

Abstract
Genetic instability is an important characteristic of cancer. While most cancers develop genetic instability at some stage of their progression, sometimes a temporary rise of instability is followed by the return to a relatively stable genome. Neither the reasons for these dynamics, nor, more generally, the role of instability in tumor progression, are well understood. In this paper we develop a class of mathematical models to study the evolutionary competition dynamics among different sub-populations in a heterogeneous tumor. We observe that despite the complexity of this multi-component and multi-process system, there is only a small number of scenarios expected in the context of the evolution of instability. If the penalty incurred by unstable cells (the decrease in the growth due to deleterious mutations) is high compared with the gain (the production rate of advantageous mutations), then instability does not evolve. In the opposite case, instability evolves and comes to dominate the system. In the intermediate parameter regime, instability is generated but later gives way to stable clones. Moreover, the model also informs us of the patterns of instability for cancer lineages corresponding to different stages of progression. It is predicted that mutations causing instability are merely “passengers” in tumors that have undergone only a small number of malignant mutations. Further down the path of carcinogenesis, however, unstable cells are more likely to give rise to the winning clonal wave that takes over the tumor and carries the evolution forward, thus conferring a causal role of the instability in such cases. Further, each individual clonal wave (i.e. cells harboring a fixed number of malignant driver mutations) experiences its own evolutionary history. It can fall under one of three types of temporal behavior: stable throughout, unstable to stable, or unstable throughout. Which scenario is realized depends on the subtle (but predictable) interplay among mutation rates and the death toll associated with the instability. The modeling approach provided here sheds light onto important aspects of the evolutionary dynamics of instability, which may be relevant to treatment scenarios that target instability or damage repair.
Keywords: Chromosomal instability, microsatellite instability, driver mutation, passenger mutation
1. Introduction
Genome instability and mutations have been identified as enabling characteristic of cancer [22]. Significant progress has been made in the last decades in our understanding of very complex and variable causes of genomic instability in cancer. It has been proposed that instability is caused by abrogated mitotic checkpoints [53], hypoxia [14, 9], chromosomal segregation errors [16], defective DNA damage repair [17], shortening of telomeres [43], reactive oxygen species [27], and oncogene-induced DNA replication stress [21]. The genetic mechanisms leading to instability can be studied by looking into the diverse array of defects associated with different components of the DNA-maintenance machinery (the “caretakers” of the genome [26]).
For several decades, scientists have been trying to answer questions related to genetic instability. What is the genetic basis of instability? What is the correlation between instability and disease course/severity? What is the role of instability in cancer initiation and progression? Ultimately the hope is that this information can be used to identify new cancer treatment targets.
In the theoretical and mathematical literature, the question that is asked most frequently is that of the role of instability in tumor progression. Is it a cause or a “side-effect” of carcinogenesis? In this context, the concept of mutator phenotype was introduced by [35, 36], and much theoretical work has been devoted to understanding the role of genetic instability in cancer initiation and progression, see e.g. [33] and the references therein. Specifically, researchers focused on chromosomal instability (CIN) and microsatellile instability (MSI), which are prevalent in many cancers (e.g., 85% of colorectal cancers (CRCs) are characterized by CIN, and the remaining15% show MSI [18]). In [42], the dynamics of CIN in colon cancer was formulated by means of a modification of a two-step model of [39]. In [28], the dynamics of instability generation was investigated in the context of sporadic and familial colon cancers. Two different pathways to instability were assumed: a number of single point mutations leading to the generation of CIN, and a two-hit process of MSI gene inactivation leading to phenotypes characterized by MSI.
Important insights into the temporal molecular dynamics of cancer, including the evolution of instability, have been obtained by comparing stochastic models with epidemiological data. [38] fitted a stochastic multistage carcinogenesis model to the large database of colorectal and pancreatic cancers in the Surveillance Epidemiology and End Results (SEER) registry. The results are consistent with the idea that chromosomal instability does not come as a first step in carcinogenesis but arises later one, after the first driver mutation has been obtained. [34] created a model that allowed multiple types of genomic instability and an arbitrary number of mutational stages. The model was fitted to US Caucasian colon cancer incidence data. It was found that the model with just one type of instability provided a very good fit to the dataset, and therefore it s unlikely that this methodology can reveal additional information about the types of instability prevalent in colon cancer.
[4] used a Wright-Fisher type model to describe the acquisition of driver mutations that triggered clonal expansions within a tumor. Cancer progression was interpreted as the result of multiple sequential mutations, each making a relatively small but positive effect on net cell growth. It was shown that, as long as the average selective advantage per mutation was on the order of 1%, carcinogenesis could occur in a biologically reasonable time even without an elevated mutation rate. An increased mutation rate reduced the waiting time to cancer accordingly. Analysis of [48] revealed that in the cases of the lung and colon cancers, the waiting time to cancer was dominated by the selective advantage per mutation and the net clonal expansion rate (and the mutation rate had less effect). [45] also used a Wright-Fisher type model to investigate under which circumstances genetic instability evolved during carcinogenesis. Both advantageous and disadvantageous mutations were included, as well as mutations that increased the cells’ mutation rate. This model demonstrated that the strength of selection for additional driver mutations determines whether or not a cancer is likely to evolve a mutator phenotype. In particular, if the selection for the driver mutations is very strong, neutral mutator mutations that evolve by drift are effectively removed from the growing clones. If the selection for the driver mutations is very week, then the clone dynamics are driven almost entirely by drift (and not clonal selection), such that selection for unstable cells is weak. Therefore, instability is most likely to be generated for an intermediate range of selection for driver mutations.
In [30], we asked the question of optimality of the rate of chromosomal loss, from the point of view of cancer. Specifically, a model of cancer initiation was formulated where two hits lead to the creation of a phenotype that escaped homeostatic control. This is reflective of the mechanism of the loss-of-finction mutation, which is observed in cancers driven by the inactivation of tumor suppressor genes. The following optimization problem was posed. On the one hand, genetic instability, which is characterized by an elevated rate of genetic change, leads to the creation of many harmful or deleterious mutations, thus lowering the effective growth rate of the cell colony. On the other hand, CIN with its increased rate of chromosomal loss can facilitate the acquisition of the second inactivation mutation, thus accelerating progress toward cancer. What is the chromosomal loss rate which leads to the fastest cancer initiation and growth? The idea is that out of many lineages that simultaneously coexist in tissues, the ones that are characterized by this optimal chromosomal loss rate are most likely to harbor cancerous growth which is ultimately observed.
The simple initial model was then refined to include the possibility of the mutation rate being a function of time [29]. What is the optimal (again, from cancer’s prospective) temporal course of instability that leads to the fastest growing tumors? This problem was solved by the methods of optimal control theory, yielding a simple and intuitive solution. It was shown that three types of scenarios can be optimal, depending on system parameters, namely, the cost of instability (the instability-associated death rate) and the benefit of instability (the increase in the rate of double-hit mutant production): (a) For high death rates and relatively low gain in mutation rates, the level of instability stays low throughout the growth. In this case instability is disadvantageous for the cancer population at all times and the best strategy is to stay stable. (b) In an intermediate case, the level of instability increases at the beginning of cancer growth and decreases as tumor cells progress more. In this case, it is advantageous for the population to keep cells with high mutation rates at first, then switch to stability. (c) For low instability-associated death rates and high gain in mutations, it is optimal for the cancer cells to stay unstable throughout the growth. All these patterns of instability are realistic; the time-dependent stable – to unstable – back to stable pattern is especially interesting, given biological evidence supporting it (see the Discussion section for more details). The ideas presented in [29] were further developed mathematically and given analytical rigor in [55, 47].
In the approach of [30, 29, 47], the instability-induced mutation rate, p, was treated as a function of time, and the optimization procedure sought the function p(t) that maximized tumor growth. The colony of cells was considered homogeneous with respect to the time-dependent mutation rate, p(t), that is, all cells were assumed to have the same mutation rate. At the same time, cellular heterogeneity is extremely common in tumors, see e.g. [51, 1]. It is rooted in the evolutionary nature of the process of tumorigenesis, and is especially prevalent in unstable tumors [50, 10]. In the present manuscript, we include genetic heterogeneity in the description on the tumor evolution, and investigate if the patterns of instability identified in [29] can still be discovered in this more complex and more realistic system.
