

Abstract
Although fundamentally significant in structural, chemical, and membrane biology, the interfacial protein-detergent complex (PDC) interactions have been modestly examined because of the complicated behavior of both detergents and membrane proteins in aqueous phase. Membrane proteins are prone to unproductive aggregation resulting from poor detergent solvation, but the participating forces in this phenomenon remain ambiguous. Here, we show that using rational membrane protein design, targeted chemical modification, and steady-state fluorescence polarization spectroscopy, the detergent desolvation of membrane proteins can be quantitatively evaluated. We demonstrate that depleting the detergent in the sample well produced a two-state transition of membrane proteins between a fully detergent-solvated state and a detergent-desolvated state, the nature of which depended on the interfacial PDC interactions. Using a panel of six membrane proteins of varying hydrophobic topography, structural fingerprint, and charge distribution on the solvent-accessible surface, we provide direct experimental evidence for the contributions of the electrostatic and hydrophobic interactions to the protein solvation properties. Moreover, all-atom molecular dynamics simulations report the major contribution of the hydrophobic forces exerted at the PDC interface. This semi-quantitative approach might be extended in the future to include studies of the interfacial PDC interactions of other challenging membrane protein systems of unknown structure. This would have practical importance in protein extraction, solubilization, stabilization, and crystallization.
Graphical Abstract
INTRODUCTION
The protein-detergent complex (PDC) interactions play a pivotal role in extraction, solubilization, and stabilization of water-insoluble membrane proteins.1–5 Therefore, they were studied by various approaches. For example, circular dichroism (CD) spectroscopy was employed to probe alterations in the secondary structure and stability of membrane proteins under diverse detergent-solubilization contexts.6 Using hydrogen-deuterium exchange, along with NMR spectroscopy and mass spectrometry, Raschle and colleagues (2016) have recently examined the time-dependent protein folding of the outer membrane protein X in proteomicelles.7 The nature of the interfacial PDC interactions was also inspected in the gas phase using ion-mobility mass spectrometry.8 Moreover, isothermal titration calorimetry (ITC) was used for the real-time probing of phase diagrams between bilayer-forming lipids and micelle-forming detergents.9–10 Differential scanning calorimetry (DSC) was adapted for the investigation of the impact of detergents on the water-soluble domains of membrane proteins.2, 5 However, the detergent-mediated solubilization and refolding of membrane proteins often lead to aggregation,11 a ubiquitous process caused by the inability of detergents to fully solvate them. There are at least three reasons for a modest progress in this research area. First, the protein aggregation substantially deteriorates the signal-to-noise ratio of most spectroscopic and calorimetric approaches. Second, the protein-free detergent micelles without an accurately determined concentration coexist with the proteomicelles in aqueous phase, adding an uncontrolled signal. Third, the quantitative assessment of the interfacial PDC interactions is impractical in the absence of a high-throughput screening (HTS) approach that utilizes a low concentration of membrane proteins.
Here, we show that we can overcome these challenges using rational membrane protein design, along with targeted chemical modification and steady-state fluorescence polarization (FP) spectroscopy,12–13 to probe the detergent desolvation transitions of membrane proteins. The FP spectroscopy was previously used to inspect: (i) the interactions of mild14 and harsh15 detergents with water-soluble proteins, (ii) the harsh detergent-induced unfolding16 and resistance of soluble proteins to denaturation,17 (iii) the detergent-mediated oligomerization of hydrophobic proteins into proteomicelles,18 and (iv) the impact of detergent on conformational changes14 and enzymatic activity19 of soluble proteins.
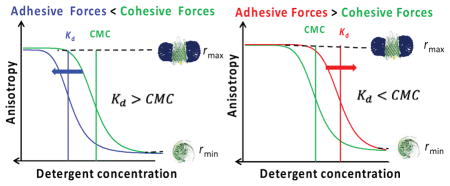
In this article, we place an emphasis on the transition of detergent desolvation of hydrophobic membrane proteins. Such a process undergoes a two-state transition, whose apparent dissociation constant, Kd, is usually within the same order of magnitude with the critical micelle concentration (CMC).2, 5, 20 The adhesive interactions occur at the specific interface between the detergent tails and hydrophobic residues on the detergent-accessible surface of the membrane protein. In addition, these interactions occur at the specific interface between the polar head groups of the detergents and water-soluble parts of the membrane protein. In contrast, the cohesive interactions are mediated by detergents, maintaining the integrity of the proteomicelle. The aberrant imbalance between these interactions produces a significant departure of the proteomicelle dissociation from the demicellization transition.
