Scholar]
- 3. Beer MS, Schmeidler J, Lesser GT et al Corticosteroid, but not NSAIDs are associated with less Alzheimer neuropathology. Neurobiol Aging 2012; 33:1258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt J, Gold R, Schönrock L, Zettl UK, Hartung HP, Toyka KV. T‐cell apoptosis in situ in experimental autoimmune encephalomyelitis following methylprednisolone pulse therapy. Brain 2000; 123:1431–41. [DOI] [PubMed] [Google Scholar]
- 5. Whitehouse MW. Anti‐inflammatory glucocorticoid drugs: reflections after 60 years. Inflammopharmacology 2011; 19:1–19. [DOI] [PubMed] [Google Scholar]
- 6. Paul C, Bolton C. Inhibition of blood–brain barrier disruption in experimental allergic encephalomyelitis by short‐term therapy with dexamethasone or cyclosporin A. Int J Immunopharmacol 1995; 17:497–503. [DOI] [PubMed] [Google Scholar]
- 7. Marchi N, Granata T, Freri E et al Efficacy of anti‐inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLOS ONE 2011; 6:e18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salassa RM, Bennet WA, Keating FR. Postoperative adrenal cortical insufficiency; occurrence in patients previously treated with cortisone. JAMA 1953; 152:1509–15. [DOI] [PubMed] [Google Scholar]
- 9. Melby JC. Systemic corticosteroid therapy: pharmacology and endocrinologic considerations. Ann Intern Med 1974; 81:505–12. [DOI] [PubMed] [Google Scholar]
- 10. Danilczuk Z, Ossowska G, Wróbel A, Lupina T. Glucocorticoids modulate behavioral effects induced by dopaminergic agonists in rats. Pol J Pharmacol 2001; 53:467–73. [PubMed] [Google Scholar]
- 11. Schäcke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol 2007; 275:109–17. [DOI] [PubMed] [Google Scholar]
- 12. Brown ES. Effects of glucocorticoids on mood, memory, and the hippocampus. Treatment and preventive therapy. Ann NY Acad Sci 2009; 1179:41–55. [DOI] [PubMed] [Google Scholar]
- 13. Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol 2007; 275:62–70. [DOI] [PubMed] [Google Scholar]
- 14. Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv 2014; 5:709–33. [DOI] [PubMed] [Google Scholar]
- 15. Deng‐Bryant Y, Readnower R, Leung LY, Tortella F, Shear D. Methods of drug delivery in neurotrauma. Methods Mol Biol 2016; 1462:89–100. [DOI] [PubMed] [Google Scholar]
- 16. Ducharme N, Banks WA, Morley JE et al Brain distribution and behavioral effects of progesterone and pregnenolone after intranasal or intravenous administration. Eur J Pharmacol 2010; 641:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chapman CD, Frey WH II, Craft S et al Intranasal treatment of central nervous system dysfunction in humans. Pharm Res 2013; 30:2475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Medina G, Ji G, Grégoire S, Neugebauer V. Nasal application of neuropeptide S inhibits arthritis pain‐related behaviors through an action in the amygdala. Mol Pain 2014; 10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen H, Chen CC, Acosta C, Wu SY, Sun T, Konofagou EE. A new brain drug delivery strategy: focused ultrasound‐enhanced intranasal drug delivery. PLOS ONE 2014; 9:e108880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minn A, Leclerc S, Heydel JM et al Drug transport into the mammalian brain: the nasal pathway and its specific metabolic barrier. J Drug Target 2002; 10:285–96. [DOI] [PubMed] [Google Scholar]
- 21. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 2003; 7:569–81. [DOI] [PubMed] [Google Scholar]
- 22. Sakane T, Akizuki M, Yamashita S, Nadai T, Hashida M, Sezaki H. The transport of drug to the cerebrospinal fluid directly from the nasal cavity: the relation to the lipophilicity of the drug. Chem Pharm Bull 1991; 39:2456–8. [DOI] [PubMed] [Google Scholar]
- 23. Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci 2000; 11:1–18. [DOI] [PubMed] [Google Scholar]
- 24. Pietrowsky R, Strüben C, Mölle M, Fehm HL, Born J. Brain potential changes after intranasal vs. intravenous administration of vasopressin: evidence for a direct nose–brain pathway for peptide effects in humans. Biol Psychiatry 1996; 39:332–40. [DOI] [PubMed] [Google Scholar]
- 25. Ali J, Ali M, Baboota S et al Potential of nanoparticulate drug delivery systems by intranasal administration. Curr Pharm Des 2010; 16:1644–53. [DOI] [PubMed] [Google Scholar]
- 26. Qin L, Wu X, Block ML et al Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007; 55:453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meneses G, Bautista M, Florentino A et al Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J Inflamm (Lond) 2016; 13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951; 193:265–75. [PubMed] [Google Scholar]
- 29. Hernandez‐Zimbron LF, Luna‐Muñoz J, Mena R et al Amyloid‐β peptide binds to cytochrome c oxidase subunit 1. PLOS ONE 2013; 7:e42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein‐reactive CD4+ T cells compared. J Immunol 1995; 1:4309–21. [PubMed] [Google Scholar]
- 31. Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 2012; 37:1491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vainchtein ID, Vinet J, Brouwer N et al In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 2014; 62:1724–35. [DOI] [PubMed] [Google Scholar]
- 33. Hoogland ICM, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflamm 2015; 12:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Catorce MN, Gevorkian G. LPS‐induced murine neuroinflammation model: main features and suitability for pre‐clinical assessment of nutraceuticals. Curr Neuropharmacol 2016; 14:155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bronzuoli MR, Iacomino A, Steardo L, Scuderi C . Targeting neuroinflammation in Alzheimer's disease. J Inflamm Res 2016; 9:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba‐1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016; 64:300–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zenaro E, Pietronigro E, Della Bianca V et al Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med 2015; 21:880–6. [DOI] [PubMed] [Google Scholar]
- 38. Yang SH, Gangidine M, Pritts TA, Goodman MD, Lentsch AB. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 2013; 40:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shah B, Khunt D, Misra M, Padh H. Non‐invasive intranasal delivery of quetiapine fumarate loaded microemulsion for brain targeting: formulation, physicochemical and pharmacokinetic consideration. Eur J Pharm Sci 2016; 91:196–207. [DOI] [PubMed] [Google Scholar]
- 40. Phukan K, Nandy M, Sharma RB, Sharma HK. Nanosized drug delivery systems for direct nose to brain targeting: a review. Recent Pat Drug Deliv Formul 2016; 10:156–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Immunohistological staining and quantification of the microglial marker ionized calcium binding adaptor molecule 1 (Iba1). Representative immunofluorescence of 30 μm coronal sections stained with Iba1, microglia (red) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue nuclei). The pictures derive from mice that received lipopolysaccharide (LPS) and 3, 12, 24 and 72 h after treatment with saline or dexamethasone (DX), either intranasally (i.n.) or intravenously (i.v.). The highest number of microglia with morphological characteristics of activated cells was observed in mice 24 h after DX administration. Bottom images represent a fivefold magnification of the region outlined in the box in the corresponding upper image.