We consider cancer evolution on a mutation-selection network that allows for “mutator” mutations (the ones that give rise to genetic instability at different levels) and also multiple driver mutations that lead to the progression to cancer. We examine the relative abundances of different subpopulations in a highly heterogeneous colony and identify the changes in the level of genetic instability, as a result of mutations and selection, at various stages of tumor progression. We find that the three patterns obtained by the method of optimization in [29] are observed in this much more complex, heterogeneous, multi-step system. In particular, we find that genetic instability may or may not evolve, or it may evolve early on during tumorigenesis and later get eliminated, giving way to a more stable population. In contrast with the methodology of [29], we do not perform an optimization procedure but instead just observe the natural evolution of various phenotypes in a heterogeneous setting. We further study the general evolutionary pattens exhibited by cell clones at different stages of carcinogenesis. We find that the early clones containing a small number of driver mutations, are likely to be dominated by stable phenotypes, and instability may or may not develop as a “passenger”. Intermediate and late clones containing more transforming mutations are more likely to have originated in an unstable cells, thus giving instability a causal role in cancer progression. We compare our results to relevant recent findings in biological literature, and discuss them in the context of treatment targets.
2. The scope of modeling
There are different types of genetic alterations contributing to genetic stability. Some of these alterations are mutations of specific genes, amplifications, deletions or rearrangements of chromosome segments, gain or loss of entire chromosomes, etc. Different genetic changes can accumulate in distinct subsets of cell populations and all can contribute the progression of cancer with different mutation rates [49, 11]. We present a wide class of models that view tumor progression as accumulation of genome alterations during a series of cell divisions. Compared to our previous work [29], the present models provide an alternative way to describe the dynamics of instability. They allow for a more detailed characterization of the process of instability generation and take into account the fact that mutations of different critical genes, different abnormalities, etc., give rise to different levels of instability and contribute to tumor progression with different rates.
2.1. General formulation
During a cell division, genomic stability is maintained by four major mechanisms [49]: high-fidelity DNA replication in S-phase, precise chromosome segregation in mitosis, error free repair of sporadic DNA damage, and a coordinated cell cycle progression. According to the classification given in [22], there are three types of defects in the caretaker genes: (i) defects in mechanisms detecting DNA damage and activating the repair machinery, (ii) defects in mechanisms that directly repaire damaged DNA, and (iii) defects in mechanisms that inactivate/intercept mutagenic molecules before they have damaged the DNA. All this results in a number of possible pathways to instability. Therefore, we will assume that there are n different types of instability that can be acquired by means of various (epi-)genetic events. Let us call these types x0, …, xn, where type x0 is stable, and the subscripts allow to distinguish different levels or types of instability. In general, the phenotypes of different stability properties are connected by means of a mutation network, which specifies what types can give rise to which other types via mutations. This results in a matrix description,
where x is a vector (x0, …, xn)T, the first term on the right corresponds to production, and the second to the death of cells. The equation above describes unlimited exponential growth; the competition dynamics will be introduced shortly. The matrix δ is a diagonal matrix with the entries being death rates of the different types,
The (n+1)×(n+1) matrix M represents reproduction and is generally dominated by the diagonal elements, with off-diagonal elements indicating mutations. For example, consider the following two matrices:
| (1) |
The matrix Mrad corresponds to the types x1, …, xn each produced by a single mutation directly from the stable type. The matrix Mseq describes a sequence of types x0, …, xn, such that each of them is produced by a mutation from the previous one. The subscripts “rad” and “seq” stand for “radial” and “sequential”, see below. The division rate of cells has been normalized to one. Clearly, many other possibilities for the mutation matrix M exist, including the processes with back mutations.
Next, we introduce the cancerous transformation. If we only include the first rate-limiting step in malignant transformation, the linear dynamics can be described by two types of population, x0, …, xn and y0, …, yn, where the first group is “benign” and the second group is “cancerous”. Suppose that the benign cells divide at a rate normalized to 1, and the cancerous cells have an elevated division rate, a. Then the linear dynamics are given by
| (2) |
where Z is the vector containing all the populations, Z = (x0, …, xn, y0, …, yn)T, and matrices , D, and R have a 2 × 2 block structure. The matrix describes cell divisions and mutations, R provides division rates, and D is the death matrix. These matrices are given by:
As before, Mx and My capture the mutation networks generating the unstable types in the benign and malignant populations, and P describes the cancerous transformation. We denote by pi the rate at which cells in the population xi undergo genetic alterations and transform into cancerous type yi as a result of instability. Then P is an (n + 1) × (n + 1) diagonal matrix with entries (p0, …, pn),
The alterations captured by this matrix include but are not limited to various forms of specific gene mutations, amplifications or rearrangements of chromosome segments, gain or lose of entire chromosomes. pi can be chosen to be the rate of basic point mutations if we assume transformation results from small scale change in DNA sequence [6] or it can be taken to represent the rate of chromosomal loss. For more detailed discussion see [30, 29].
We assume that the cell types xi are characterized by higher levels of genetic instability for larger i. Therefore, the mutation rates pi comprise an increasing function of i. On the other hand, genetic instability can lead to high death toll in the population [11]. Therefore, the death rates denoted by di also comprise an increasing sequence. This is discussed in more detail in the following sections. Figure 1 shows schematic representations of the two types of models corresponding to the two matrices in (1).
Figure 1. Diagram for two simple models.
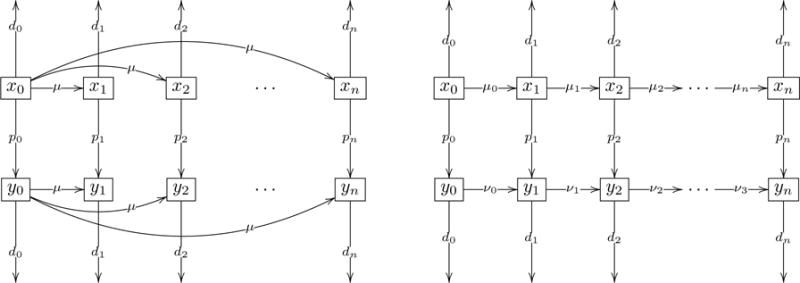
Variables xi, 0 ≤ i ≤ n represent non-cancerous populations and yi, 0 ≤ i ≤ n represent the cancer populations. The index indicates the level of instability. Parameters di represent death rates of populations xi and yi, parameters pi represent instability induced mutation rates, μ, μi, 0 ≤ i ≤ n and νi, 0, ≤ i ≤ n represent mutations generating the unstable types. (a) Radial mutation network model. (b) Sequential mutation network model.
So far we only presented equations that describe exponential growth of the populations. In order to account for homeostatic control and competition among the cells, we will use two alternative nonlinear models: (a) the quasispecies-type equations, and (b) the logistic growth equations. Qualispecies equations can be obtained from an exponential growth model, Ż = QZ by subtracting a term that represents competition and is related to the mean fitness of the types:
where
with the four multipliers on the right having dimensions 1 × 2(n + 1), 2(n + 1) × 2(n + 1), 2(n + 1) × 2(n + 1), and 2(n + 1) × 1. This definition of ϕ is slightly different from the usual one. Quantities and characterize the degree of participation of the corresponding population in the competition dynamics. For example, if all the types are under homeostatic control, then we take for all i, and the term ϕ equals the mean fitness of all types (this is the conventional definition of ϕ). We however assume that populations y0, …, yn are malignant, and unlike populations x0, …, xn, they are not under homeostatic control, but expand indefinitely. This corresponds to for all i.
The second way to introduce competition is to use the logistic growth model by replacing the matrix R in equation (2) with the following,
Here, we set
and the quantities , are carrying capacities of the corresponding populations.
The above formalisms can be generalized in a straightforward way if we were to take into account more driver mutations leading to cancer development. This corresponds to more rows in figure 1 and is described in section 3.2.