For exploring the PDC interactions exposing β-barrel surfaces, we chose the outer membrane protein G (OmpG)21 and three extensive truncation derivatives of ferric hydroxamate uptake component A (FhuA)22 of E. coli (Fig. 1). We demonstrate that robust β-barrel proteins, which tolerate extensive changes in charge distribution across the solvent-accessible surface, exhibit drastic alterations in the interfacial PDC interactions. In some instances, these major modifications culminated with the transition from excellent to poor solubilization properties due to variations from strong to very weak adhesive interactions. For example, the zwitterionic detergents solubilized well the acidic β barrels, but exhibited weak adhesive contacts with the basic β barrels, performing poorly in solubilizing the latter proteins. Moreover, hydrophobic interactions played a major role in the PDC. This was clearly supported by the full-atomistic molecular dynamics (MD) simulations with an uncharged maltoside-containing detergent. Finally, we show that such a semi-quantitative experimental approach might be extended to other challenging membrane protein systems of different subunit stoichiometry or unknown structure, suggesting its potentiality to produce impactful transformations in the areas of membrane chemical biology.
Figure 1. Cartoons showing the backbone homology structures of the four β-barrel proteins inspected in this work.

(A) OmpG; (B) FhuA ΔC/Δ5L; (C) FhuA ΔC/Δ5L_25N; and (D) FhuA ΔC/Δ7L_30N. Positions of the fluorophore attachment are marked by yellow. All negative charge neutralizations with respect to FhuA ΔC/Δ5L are indicated in red. Moreover, there are three additional lysine mutations in the β turns of FhuA ΔC/Δ7L-30N that were marked in blue, out of which two are negative-to-positive charge reversals. The top of each cartoon shows the protein abbreviated name and its respective isoelectric point.
METHODS
Cloning, expression, and purification of FhuA ΔC/Δ5L
The fhua Δc/Δ5l gene lacking the regions coding for the cork domain (C) and five extracellular loops L3, L4, L5, L10, and L11, was produced through de novo synthesis (Geneart, Regensburg, Germany).23–24 fhua Δc/Δ5l_t7 was created by inverse PCR using pPR-IBA1-fhua Δc/Δ5l-6×His+ plasmid as a template. The PCR product was self-ligated to create pPR-IBA1-fhua Δc/Δ5l_t7-6×His+. The β turn T7 (V331PEDRP336) was replaced with a single cysteine-containing, flexible, GS-rich peptide loop (GGSSGCGSSGGS) for the fluorophore attachment. Protein expression was conducted, as previously published.25–26
Refolding of FhuA ΔC/Δ5L
We employed a rapid-dilution refolding protocol.27–28 Briefly, 40 μl of 6×His+-tag purified and guanidinium hydrochloride (Gdm-HCl)-denatured FhuA protein was 50-fold diluted into 200 mM NaCl, 50 mM HEPES, pH 7.4 solutions at 4°C, which included detergents at concentrations above their CMC (Table 1). Different starting detergent concentrations were used, as follows (when multiple concentrations are given, the lower concentrations were needed to get dilutions with a low enough detergent concentration to cover the required range): (i) 5 and 20 mM starting detergent concentration for n-decyl-β-D-maltopyranoside (DM), n-undecyl-β-D-maltopyranoside (UM), and n-dodecyl-β-D-maltopyranoside (DDM); (ii) 50 mM 4-cyclohexyl-1-butyl-β-D-maltoside (CYMAL-4); (iii) 50 mM n-octyl-β-D-glucoside (OG); (iv) 50 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate] (CHAPS); and (v) 20 mM 1-lauroyl-2-hydroxy-sn-glycero-3-phosphocholine (LysoFos). All detergents were purchased from Anatrace (Maumee, OH). To avoid hydrolysis and oxidation,29 detergent solutions were freshly prepared.
Table 1.