2.2. Radial mutation network model
As it is described above, there are many possibilities for the particular form of the mutations network that connects types xi (and yi) together. One possibility is presented in diagram 1(a), see matrix Mrad, equation (1). There, cell types of various levels of instability can be produced by means of single mutation events from the original type, x0. This gives rise to a fan-like, or radial, mutation network. The mutation network topology must be coupled with further assumptions on the system dynamics. As the first possibility, we will use the quasispecies-type equations. In this case, the processes of growth and mutations illustrated in diagram 1(a) are given by the following equations:
| (3) |
| (4) |
| (5) |
| (6) |
The term containing ϕ, with
| (7) |
accounts for the homeostatic control present in a system of xi cells. In the absence of cancerous mutations the total number cells in the populations xi near equilibrium stays constant and is normalized to 1. yi cells break out of regulation and grow exponentially. The term ϕ in the equations for yi is added to represent a partial, non-symmetric, homeostatic control that may play some role at the beginning of the growth of yi cells. Later on that term becomes simply a correction to the growth rate of the yi cells, similar to [29].
This way of modeling the above dynamics with quasispecies-type equations is not unique. We also considered the model with the ϕ−term removed from the equations for the cancerous populations yi in (5, 6). The results obtained are qualitatively similar.
Another way of formulating the dynamics on diagram 1(a) is to assume logistic growth in both non-cancerous and cancerous cell populations. With logistic growth the equations (3,6) will change as follows
| (8) |
| (9) |
| (10) |
| (11) |
where , and F are defined in section 2.1. All these models give similar outcomes, as discussed in the following sections.
2.3. Sequential mutation network model
The second mutation network considered here is depicted in figure 1(b) and expressed by matrix Mseq, equation (1). It assumes that high levels of genetic instability are results of accumulation of genetic alterations in a cell during a series of cell divisions. As before, x0 is the population size of non-cancerous stable cells.
We group the offspring of type x0 that have a single mutation of a critical gene as type x1. Unlike the radial mutation network model, where different levels of instability in populations xi result from different types of point mutations of stable cells, here the increased level of instability in xi is a result of mutations of population xi−1 that occur with rate μi−1. Cancerous cells can undergo similar subsequent point mutations with rate νi. Under the quasispecies-like dynamics, the model is given by the following ODEs:
| (12) |
| (13) |
| (14) |
| (15) |
ϕ is given in (7). Again the dynamics described above can be formulated without the ϕ− term in the yi equations in (14, 15). Another possibility is to replace the expression for ϕ with the following term
| (16) |
Similar to the radial mutation network model, we can also assume logistic growth for non-cancerous and cancerous cell populations. Changing the model will lead to quantitative changes of the outcome as shown below, but the results remain qualitatively robust.
3. Results
3.1. One driver mutation
In this section we investigate how the level of instability changes during the process of tumor growth in the radial and sequential mutation network models (figure 1), described in (3,6) and (12,15). To analyze the dynamics of genetic instability of the system, we first examine the changes in the population sizes of cancer subpopulations yi depending on the rate of instability pi and the death rate di.
The death rates di depend upon the level of instability of cells. If a population is characterized with a small amount of genetic instability the chance of accumulating deleterious mutations is relatively small. On the other hand, higher levels of instability increase the generation rate of deleterious mutations, which can reduce the cells’ fitness and lead to death. Therefore, di can be described by a monotonically increasing (non-decreasing) function of i. The behavior of the radial and sequential models were studied with various functions for the death rates. As all increasing functions of i give similar results, we will mostly illustrate our analysis using the following linear function for the death rates of unstable populations:
| (17) |
The magnitude D in equation (17) characterizes the maximum death rate resulting from genetic instability that is possible in the system. The effect of death on the solution was investigated by considering several values of the magnitude D. The reproduction rate of stable cells is normalized to one, and their death rate is taken to be 10−4.
In stable cells, inactivating point mutations have been estimated to occur at a rate of 10−7 per gene per cell division, μ = 10−7. We assume that in stable cells the mechanism of cancerous mutations is of the same form as basic mutations and let p0 be the basic mutation rate 10−7. As cells undergo various forms of genetic alterations, they acquire higher rates for cancerous mutations and the probabilities (per cell division) of these mutations in unstable cells are known to range from 10−7 to 10−2 [32, 40]. We assign the rate of genetic instability, p1, in this range, assuming that the initial transition from stable cells to an unstable population can increase the rate of cancerous mutations by a few orders of magnitude. The rest of instability rates are represented by an increasing function of i. In particular we let p2, p3, …, pn be given by the following sequence:
| (18) |
An example of solution curves of the cancerous and non-cancerous populations is presented in Figure 2. Plotted are the simulated time-series of the various population levels; stable populations x0 and y0 are depicted by thick black lines in panels (a) and (b), and populations with higher levels of genetic instability are represented by gray and dashed lines. We can see in Figure 2(a) that the non-cancerous cells, which do not escape homeostasis, remain genetically stable during tumor progression. Cancerous cells (Figure 2(b)) grow exponentially and are also dominated by stable cells. For the specific parameter set, at the beginning of growth, a certain fraction of cancer cells becomes unstable (the line corresponding to i = 3 dominates the unstable population). As tumor progresses further, the system switches to having more cells in the populations that are less unstable (the line corresponding to i = 1 becomes dominant). This is one possible scenario for the dynamics of genetic instability. As explained below, depending on the parameter regime other scenarios may be observed.
Figure 2. Plots of cancerous and non-cancerous populations for radial mutation network, equations (3–6).
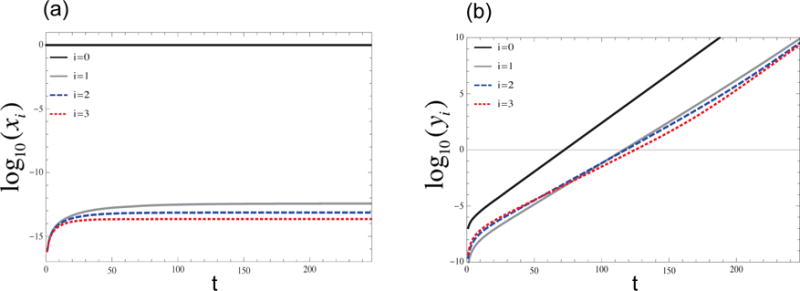
(a) The populations xi, log10D = −1.2. Non-cancerous populations stay relatively stable for all values of t. (b) Cancerous population for an intermediate death rate, log10D = −1.2. In this case the more unstable populations are larger at the beginning of growth, but as tumor progress the system switches to higher number of cells in more stable populations. The rest of parameters are d0 = 10−4, p0 = 10−7, p1 = 10−4, a = 1.2, μ = 10−7, n = 3.
In order to quantify the dynamics of genetic instability, we will use the instability index,
| (19) |
which is simply the logarithm of the mean mutation rate of the population measured with respect to the driver mutations (quantity pi for each instability type). Analysis of the solutions of the two models in figure 1(a,b) for a variety of biologically meaningful parameter values revealed three possible scenarios for temporal behavior of the instability index. Interestingly, these three scenarios coincide with the ones found by a completely different methodology in [29]. They are listed below:
For some parameter values the level of instability does not evolve and stays low throughout growth, Figure 3(a) plot ➀.
Figure 3(a) plot ➁ presents a parameter regime where the level of instability increases initially and decreases as tumor continues to grow.
In some cases, the level of instability increases and remains high at all times, see Figure 3(a) plot ➂.
Figure 3. The dynamics of the instability index for models with one driver mutation and many driver mutation.
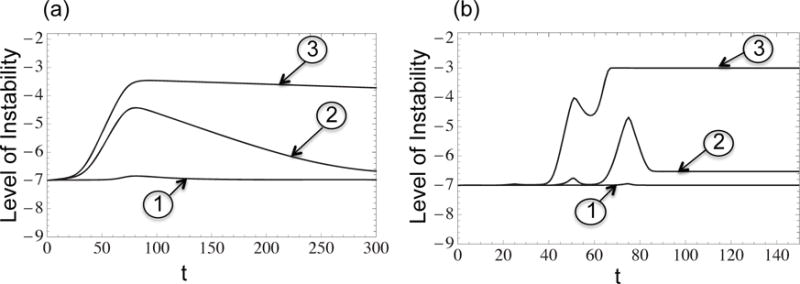
➀, ➁, ➂ correspond to the three cases in section 3.1. (a) One driver mutation model with quasispecies equations, radial mutation network: n = 3, a = 1.2, μ = 10−7, p0 = 10−7, p1 = 10−5, p2 = 10−3.5, p3 = 10−2, d0 = 10−4, d1 = 10−3, 10−2.5, 10−3.5, d2 = 10−1.5, 10−2, 10−3, d3 = 10−0.5, 10−2.5 for cases ➀, ➁, ➂ respectively. (b) Many driver mutation model with logistic growth equations, radial mutation network: n = 3, m = 6, , , , , ai = 1 + 0.05i, 0 ≤ i ≤ 6, μ = 10−7, p0 = 10−7, p1 = 10−5, p2 = 10−3, p3= 10−2, d0 = 10−4, d1 = 10−0.5, 10−1, d2 = 10−0.2, 10−0.3, 10−0.5, d3 =10−0.1, 10−0.15, 10−0.2, for cases ➀, ➁, ➂ respectively.
In this section we investigate how these three cases depend on the parameter regimes. In particular, we are interested to determine for what values of mutation rates pi and death rates di the system falls into the intermediate case, where instability is transient: it comes up at an early stage and decreases later on.
Manipulation of the death rates di given in (17) leads to the conclusion that for a wide choices of instability rates, it is possible to transition between the three scenarios by only changing the magnitude of the death rates D. An example of such a transition is illustrated in Figure 3(a). When fitness reduction due to instability is small, the cancer population will most likely keep the level of instability high throughout growth, Figure 3(a) ➀. On the other hand, if we assume higher penalty for instability (large D) cancer will remain stable at all times, Figure 3(a) ➂. There also is the intermediate case given in Figure 3(a) ➁, where the death rate is not too large and instability speeds up tumor growth at first, but appears to become a “burden” for the cancer ells after the initial growth.
Which of the three scenarios is observed for a particular system? In the model of [29], the strongest determiners of instability were the mutation rate and the death rate of cells. Using this lead, here we study the effect of the mutation rates pi by determining if the system undergoes scenarios 1, 2, or 3 for various points in the (p1, D) plane. We start with the simple case n = 1. Figure 4 presents a typical two-dimensional phase diagram, which quantifies the three cases. To obtain this figure, for each parameter pair (D, p1) (see equations 17, 18), we simulated the ODEs until the tumor population reached a maximum fixed size of 1014. For each simulation, we have to distinguish which of the three behavior classes (Figure 3) is observed. Because there is a continuum of dynamical patterns, we designed a rule to classify the time series of the instability index into one of the three cases. In particular, we consider any increases of the instability curve that are less than an order of magnitude as insignificant changes, and categorize these types of plots as scenario 1. When the increase in the instability level is higher than an order of magnitude and stays high at the time of simulation termination, the system is in scenario 3. Otherwise the behavior is classified as scenario 2.
Figure 4. Level of instability depending on the parameters D and p1 for radial mutation network model.
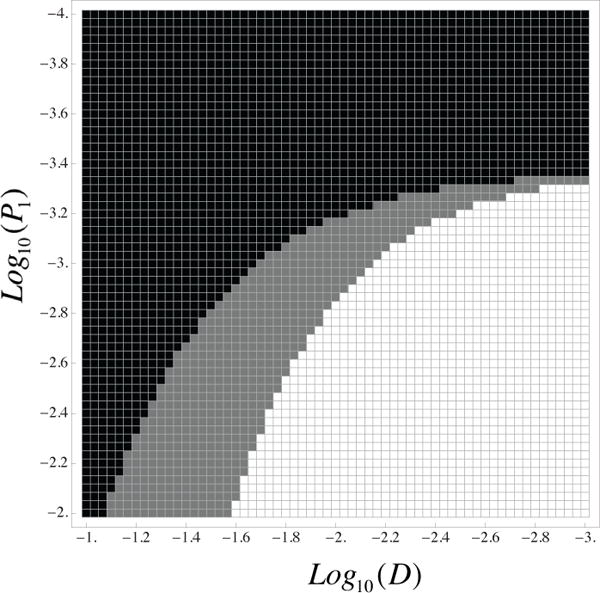
Black corresponds to low levels of instability at all times, white to high level of instability at all times, and gray to high level of instability at first then switching to stability. The parameters are d0 = 10−4, p0 = 10−7, a = 1.2, μ = 10−7, n = 1.
We can see that small values of pi and large values of di correspond to the regime where genetic instability is never achieved (black cells). On the other hand, when the death rates are small and instability related mutation rates are large, instability is generated at an early stage and persists throughout the growth phase (white cells in the diagram). The intermediate case is represented by gray cells. In this regime, raised mutation rates result in significant number of mutations at the beginning of growth. As cancer grows exponentially, after initial growth new cancerous mutations become insignificant and populations with smaller death rates grow to higher numbers. The results are similar for larger values of n.
Intuitively, the behavior of the instability index can be explained in the three regimes. If the penalty for instability (the increase in the death rate) is relatively high, and the gain (the increased rate of mutant production) is small, unstable cells lose out in the evolutionary competition, and the system undergoes scenario 1. On the other hand, if the penalty is relatively small, and the added mutation rate is high, the colony behaves according to pattern 3. Finally, in the intermediate case, scenario 2 is observed. For these parameters, unstable cells are generated relatively fast, thus the instability index experiences a temporary rise. As the colony continues to grow, the unstable colony decreases relative to the stable population because of its decreased net growth rate.
To check the robustness of the model, we compared the qualitative shapes of the instability curve for several dynamical formulations of the model. Figure 5 presents the instability curves for the quasispecies-type equations with partial homeostatic control for y (Figure 5(a)); the quasispecies-type equations without homeostatic control for y (Figure 5(b)); and the logistic growth equations (Figure 5(c)). Keeping all the parameters fixed and only varying the magnitude of the death rate, we are able to obtain all three scenarios for the instability index described above, for all three models. One major difference between Figure 5(a) and (b) is that even though the population experiences similar changes in the level of instability, when the homeostatic control term ϕ is omitted from equations (5–6) the cancer cell population reaches its maximum value faster. This result is very intuitive as cancer cells grow faster without the partial control presented by ϕ. We get similar results in the case of logistic growth shown in Figure 5(c).
Figure 5. Three scenarios for different dynamic versions of the radial mutation network model.
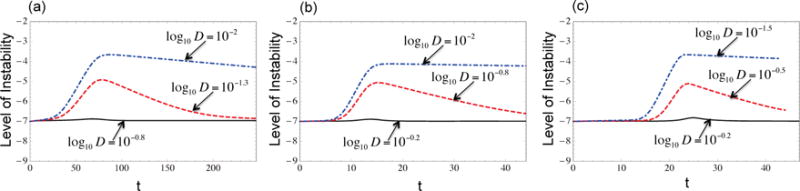
(a) Quasispecies-type equations with partial homeostatic control for y, (b) Quasispecies-type equations without homeostatic control for y. (c) Logistic growth equations. For all these models it is possible to get all three scenarios by only changing the magnitude of the death rates D. In (b) and (c) the cancer population reaches its maximum value faster. The parameters d0 = 10−4, p0 = 10−7, p1 = 10−4, a = 1.2, μ = 10−7, n = 4.
Similar simulations were run for the sequential mutation network model. Again we let the death rates di be represented by the linear function given in (17), and for cancerous mutation rates we use (18). We have investigated two different assumptions for the mutation rates μi, νi. First, we set all the rates equal to each other, μi = νi = 10−7 for all i ≥ 1. This assumes that the molecular mechsms of acquiring different levels of instability are different from mechanisms of cancer initiation (characterized by rates pi). For example, μi could be rates of point mutations, and pi are rates of chromosomal loss. This situation is similar to that investigated in [30, 29]. For this choice of mutation rates, we were able to observe all three cases discussed above and the results were similar to Figure 4. The difference between this model and the radial mutation network model is that in the latter case, scenarios ➁ and ➂ of Figure 3 can be obtained more easily (for larger ranges of the death rates and mutation rates). The reason for this difference is that in a sequential model, for a cell to acquire growth inducing alterations of the genome, it has to undergo a chain of mutations, whereas in the radial model, the increase in the level of instability results from a single mutation of stable cells. This difference is more apparent when n is large.