Physical properties of the detergents examined in this work.
| Detergent | FW (Da)a | Head group | Aggregation number, Naggb | CMC (mM)b | Micellar Weight, MWm (kDa) | References |
|---|---|---|---|---|---|---|
| n-dodecyl-β-D-maltoside (DDM) | 511 | Non-ionic | ~78–149 | ~0.17 | 70 | 29 |
| n-undecyl-β-D-maltoside (UM) | 497 | Non-ionic | ~71 | ~0.59 | 35 | 29 |
| n-decyl-β-D-maltoside (DM) | 483 | Non-ionic | ~69 | ~1.8 | 33 | 29 |
| 4-cyclohexyl-1-Butyl-β-D-maltoside (CYMAL-4) | 481 | Non-ionic | ~25 | ~7.6 | 12 | 63 |
| n-octyl-β-D-glucoside (OG) | 292 | Non-ionic | ~27–100 | ~25 | 25 | 29 |
| 3-[(3-cholamidopropyl)- dimethylammonio]-1-propane sulfonate] (CHAPS)c | 615 | Zwitterionic | ~10 | ~5.9c | 6 | 29 |
| 1-lauroyl-2-hydroxy-sn-glycero-3- phosphocholine (LysoFos)d | 440 | Zwitterionic | 80d | ~0.7e | 35 | 1, 64 |
| n-dodecyl-N,N′-dimethylglycine (LD)d | 271 | Zwitterionic | NAf | ~1.5g | NAf | 65 |
Formula weights of the detergent monomers (FW) were reported by Anatrace (https://www.anatrace.com/).
CMC values or aggregation numbers, Nagg, in water were reported by Anatrace (https://www.anatrace.com/).
CMC value of CHAPS in 200 mM NaCl is 5.9 mM.66
The Nagg for LysoFos used in this work is ~80.1
CMC value of LysoFos in 140 mM NaCl, 20 mM Tris-HCl, pH 7.2 is 0.7 mM.64
NA stands for not available.
Detergent monomers are neutral at pH > 6.65
Fluorescent labeling of the FhuA derivatives
10 μM FhuA derivatives (Table 2) were each incubated with 200 μM Texas Red C2 maleimide (Thermo Fisher Scientific) overnight at room temperature. The incubation buffer contained 200 mM NaCl, 50 mM Tris, 1 mM TCEP, pH 8.0, and 6 M Gdm-HCl. Proteins were separated from the unreacted fluorophore by Ni2+-NTA column chromatography in the same buffer, but with a 10–200 mM imidazole step gradient. Using ε595 = 104,000 M−1cm−1 for Texas Red C2 and a correction factor of 0.26 x ε595 to account for the fluorophore absorbance at 280 nm, labeling stoichiometry was determined as ~0.3–0.8 labels/protein.
Table 2.
Biophysical properties of the β-barrel proteins used in this study.67
| Proteina | pI/Charge state | GRAVYc | Aliphatic indexd | Negative residues | Positive residues | Total number of residuese |
|---|---|---|---|---|---|---|
| WT-OmpG | 4.4/acidic | −0.798 | 55.87 | 55 | 22 | 281 |
| FhuA ΔC/Δ5Lb | 5.7/acidic | −0.550 | 60.42 | 57 | 48 | 505 |
| FhuA ΔC/Δ5L_25N | 9.3/basic | −0.563 | 58.31 | 34 | 43 | 473 |
| FhuA ΔC/Δ7L_30N | 9.6/basic | −0.574 | 57.42 | 27 | 42 | 426 |
All proteins have a 6His+ tag at the C terminus.
This engineered FhuA includes a 33-residue signal peptide at the N terminus.
The GRAVY hydrophobicity parameter was calculated by adding individual hydropathy indexes68 of each residue and dividing by the total number of residues. Increasing positive GRAVY number shows a more hydrophobic protein.
The aliphatic index is given by the relative volume of aliphatic chain-containing residues.69
The total number of residues includes those amino acids from the Gly/Ser-rich containing polypeptide loop and 6×His+ tag.
Expression and purification of OmpG D224C
A cysteine was engineered on extracellular loop L6 of OmpG using single-site mutagenesis PCR. OmpG D224C was expressed, purified, and refolded as previously described.30 Proteins were expressed in E. coli BL21 (pLys) cells, which were transformed with the plasmid pT7-OmpG D224C. Cells were grown in LB media at 37°C until the OD600 reached a value of 0.6, at which time they were induced by 0.5 mM IPTG. Cells were harvested three hours later and lysed in lysis buffer (50 mM Tris·HCl, pH 8.0, 150 mM NaCl, 200 μg/ml lysozyme, 1 mM EDTA, 3 mM TCEP) via sonication. The lysate was centrifuged at 19,000 g for 30 min before washing once with 30 ml of a buffer containing 50 mM Tris·HCl, pH 8.0, 1.5 M urea, 3 mM TCEP. Then, the OmpG D224C-containing inclusion bodies were dissolved in 30 ml of buffer containing 50 mM Tris·HCl, pH 8.0, 8 M urea, 3 mM TCEP and passed through a 0.45 μm filter before FPLC purification. Protein purification was accomplished using a 5ml Q-ionic exchange column (GE Healthcare Life Sciences, Pittsburg, PA) and eluted in a buffer containing 50 mM Tris·HCl, pH 8.0, 8 M Urea, 3 mM TCEP, 500 mM NaCl by applying a salt gradient.
Fluorescent labeling of the OmpG D224C
After purification, OmpG D224C was incubated in 10 mM TCEP for 30 min on ice. Then, TCEP was removed using a desalting column, which was equilibrated with buffer containing 50 mM HEPES, pH 7.0, 150 mM NaCl, 8 M urea. The reduced protein was incubated in Texas Red C2-maleimide (Thermo Fisher Scientific), in a molar protein:fluorophore ratio 1:20 either at room temperature for 2 hours or at 4°C overnight. The reaction mixture was passed through the desalting column to eliminate all unreacted reagents. The chemically modified OmpG D224C sample was snap-frozen in liquid nitrogen and stored at −80°C. To test the folding properties of the labeled protein, an aliquot of the protein sample was diluted with the refolding buffer containing 50 mM Tris·HCl, pH 9.0, 3.25% OG until the final urea concentration reached 3.0 M. Samples were then incubated at 37 °C for 3 days. The refolding efficiency of Texas Red-labeled OmpG D224C was determined using the heat-modifiability assay through the SDS-PAGE analysis.27, 31
Expression and purification of SELENOK U92C and SELENOS U188S
The cloning, expression, and purification of Homo sapiens SELENOK U92C (UniProtKB Q9Y6D0) and SELENOS U188S (UniProtKB Q9BQE4) used in this study were described previously.32 In short, SELENOK U92C was cloned into a pMAL-C5X vector (New England Biolabs, Ipswich, MA) and fused to maltose-binding protein (MBP). A 6×His+ tag was introduced between residues I3 and E4 of MBP to facilitate purification. A short linker NSSS with a tobacco etch virus (TEV) protease cleavage site (ENLYFQG) was used to connect the two proteins. In addition, an eight-amino acid-StrepII tag (WSHPQFEK) was inserted between the TEV protease cleavage site and SELENOK U92C to assist the purification. Following cleavage of the fusion protein by TEV protease, SELENOK U92C retained in its N-terminus the sequence GWSHPQFEK. MBP-SELENOK U92C was purified by amylose affinity chromatography. Then the fusion partner MBP was cleaved off by TEV protease. SELENOK U92C was further purified by Strep-Tactin affinity column (GE Healthcare Life Sciences). All purification steps were carried out in buffers supplemented with 1.3 mM DDM, which represented the starting detergent concentration for the follow-up dilutions. Protein purity, as assessed by 16% TRICINE-SDS-PAGE, was greater than 95% (Supporting Information, Fig. S1). Similarly, SELENOS U188S, with the native selenocysteine at position 188 mutated to serine, was cloned in the same way into the pMAL-C5X vector and fused with MBP.33 A short linker NSSS and a TEV cleavage site, ENLYFQS, was used to connect the two proteins. Following cleavage with TEV protease, only a serine was present before the first native amino acid. Expression and purification of SELENOS U188S was similar to the procedure above with the only difference that instead of the Strep-Tactin affinity chromatography SELENOS U188S was purified by a HisTrap FF column (GE Healthcare Life Sciences) to remove the 6×His+ tagged-MBP and TEV protease.34 The flow through containing the purified SELENOS U188S was collected. The protein purity, as determined by SDS-PAGE, was greater than 95%.