As an alternative choice for the mutation rates μi, νi, we assumed that genetically unstable cell populations are more prone to further genetic changes that will result in more instability. Therefore, we took μi = νi = pi. The results are almost identical to the above choice of μi, νi. After comparing these and other choices for μi, we concluded that basic mutation rates do not change the dynamics of the system significantly.
Figure 6 shows how the shape of the instability curve depends on the number of populations present. As expected, both radial and sequential models reach higher levels of instability when n is larger. This is because higher cancerous mutation rates compensate for the smaller sizes of more unstable populations.
Figure 6. The level of instability depending on the number of unstable types, n for radial mutation network.
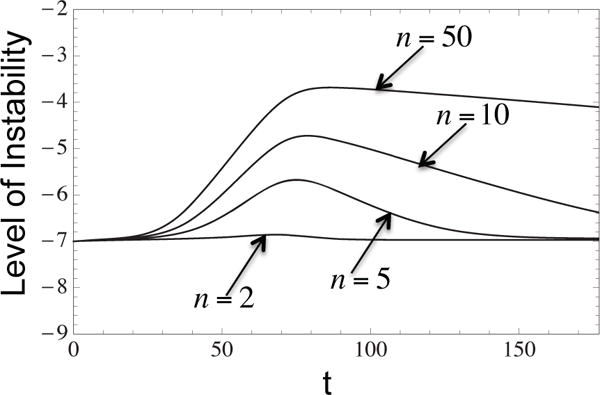
Displayed are plots for n = 2, 5, 10, 50. Higher numbers of populations correspond to more instability in the system. The parameters are log10 D = −0.7, d0 = 10−4, p0 = 10−7, a = 1.2, n = 4, μi = pi for i ≥ 1.
3.2. Multiple driver mutations
Typically, tumor growth is initiated by one or more mutations, which, in collaboration with the environment, give the cell selective advantages, resulting in the expansion of the clone derived from this cell. Successive advantageous mutations (the driver mutations) cause waves of clonal expansion, followed by growth plateaus, as discussed e.g. in[54]. In this section we consider this stepwise nature of tumorigenesis and assume that cells acquire multiple driver mutations giving rise to tumor growth. These mutations result in subpopulations that are characterized by more aggressive properties than the parent populations, and have higher growth rates. We examine the solution curves of the system and analyze the changes in the level of instability in the radial and sequential mutation network models.
To describe the multi-step dynamics, we first use an extension of the logistic growth equations , presented in section 2. Here, . The lower index varies from 0 to n and corresponds to the degree of instability. The upper index varies from 0 to m and describes the stage in the multistage carcinogenesis; it corresponds to the number of driver mutations carried by the cell. Therefore, are benign populations for 0 ≤ i ≤ n and are cancerous populations for 0 ≤ i ≤ n, 1 ≤ j ≤ m. is given by a block diagonal matrix with blocks , 0 ≤ j ≤ m. and R is a block diagonal matrix with blocks , 0 ≤ j ≤ m, defined in section 2.
This mutation network (assuming the radial mutation structure) is presented in Figure 7; sequential mutation network gives similar results. We assume that cells with the same number of driver mutations undergo similar growth limitations independent of their instability level, and let the carrying capacity and growth rate of populations at each step be the same. With further mutations, cells acquire higher growth rates and are subject to less regulation (can achieve a higher population size).
Figure 7.
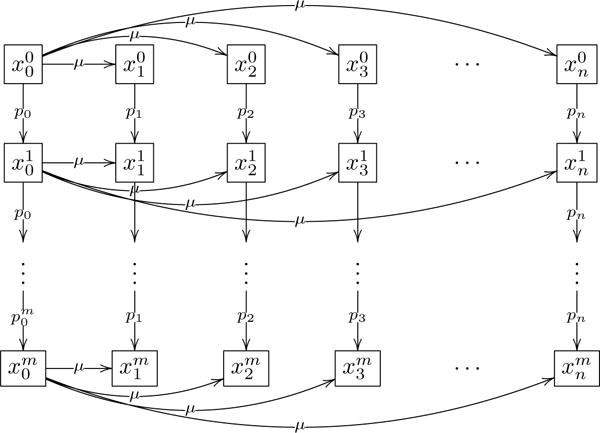
Radial mutation network involving multiple driver mutations.
Figure 3 plots three typical scenarios of the instability index dynamics in the case of multiple driver mutations. Again, as in the case of a one-step model, there are three scenarios: (1) the tumor remains stable throughout the course of its growth; (2) instability is generated but later declines; (3) instability is generated and the level of instability remains high all the way through. These three behaviors are demonstrated in Figure 3(b) graphs ➀, ➁, ➂ respectively. The patterns observed in the multiple driver mutant system are very similar to the ones described for a simpler, single-step system of Section 2.2. Again, high penalty and low gain associated with genetic instability result in a mostly stable population (scenario 1), the opposite regime leads to scenario 3 (a high level of instability throughout the growth of the tumor), and intermediate parameters correspond to a temporary rise of instability.
There are however some differences between the one-step and multiple-step models, for example, the existence of additional local maxima (or “bumps”) in the instability index curve in the case of multiple driver mutations. In order to understand the differences and get a sense of the reasons for the observed dynamics, we turn to the detailed dynamics of various sub-populations, see figure 8.
Figure 8. Temporal dynamics of different sub-populations with multiple driver mutations.
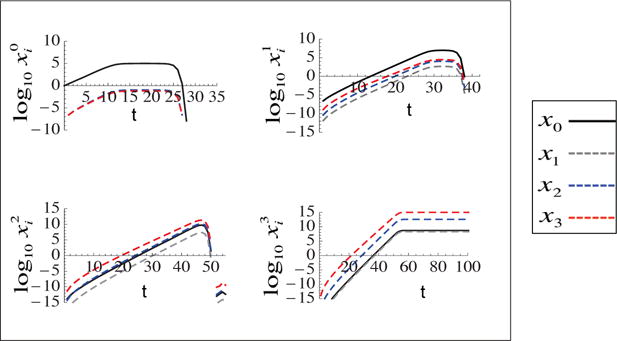
Radial mutation network with logistic growth equations. Each plot corresponds to a single step in carcinogenesis and plots the subpopulations at different instability levels. m = 3, n = 3, d0 = 10−4, d1 = 10−4, d0 = 10−3, d2 = 10−2, d3 = 10−1, p0 = 10−7, p1 = 10−5, p2 = 10−3.5, p3 = 10−2, , , , , a0 = 1, a1 = 1.05, a2 = 1.1, a3 = 1.15, a4 = 1.2.
Figure 8 plots the temporal dynamics of various sub-populations, grouped by the number of driver mutations that they carry. For example, the upper left graph depicts non-cancerous populations at different levels of stability (and correspond to the first row in the diagram of figure 7). The upper right plot shows the populations that have acquired one driver mutation (and correspond to the second row in the diagram of figure 7), and so on. The different lines in each plot correspond to the different levels of instability, with the black line depicting the stable subpopulations. We observe that each population with a given number of driver mutations grows until a certain level (given by the carrying capacity associated with that phenotype) and then decays, giving rise to the next step in the cancerous evolution, which brings in the more aggressive and faster growing type with an extra driver mutation. Because the process of replacement of one population by another is very fast, it causes drastic changes in the instability level of the system for a short period of time. This behavior is at the core of the observed “bumps” visible in the instability index curve, figure 3. The bumps mark the step-wise progression of the ever-changing cancerous colony.
Next, we notice in figure 8 that the non-cancer population and the population at step one are predominantly stable throughout tumor growth (the black lines dominate). For the specific parameter values used in the figure, cells with two and three driver mutations are dominated by unstable populations. In general, the number of predominantly stable stages depends on the system parameters; the first two stages however always remain stable, and, for m > 1, the following stages may become predominantly unstable.
An interesting question is whether instability can be considered a “driver” or a “passenger” phenomenon in carcinogenesis. Technically, it cannot be defined as a “driver” mutation, because it usually does not provide a cell with a selective advantage. It can however, under some circumstances, be regarded as one of the causal events in carcinogenesis. It is evident that for the parameters of figure 8, the core of the tumor grows out of a genetically unstable cell (level 3 population is dominated by the most unstable cells, the bottom right graph). Therefore one might argue that the presence of an unstable pool of cells prior to the growth of the largest clonal wave was a necessary event in the tumor’s natural history. In this sense, instability can be considered to play a causal role. Our simulations demonstrate that the strongest determiners of whether or not tumor will grow from unstable populations are the balance of the increased mutation and death rates associated with the instability. Other parameters such as the growth regulation factors encoded in the growth rates and the carrying capacity of the various stages may also pay a role. In general, instability is more likely to play a causal role if the carcinogenesis process involves many steps (relatively large values of m).