Fluorescent Labeling of SELENOK U92C and SELENOS U188S
40 μM SELENOK U92C or SELENOS U188S were reduced by addition of 5 mM dithiothreitol (DTT) and incubated at room temperature for 20 min. DTT was then removed using a desalting column (5 mL HiTrap desalting column, GE Healthcare Life Sciences). Labeling reactions were carried out using 50 μM SELENOK U92C or SELENOS U188S in the reaction buffer (50 mM sodium phosphate, 200 mM NaCl, 0.067% DDM, 1 mM EDTA, pH 7.5) supplemented with 1 mM Texas Red C2- maleimide (Setareh Biotech, Eugene, OR) and incubated at room temperature for 1 h. Excess Texas Red C2 maleimide was removed by dialysis in the dark against the reaction buffer. SELENOK U92C was specifically labeled on the C92 position, as this is the only cysteine in the protein. SELENOS U188S was only labeled on the C174 position since the other cysteine is located in the trans-membrane helix and was proven to be inaccessible for fluorescent labeling.
Anisotropy measurements
For FP measurements, we used a SpectraMax I3 plate reader (Molecular Devices, Sunnyvale, CA) equipped with the Paradigm detection cartridge for Rhodamine FP spectroscopy.35 The excitation and emission wavelengths were 535 and 595 nm, respectively. A Texas Red fluorophore was covalently attached to an engineered cysteine sulfhydryl, because of its optical stability over a broad range of experimental circumstances.36 The attachment site was chosen on the water-soluble domains of the membrane proteins, because of the hydrophilic nature of this bright fluorophore.37 The FP recordings were carried out using 96-well Costar assay plates (Corning Incorporated, Kennebunk, ME). The fluorescence anisotropy depends on the time-dependent orthogonal, Io(t), and parallel, Ip(t), emission intensities, as follows:36, 38
| (1) |
Here, G is a sensitivity correction factor for the detection modes when emission polarizers are oriented vertically and horizontally.
| (2) |
IHH denotes the intensity with both the excitation and emission polarizers in a horizontal orientation. IHV indicates the intensity with the excitation and emission polarizers oriented horizontally and vertically, respectively. The FP data were processed as average ± SD over a number of at least three independent acquisitions. The robustness of the acquired data was illustrated in figures through vertical SD bars.
We executed steady-state anisotropy recordings with diluted refolded protein samples within individual wells, while keeping the final protein concentration constant at either 28 nM (β-barrel proteins) or at 200 nM (α-helical proteins). For all proteins, this was accomplished by diluting the refolded protein sample within individual wells with buffer containing detergents at various concentrations. The final detergent concentration in the protein samples for anisotropy measurements was derived using the following equation:
| (3) |
where V and Cf denote the well volume and the final detergent concentration of the protein sample for anisotropy measurements, respectively. Cs and Cd indicate the detergent concentrations of the refolded protein (starting concentrations) and diluting buffer, respectively. Vs and fs are the volume and fractional volume (Vs/V) of the refolded protein sample at a starting detergent concentration, respectively. Vd and fd are the volume and fractional volume (Vd/V) of the diluting buffer containing detergents at different concentrations, respectively. In this way, we were able to prepare samples containing detergents in a broad range of concentrations below and above their CMC.
We verified that potential self-quenching of Texas Red does not produces a time-dependent reduction in the FP output of the protein-Texas Red conjugate. Therefore, we performed control time-dependent anisotropy experiments, as follows: (i) at the beginning of the measurements at detergent concentrations much greater than their CMCs (Supporting Information, Fig. S2); and (ii) after 24 hours, reaching the endpoints of the detergent desolvation reaction (Supporting Information, Fig. S3). In both cases, we found no time-dependent alterations of the anisotropy readout. The FP anisotropy measurements were conducted under equilibrium conditions. The incubation time for the equilibration of protein samples after detergent dilution was 15 min. Then, a time-dependent kinetic read of the fluorescence anisotropy was acquired at the beginning of the detergent desolvation reaction. To assure uniform recording conditions, we collected the endpoints after 24 h, a period in which the protein samples, incubated at different detergent concentrations, were covered and placed at 4°C. These endpoints were used to achieve the detergent dissociation isotherms. Protein aggregation increased over time upon drastic detergent depletion, but without affecting the signal-to-noise ratio of the anisotropy endpoints. The Hill-Langmuir dissociation-isotherm curves were fitted by:39
| (4) |