The reasons for these patterns can be understood from the model analysis. First let us concentrate of populations and in figure 7, which are unstable and stable non-cancerous cells. It is clear that because the unstable population is produced from the wild type population and has a lower growth rate due to the deleterious mutations experienced by unstable cells. Therefore, it is expected that the non-cancerous population is dominated by stable sub-populations. Let us next compare the dynamics of populations and in figure 7; these are unstable and stable cells carrying one driver mutations. Population is produced at a relatively high rate (pn > p0), but it is produced by a smaller pool of cells and it grows slower than population , because it has an increased, instability-related death rate.
It turns out that the gain in production rate is typically not sufficient to offset the loss in the growth exponent, and population remains smaller than for all realistic parameter regimes. More precisely, if the mutation rates of the unstable types, pi, are below 10−2, which is a biologically plausible maximum [32], and if the basic mutation rate, μ, is under 10−6, then the majority of the cell populations that carry a single driver mutation are stable. Only raising the mutation rate μ to about 10−5 results in the unstable clone overtaking the growing colony at this first stage of carcinogenesis. This result is consistent with the findings of [54].
Even though among the carriers of a single driver mutation, the stable population comprises the majority, the relative fraction of the unstable population among these cells is higher than that among the non-cancerous cells. This happens due to the large difference in the mutation rates pn and p0, and it is important to understand the further development of tumor dynamics. Consider the next stage of carcinogenesis. Compared with population , population is created by a smaller pool of cells , but as mentioned above, the relative fraction of the producing population is higher. For some parameter combinations, the disadvantage resulting from the lowered growth rate of can be compensated by their fast production (pn ≫ p0), resulting in the dominance of the unstable population at this level (see the bottom graphs of figure 8).
This argument outlines an important difference between the one-stage and multi-stage model. In the single driver mutation model described in Section 2.2, even though genetic instability may evolve in the course of cancer evolution, the population that eventually takes over is stable (see black line on Figure 2(b)). In other words, cells with elevated mutation rates that come up in the course of tumorigenesis are dead ends, which themselves do not give rise to tumor growth. Contrary to this, in the model with multiple driver mutations, instability may play a causal role, giving rise to the population that eventually wins the evolutionary competition.
Depending on the interplay of the instability-related changes in the mutation and death rate of cells, we observe three evolution patterns for cancer populations with two or more driver mutations. Populations that have more than one driver mutation can originate and evolve according to exactly three different scenarios: (i) originate from stable cells and stay stable throughout growth, (ii) originate from unstable populations then stabilize as cancer progresses, or (iii) originate from unstable cells and remain unstable.
If instability results in a modest increase in the intrinsic mutation rate and a relatively large increase in the death rate, then instability is disadvantageous. In this case tumor starts to grow in a stable population and scenario (i) is observed. On the other hand, when instability yields a high increase in mutation rates, unstable driver mutations may play a causal role in cancer formation. Here, populations with more than one driver mutations initially start to grow from an unstable cell. Depending on the relative increase in the instability-related death rate, we observe behavior described in scenarios (ii) and (iii) above. In case (ii), the death rates of unstable populations are increased significantly, and after the initial growth the stable populations come to dominate the colony. Case (iii) corresponds to a relatively small increase in death rates. In this case the price paid because of the deleterious mutations is insignificant compared to the advantages gained from high mutation rates.
Finally we comment on the relationship between the instability index curve and the more detailed, population dynamic curves considered above. High levels of instability at the end of growth, Figure 3(b) ➂, almost always indicate that tumor grows from unstable cells. Similarly, if instability does not evolve, Figure 3(b) ➀, then most likely instability does not play a causative role in tumor formation. If, however, instability of the system grows similar to Figure 3(b) ➁, the instability curve does not necessarily reflect whether or when the majority of the cells is unstable. It merely describes the temporal dynamics of the relative fraction of unstable cells. For example, acquiring a relatively low percentage of very unstable cells can visibly increase the instability index, without the majority of the population even acquiring the unstable status.
3.3. Including back mutations
In the description so far we excluded back-mutations and studied the dynamics of instability from the viewpoint of competition among subclones. What happens if back-mutations are included in the model? There are different possibilities for modeling back mutations. If instability is acquired as an inactivation mutation, then the back mutation rate should be about two orders of magnitude lower compared to the mutation rate giving rise to the instability. If instability emerges as a result of an activation mutation, then the rate of back mutations should be similar to that of forward mutations. When including back mutations, we have tried both scenarios. Clearly, a larger effect is expected in the case where back mutations have a similar order of magnitude to forward mutations. The results of the corresponding simulations are presented in figure 9.
Figure 9. The dynamics of the instability in the presence of back mutations.
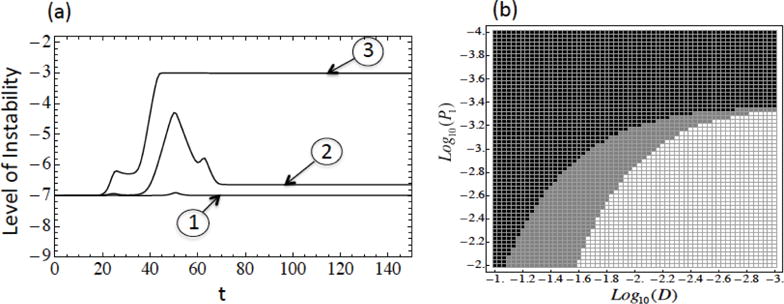
(a) Radial mutation network with multiple driver mutations. ➀, ➁, ➂ correspond to the three cases in section 3.1. n = 3, m = 6, , , , , ai = 1 + 0.05i, 0 ≤ i ≤ 6, μ = 10−7, p0 = 10−7, p1 = 10−5, p2 = 10−3, p3 = 10−2, d0 = 10−4, d1 = 10−0.5, 10−1, 10−2, d2 = 10−0.3, 10−0.4, 10−1, d3 = 10−0.1, 10−0.27, 10−0.2, for cases ➀, ➁, ➂ respectively. (b) Level of instability depending on the parameters D and p1 for radial mutation network model with a single driver mutation. Black corresponds to low levels of instability at all times, white to high level of instability at all times, and gray to high level of instability at first then switching to stability. The parameters are d0 = 10−4, p0 = 10−7, a = 1.2, μ = 10−7, n = 1.
Figure 9(a) should be qualitatively compared with figure 3, which shows the dynamics of instability in the absence of back mutations. Figure 9(b) should be compared with figure 4, which shows the phase diagram of different instability regimes in the parameter space of mutation and death rates. One can see that the results in the presence of back mutations are very similar to those obtained in the absence of back mutations.
The fact that the clonal dynamics are very similar with and without back mutations is in itself very interesting. It suggests that the contribution of clonal dynamics and evolutionary competition to the observed changes of the overall tumor phenotype is very important, and in the deterministic description of this paper it can even mask the absence of back mutations in the system. In a stochastic model, the possibility of re-creating a stable phenotype in the case of its spontaneous extinction by means of back mutations will be a necessary modeling component, because back mutations provide a direct way for an individual cell to transform from unstable to stable. Our approach however suggests that the indirect mechanism of stable-to unstable-to stable conversion by means of clonal dynamics appears at least as significant.
4. Discussion and conclusions
We have formulated a class of mathematical models to study the temporal changes of the level of instability in tumor progression. In our models we consider mutations of different critical genes and other types of genetic changes, such as rearrangements or loss of chromosomes. The noncancerous and malignant populations are divided into sub-populations characterized by different levels of instability. These sub-populations can acquire further genetic changes resulting in further growth advantages, leading to escape from homeostatic control.