rmin and rmax denote the minimum and maximum values of anisotropy, respectively.10 p and Kd indicate the Hill coefficient and the apparent dissociation constant, respectively. The major assumption of this fitting procedure is that the protein surface shows specific binding sites for detergent monomers. The steepness of the two-state transition of detergent desolvation at half detergent saturation, q, was calculated by the following equation:
| (5) |
MD simulations of the interactions of DDM with β-barrel proteins
All simulations were performed using the molecular dynamics program NAMD2,40 periodic boundary conditions, and a 2-fs timestep. The CHARMM36 force field41 was used to describe proteins, detergents, TIP3P water, and ions. The CUFIX corrections were applied to improve description of charge-charge interactions.42–43 RATTLE44 and SETTLE45 algorithms were applied to describe covalent bonds that involved hydrogen atoms in proteins, detergents and water molecules. Particle-Mesh-Ewald (PME)46 algorithm was used to evaluate the long-range electrostatic interaction on a 1 Å-spaced grid; the full electrostatics calculation was performed every three timesteps. Van der Waals interactions were evaluated using a smooth 10–12 Å cutoff. Atomic coordinates of the four β-barrel proteins, OmpG (PDB entry 2IWV21), FhuA ΔC/Δ5L (PDB entry 1BY522), FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N, were obtained from the Protein Data Bank. Structures containing deletions and mutations were built by modifying the wild-type structure. For each β-barrel protein, two systems were constructed differing by the initial placement of the DDM molecules. The cubic arrangement of DDM was realized by placing 21 DDM molecules around the protein with the average protein-to-DDM distance of 5.7 nm, whereas in the planar arrangement, the DDM molecules were placed within a plane passing through the geometrical center of the protein. The systems were solvated using the VMD’s Solvate plugin. Waters overlapping with the proteins and DDM molecules were removed. Sodium and chloride ions were added to neutralize the system and bring the ion concentration to 200 mM. The final systems contained approximately 172,000 atoms. The initial DDM concentration was 20 mM. One additional FhuA ΔC/Δ5L system was built to containing 105 DDM molecules in the same electrolyte volume, which corresponded to a concentration of 100 mM DDM with cubic arrangement. Each system was minimized for 9600 steps using the conjugate gradient method, then equilibrated for ~230 ns in the constant number of atoms, pressure and temperature ensemble. The Nose-Hoover Langevin piston pressure control47–48 was used to maintain the pressure of the system at 1 atm by adjusting the system’s dimension. Langevin thermostat49 was applied to all the heavy atoms of the system with a damping coefficient of 0.1 ps−1. All the trajectories were analyzed by using VMD.50
RESULTS
Rationale for data acquisition, analysis, and interpretation
For a satisfactorily solubilizing detergent, we determined that at concentrations much greater than the CMC (Table 1) the FP anisotropy reached a concentration-independent maximum value, rmax (Fig. 2). In contrast, at detergent concentrations comparable with or below the CMC, the FP anisotropy followed a decrease to a concentration-dependent value, r(c) < rmax. Moreover, at detergent concentrations much lower than the CMC the FP anisotropy decreased to a concentration-independent minimum value, rmin. OmpG51 and FhuA23, 35 proteins exhibit an overwhelming preponderance of anti-parallel β-sheet structure in solution under detergent-refolding conditions. At detergent concentrations well below their CMC, a decrease in the FP anisotropy was produced by the dissociation of the detergent monomers from the protein, resulting in a reduction in the hydrodynamic radius, Rh, of the PDC, and a corresponding increase in its tumbling rate. This interpretation was also supported by the observation that at detergent concentrations well above their CMC, no significant change in the FP anisotropy readout was noted (Supporting Information, Fig. S2).
Figure 2. Graphic illustrating the three hypothetical scenarios of the balance between adhesive and cohesive interactions of PDCs.

(A) The detergent-protein interactions are weaker than the detergent-detergent interactions that keep the proteomicelle molecules together; (B) The detergent-protein interactions are stronger than the detergent-detergent interactions; (C) The two types of interactions are of similar magnitude.
Therefore, the detergent-solubilized membrane proteins featured a maximum anisotropy, rmax, whereas the detergent-desolvated proteins exhibited a minimum anisotropy, rmin (Fig. 2). Of course, deviations from this rule occurred under poor detergent solubilization conditions, even if the detergent concentration was much greater than the CMC. For each case, the mid-point of the transition of detergent desolvation, Kd, was compared with CMC. If Kd > CMC, then the cohesive interactions were greater than the adhesive interactions (Fig. 2A), and vice-versa, if Kd < CMC (Fig. 2B). The adhesive and cohesive interactions were comparable to each other when Kd ≅ CMC (Fig. 2C).