By measuring the level of instability in the system at each time point, we find that there are several typical patterns that are observed with respect to the evolution of genetic instability. We found that for small death rates and large mutation rates, the system generates and keeps high levels of instability throughout the growth. The opposite scenario is observed when the cells pay a large penalty for being unstable. When the death rate is large and the mutation rates resulting from instability are relatively small, instability does not evolve. Finally, the third scenario is observed when the death rates and the mutation rates are in balance. In this case, the lineage generates high levels of instability at first, and then switches to a more stable state as tumor progresses further. An overview of different scenarios is presented in table 1.
Table 1.
Typical patterns for evolution of genetic instability and evolution of individual clonal waves. Results with □ are observed for models with both one and multiple driver mutations. Results with ◇ are observed only for models with multiple driver mutations.
| Typical patterns for evolution of individual clonal waves | Typical patterns for evolution of genetic instability |
|---|---|
|
| |
|
□1. Stable throughout growth Results from low mutation rates and high death rates Instability is a “passenger” phenomenon |
□1. Instability does not evolve Observed when the death rates are large and mutation rates are low |
|
◇2. Originates from unstable cells then stabilizes Results from high mutation rates and moderate death rates Instability is a causal event |
□2. Instability is generated but gives way to more stable clones Observed when the death rates and mutation rates are in balance |
|
◇3. Originates from unstable cells and remains unstable Results from high mutation rates and low death rates Instability is a causal event |
□3. Instability evolves and comes to dominate the system Observed when the death rates are low and mutation rates are large |
This latter scenario, characterized by a return of stability to a previously unstable tumor, has been documented in the literature. [8] argue that, while instability plays an important role in early stages of carcinogenesis, subsequent survival and perpetuation of the malignant clones require the resumption of mitotic stability. In the context of the functioning of the cell centrosomes, the authors suggest two possible mechanisms, by which stability can be regained: (i) selective inactivation of the extra centrosomes (“deamplification”) or (ii) their coalescence into two functional spindle poles, which correct the problem of centrosome excess. A relevant phenomenon termed “genetic convergence” was first described in [23]. In an in vitro study [12], it is shown that centrosomal changes continue to occur as tumors progress, leading to conversion to relative chromosomal stability. In several cell types, extensive chromosome instability and centrosome hyperamplification were observed during early to mid passages, while at late passages, a distinct subpopulation of cells became dominant in each culture, and centrosome hyperamplification and chromosome instability were suppressed in these cells. The authors argue that at “a certain time point when cells have acquired a chromosome composition that provides appropriate growth advantage in culture, maintenance of the chromosome composition becomes a selection force.” Evidence of genomic convergence was also found in the context of prostate cancer [52]. In [41], CIN and aneuploidy of cells have been studied. It was shown that once diploid cells become aneuploid, they also appear to become chromosomally stable, which provides a mechanism of instability reversal, see also [3]. Another possible mechanism of return of stability is related to the function of c-Myc. It has been suggested (see [20]) that intermediate c-Myc expression levels may promote genomic stability, consistent with the observation that DNA damage is reduced when c-Myc is inhibited by HIF-1α [25, 31]. In the context of telomere-induced instability, similar temporal patterns of instability dynamics have been reported. It has been argued by [13] that the level of genetic instability in breast cancers first increases, reaches a peak and then decreases as the cancer passes through telomere crisis. Paper [44] studies intestinal carcinoma in mice and humans, and reports data which are consistent with a similar model: telomere dysfunction promotes chromosomal instability, which in turn drives carcinogenesis at early stages. Later on, telomerase activation restores stability to allow further tumor progression, see also [46, 2, 19].
The multi-step model we study in the paper provides some important insights into the complexities of an evolving, heterogeneous cancerous population, which at all times consists of different sub-populations at different stage of carcinogenesis and having different levels of instability. These populations are at competition with each other, which is made more intricate by the different degrees of escape from homeostatic control and regulation exhibited by different cellular phenotypes. There are some patterns, however, that appear quite general within the constraints of the model. Mutations causing instability are “passengers” in tumors that have undergone only a small number of malignant mutations. Further down the path of carcinogenesis, unstable cells are more likely to give rise to the winning clonal wave that takes over the tumor and carries the evolution forward. Further, each individual clonal wave (cells harboring a fixed number of malignant driver mutations) experiences its own evolution of instability. It can fall under three types of temporal behavior: stable throughout, unstable to stable, or unstable throughout. Which scenario is realized again depends on the subtle (but predictable) interplay among mutation rates and the death toll associated with the instability.
The knowledge gathered on genetic instability in cancer can be helpful in developing therapeutic approaches [36]. The notion of chromosomal instability is closely related to the functioning (or malfunctioning) of DNA damage repair pathways, which is highly relevant to cancer therapy. For example, radiation therapy as well as many chemotherapeutic agents are effective in treatment due to their abilities to cause DNA damage. This highlights the connection between repair capability and therapeutic outcome in many treatment scenarios. Targeted inhibition of an appropriate DNA repair pathway in cancer cells is a promising strategy to increase treatment efficacy [5, 37, 7]. Inhibitors of DNA repair could increase the efficacy of DNA-damaging anticancer drugs; small-molecule inhibitors of DNA repair have been combined with conventional chemotherapy drugs in several phase I-II clinical trials [24].
There are efforts underway to manipulate the DNA damage responses to selectively induce tumor cell death through catastrophic genomic instability. It has also been proposed [24] that DNA repair inhibitors could be used as single agent therapies. They can prevent the repair of replication lesions present in tumor cells and convert them into fatal replication lesions that specifically kill cancer cells. A potential advantage of such therapy is its selectivity for tumor cells, which would result in fewer side effects.
Therapeutic potential of DNA damage repair (DDR) has also been discussed in the context of synthetic lethality; one example is the use of poly(ADP-ribose) polymerase (PARP) inhibitors in homologous recombination repair (HRR)-defective tumors [15]. Another possible application for DDR inhibitors is to block apoptotic events, such as those mediated by CHK2 and p53, thus alleviating toxicities to normal tissues.
The modeling approach provided here sheds light onto some aspects of the evolutionary dynamics of instability, which may be relevant to the above treatment scenarios. While the mean mutation rate of the lineage can be highly elevated, the cell population can be extremely heterogeneous, and the majority of cells can actually be stable (that is, their DDR system intact). Moreover, the composition of the population with respect to instability and DDR deficiency can change significantly during tumor progression. Therefore, investigating the particular instability type that develops and analyzing its effect on both the mutation rate and the death rate of cells may allow us to understand what pattern of instability is expected, and to what degree the population is heterogeneous. This may guide the choice of a targeting strategy, depending on the disease stage. For example, if a high degree of tumor heterogeneity is predicted, where subclones differ significantly by their instability status, a poorer disease prognosis is expected.
In general, intratumoral heterogeneity poses a significant challenge to targeted therapies [10]. In particular, it is believed that drug development strategies may benefit from targeting clonally dominant events [51]. Examples of such “founder” events include EGFR and KRAS mutations in lung cancer, and KRAS and BRAF mutations in colorectal carcinoma. The framework developed here provides a method of quantitative analysis of tumor dynamics under different types of treatment. Under what circumstances targeting a given event could be beneficial? What are the consequence (especially from the point of view of tumor evolution) or targeting different pathways? Can removing a certain subclone lead to unwanted consequences by changing clonal competition dynamics? These are some of the most pressing applications of the present framework.
The model studied here is a non-spatial deterministic model, and as such, it has many limitations. For example, including stochasticity is the next obvious step in the analysis. Spatial effects should also be taken in the account, e.g. by means of a hybrid multi-scale approach. We regard the present model as a conceptual formulation of a question and a sketch of an answer. We expect the clear patterns predicted by the current model to persist beyond the limitations of ordinary differential equations. If however future, more complex models show deviations from our predictions, it will be most instructive to analyze the possible reasons for this. Such work, which would rely on stepwise incremental complexification of the modeling approach, is likely to lead to further interesting insights and improved understanding of each component of the evolution of instability in cancer.