Alterations in the charge distribution of the solvent-accessible surface of β-barrels
To further examine the impact of electrostatic adhesive interactions on Kd, we examined four β-barrel proteins of varying charge distribution on the solvent-accessible surface (Table 2). These were OmpG21 and three derivatives of FhuA22 of E.coli, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N (Fig. 1).24 FhuA ΔC/Δ5L is a truncation FhuA mutant lacking the 160-residue, N-terminal cork domain (C) and extensive parts of the extracellular loops L3, L4, L5, L10, and L11. FhuA ΔC/Δ5L_25N features 25 negative charge neutralizations on the extracellular loops and periplasmic β turns with respect to FhuA ΔC/Δ5L. FhuA ΔC/Δ7L_30N was derived by additional four loop truncations, L4, L5, L7, and L8, with respect to the FhuA ΔC/Δ5L scaffold, and with a total of 30 negative charge neutralizations with respect to FhuA ΔC/Δ5L. These charge neutralizations were conducted by replacing D and E with N and Q, respectively. In this way, we accomplished an extensive change in the balance between positive and negative residues on the solvent-accessible surface. Therefore, at physiological pH negative residues were dominant in OmpG and FhuA ΔC/Δ5L, making these proteins acidic (pI < 7.0), whereas positive side chains are prevalent in FhuA ΔC/Δ5L_25N and FhuA ΔC/Δ7L_30N, making these proteins basic (pI > 7.0) (Table 2).
The balance of adhesive and cohesive interactions of the β barrel-containing proteomicelles Neutral detergents
Fig. 3 shows the transitions of detergent desolvation with four maltoside-containing neutral detergents, as follows: DDM (Fig. 3A), UM (Fig. 3B), DM (Fig. 3C), CYMAL-4 (Fig. 3D) (Supporting Information, Table S1). In these panels, we showed the basal anisotropy readout, r1 =0.16, recorded with FhuA ΔC/Δ5L when fully unfolded (e.g., in most rotationally diffusive state) using 6 M Gdm-HCl (Table 3). The only distinction among DDM, UM, and DM is the length of their hydrophobic tail, with 12, 11, and 10 alkyl carbons, respectively. CYMAL-4 is also a maltoside-containing detergent, but containing a very short hydrophobic tail (e.g., 4 alkyl carbons) and a cyclohexyl group. When all four proteins were incubated in DDM, we noted that the three FhuA protein mutants exhibited Kd values greater than the CMC, suggesting that the cohesive forces outperformed the adhesive forces, a finding that was not encountered with OmpG (Supporting Information, Table S2). The FhuA mutants showed a shift in the UM desolvation-induced transition towards stronger adhesive interactions (Fig. 3B). Interestingly, the DM desolvation-induced transition recoded with the basic FhuA proteins was very sharp and featured the largest positive Hill cooperativity p values of ~27 (Fig. 3C and Table 4), contrasting to those noted with weakly adhesive acidic β barrels. Moreover, all four β-barrel proteins exhibited stronger adhesive than cohesive interactions with CYMAL-4 (Fig. 3D and Table 4). Therefore, at physiological pH conditions, we found that for basic β barrels the adhesive interactions increased with respect to cohesive interactions in the order DDM → UM → CYMAL-4 → DM (Supporting Information, Table S2; last column).
Figure 3. Dose-response changes in fluorescence anisotropy for neutral maltoside-containing detergents.

(A) n-dodecyl-β-D-maltoside (DDM); (B) n-undecyl-β-D-maltoside (UM); (C) n-Decyl-β-D-maltoside (DM); (D) 4-Cyclohexyl-1-butyl-β-D-maltoside (CYMAL-4). All anisotropy measurements were conducted out in 200 mM NaCl, 50 mM HEPES, pH 7.4, and at room temperature. The anisotropy data were recorded by adding overnight detergent-refolded protein to a bath of varying detergent concentration, but keeping the final protein concentration at 28 nM. Starting detergent concentrations were above the CMC. Thereafter, they were reduced at concentrations below the CMC (Experimental Methods). Time-dependent anisotropy measurements were conducted directly after dilution of the refolded protein sample at respective detergent concentration. Vertical bars represent the magnitudes of the CMC and Kd of the PDCs of varying isoelectric point of the proteins. The horizontal dashed bar represents the minimum anisotropy value, r1 = ~0.16, obtained with FhuA ΔC/5L in 6 M Gdm-HCl (Table 3). This anisotropy value corresponds to the most rotationally diffusive FhuA ΔC/Δ5L.