Acknowledgments
NK gratefully acknowledges the support of NIH grant 1 U01 CA187956-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annual Review of Pathology: Mechanisms of Disease. 2013;8:277–302. doi: 10.1146/annurev-pathol-020712-163923. [DOI] [PubMed] [Google Scholar]
- 2.Artandi SE, DePinho RA. A critical role for telomeres in suppressing and facilitating carcinogenesis. Current opinion in genetics & development. 2000;10(1):39–46. doi: 10.1016/s0959-437x(99)00047-7. [DOI] [PubMed] [Google Scholar]
- 3.Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore–microtubule dynamics. Nature cell biology. 2009;11(1):27–35. doi: 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, Nowak MA. Genetic progression and the waiting time to cancer. PLoS computational biology. 2007;3(11):e225. doi: 10.1371/journal.pcbi.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nature Reviews Cancer. 2011;11(4):239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 6.Benhamou S, Sarasin A. Variability in nucleotide excision repair and cancer risk: a review. Mutation Research/Reviews in Mutation Research. 2000;462(2):149–158. doi: 10.1016/s1383-5742(00)00032-6. [DOI] [PubMed] [Google Scholar]
- 7.Bouwman P, Jonkers J. The effects of deregulated dna damage signalling on cancer chemotherapy response and resistance. Nature Reviews Cancer. 2012;12(9):587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 8.Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends in cell biology. 2001;11(1):18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 9.Bristow RG, Hill RP. Hypoxia and metabolism: hypoxia, dna repair and genetic instability. Nature Reviews Cancer. 2008;8(3):180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 10.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 11.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends in Genetics. 1999;15(12):M57–M60. [PubMed] [Google Scholar]
- 12.Chiba S, Okuda M, Mussman JG, Fukasawa K. Genomic convergence and suppression of centrosome hyperamplification in primary p53−/− cells in prolonged culture. Experimental cell research. 2000;258(2):310–321. doi: 10.1006/excr.2000.4916. [DOI] [PubMed] [Google Scholar]
- 13.Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, et al. In situ analyses of genome instability in breast cancer. Nature genetics. 2004;36(9):984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- 14.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex dms and hsrs. Molecular cell. 1998;2(2):259–265. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- 15.Curtin NJ. Dna repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer. 2012;12(12):801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 16.Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S-O, Gonzalez S, Crum CP, Münger K. The human papillomavirus type 16 e6 and e7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proceedings of the National Academy of Sciences. 2000;97(18):10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duensing S, Münger K. The human papillomavirus type 16 e6 and e7 oncoproteins independently induce numerical and structural chromosome instability. Cancer research. 2002;62(23):7075–7082. [PubMed] [Google Scholar]
- 18.Fearon ER. Molecular genetics of colorectal cancer. Annual Review of Pathology: Mechanisms of Disease. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 19.González-Suárez E, Samper E, Flores JM, Blasco MA. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nature genetics. 2000;26(1):114–117. doi: 10.1038/79089. [DOI] [PubMed] [Google Scholar]
- 20.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B, et al. Hif-α effects on c-myc distinguish two subtypes of sporadic vhl-deficient clear cell renal carcinoma. Cancer cell. 2008;14(6):435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced dna damage model for cancer development. science. 2008;319(5868):1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 22.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Heim S, Mandahl N, Mitelman F. Genetic convergence and divergence in tumor progression. Cancer research. 1988;48(21):5911–5916. [PubMed] [Google Scholar]
- 24.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. Dna repair pathways as targets for cancer therapy. Nature Reviews Cancer. 2008;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 25.Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability?a calculated mechanism underlying tumor progression. Journal of Molecular Medicine. 2007;85(2):139–148. doi: 10.1007/s00109-006-0133-6. [DOI] [PubMed] [Google Scholar]
- 26.Kinzler KW, Vogelstein B. Gatekeepers and caretakers. Nature. 1997;386(6627) doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 27.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 28.Komarova NL, Lengauer C, Vogelstein B, Nowak MA. Dynamics of genetic instability in sporadic and familial colorectal cancer. Cancer biology & therapy. 2002;1(6):685–692. doi: 10.4161/cbt.321. [DOI] [PubMed] [Google Scholar]
- 29.Komarova NL, Sadovsky AV, Wan FY. Selective pressures for and against genetic instability in cancer: a time-dependent problem. Journal of The Royal Society Interface. 2008;5(18):105–121. doi: 10.1098/rsif.2007.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komarova NL, Wodarz D. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(18):7017–7021. doi: 10.1073/pnas.0401943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshiji M, To KKW, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. Hif-1α induces genetic instability by transcriptionally downregulating mutsα expression. Molecular cell. 2005;17(6):793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386(6625):623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 33.Little MP. Cancer models, genomic instability and somatic cellular darwinian evolution. Biology Direct. 2010;5:19. doi: 10.1186/1745-6150-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Little MP, Vineis P, Li G. A stochastic carcinogenesis model incorporating multiple types of genomic instability fitted to colon cancer data. Journal of theoretical biology. 2008;254(2):229–238. doi: 10.1016/j.jtbi.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 35.Loeb LA. A mutator phenotype in cancer. Cancer research. 2001;61(8):3230–3239. [PubMed] [Google Scholar]
- 36.Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nature Reviews Cancer. 2011;11(6):450–457. doi: 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lord CJ, Ashworth A. The dna damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 38.Meza R, Jeon J, Moolgavkar SH, Luebeck EG. Age-specific incidence of cancer: Phases, transitions, and biological implications. Proceedings of the National Academy of Sciences. 2008;105(42):16284–16289. doi: 10.1073/pnas.0801151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moolgavkar SH, Dewanji A, Venzon DJ. A stochastic two-stage model for cancer risk assessment. i. the hazard function and the probability of tumor. Risk Analysis. 1988;8(3):383–392. doi: 10.1111/j.1539-6924.1988.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 40.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156(1):297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholson JM, Cimini D. Cancer karyotypes: survival of the fittest. Frontiers in oncology. 2013;3 doi: 10.3389/fonc.2013.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih IM, Vogelstein B, Lengauer C. The role of chromosomal instability in tumor initiation. Proceedings of the National Academy of Sciences. 2002;99(25):16226–16231. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plug-DeMaggio AW, Sundsvold T, Wurscher MA, Koop JI, Klingelhutz AJ, McDougall JK. Telomere erosion and chromosomal instability in cells expressing the hpv oncogene 16e6. Oncogene. 2004;23(20):3561–3571. doi: 10.1038/sj.onc.1207388. [DOI] [PubMed] [Google Scholar]
- 44.Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nature genetics. 2001;28(2):155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 45.S Datta R, Gutteridge A, Swanton C, Maley CC, Graham TA. Modelling the evolution of genetic instability during tumour progression. Evolutionary applications. 2013;6(1):20–33. doi: 10.1111/eva.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samper E, Flores JM, Blasco MA. Restoration of telomerase activity rescues chromosomal instability and premature aging in terc−/− mice with short telomeres. EMBO reports. 2001;2(9):800–807. doi: 10.1093/embo-reports/kve174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Tapia C, Wan FY. Fastest time to cancer by loss of tumor suppressor genes. Bulletin of mathematical biology. 2014;76(11):2737–2784. doi: 10.1007/s11538-014-0027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schöllnberger H, Beerenwinkel N, Hoogenveen R, Vineis P. Cell selection as driving force in lung and colon carcinogenesis. Cancer research. 2010;70(17):6797–6803. doi: 10.1158/0008-5472.CAN-09-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen Z. Genomic instability and cancer: an introduction. Journal of molecular cell biology. 2011;3(1):1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 50.Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proceedings of the National Academy of Sciences. 2011;108(16):6468–6473. doi: 10.1073/pnas.1005529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer research. 2012;72(19):4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ta HQ, Gioeli D. The convergence of dna damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocrine-related cancer. 2014;21(5):R395–R407. doi: 10.1530/ERC-14-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thompson DA, Belinsky G, Chang T, Jones DL, Schlegel R, Münger K. The human papillomavirus-16 e6 oncoprotein decreases the vigilance of mitotic checkpoints. Oncogene. 1997;15(25):3025–3035. doi: 10.1038/sj.onc.1201495. [DOI] [PubMed] [Google Scholar]
- 54.Tomlinson IP, Novelli M, Bodmer W. The mutation rate and cancer. Proceedings of the National Academy of Sciences. 1996;93(25):14800–14803. doi: 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan FY, Sadovsky AV, Komarova NL. Genetic instability in cancer: an optimal control problem. Studies in Applied Mathematics. 2010;125(1):1–38. [Google Scholar]