Table 3. The recorded minima and maxima of the anisotropy with neutral and zwitterionic detergents and β-barrel proteinsa.
This table also illustrates the rotational diffusion coefficients as well as alterations in hydrodynamic radii of the proteomicelles during the detergent desolvation transitions.
| DMb | rmin c | rmax c | Dr slow (107 s−1)d | Dr fast (107 s−1)d | Rh max (nm)e | ΔRh (nm)f |
| OmpG | 0.214 ± 0.005 | 0.327 ± 0.001 | 0.89 ± 0.02 | 3.5 ± 0.2 | 2.6 | 0.95 ± 0.04 |
| FhuA ΔC/Δ5L | 0.219 ± 0.005 | 0.360 ± 0.001 | 0.44 ± 0.01 | 3.3 ± 0.2 | 3.3 | 1.6 ± 0.1 |
| FhuA ΔC/Δ5L_25N | 0.166 ± 0.003 | 0.343 ± 0.002 | 0.66 ± 0.03 | 5.6 ± 0.2 | 3.0 | 1.5 ± 0.1 |
| FhuA ΔC/Δ7L_30N | 0.168 ± 0.007 | 0.312 ± 0.001 | 5.5 ± 0.4 | 1.0 ± 0.1 | 2.4 | 1.0 ± 0.1 |
| CYMAL-4b | rmin c | rmax c | Dr slow (107 s−1)d | Dr fast (107 s−1)d | Rh max (nm)e | ΔRh (nm)f |
| OmpG | 0.163 ± 0.001 | 0.326 ± 0.001 | 0.90 ± 0.01 | 5.8 ± 0.1 | 2.6 | 1.2 ± 0.1 |
| FhuA ΔC/Δ5L | 0.242 ± 0.001 | 0.367 ± 0.001 | 0.36 ± 0.01 | 2.6 ± 0.1 | 3.6 | 1.7 ± 0.1 |
| FhuA ΔC/Δ5L_25N | 0.166 ± 0.001 | 0.341 ± 0.001 | 0.69 ± 0.01 | 5.6 ± 0.1 | 2.9 | 1.4 ± 0.1 |
| FhuA ΔC/Δ7L_30N | 0.168 ± 0.025 | 0.345 ± 0.004 | 0.63 ± 0.05 | 5.5 ± 1.2 | 2.9 | 1.5 ± 0.2 |
| OGb | rminc | rmaxc | Drslow (107 s−1)d | Drfast (107 s−1)d | Rh max (nm)e | ΔRh (nm)f |
| OmpG | 0.153 ± 0.002 | 0.306 ± 0.001 | 1.2 ± 0.1 | 6.4 ± 0.1 | 2.4 | 1.0 ± 0.1 |
| FhuA ΔC/Δ5L | 0.162 ± 0.002 | 0.291 ± 0.002 | 1.5 ± 0.1 | 5.8 ± 0.1 | 2.2 | 0.80 ± 0.02 |
| FhuA ΔC/Δ5L_25N | ~0.15 | ~0.17 | ~5.2 | ~6.6 | NDh | NDh |
| FhuA ΔC/Δ7L_30N | ~0.15 | ~0.17 | ~5.2 | ~6.6 | NDh | NDh |
| CHAPSb | rmin c | rmax c | Dr slow (107 s−1)d | Dr fast (107 s−1)d | Rh max (nm)e | ΔRh (nm)f |
| OmpG | NDh | 0.318 ± 0.002 | 1.0 ± 0.1 | NDh | 2.5 | NDh |
| FhuA ΔC/Δ5L | 0.172 ± 0.048 | 0.292 ± 0.004 | 0.53 ± 0.2 | 1.5 ± 0.1 | 2.2 | 0.77 ± 0.18 |
| FhuA ΔC/Δ5L_25N | ~0.15 | ~0.17 | ~5.2 | ~6.6 | NDh | NDh |
| FhuA ΔC/Δ7L_30N | ~0.16 | ~0.17 | ~5.2 | ~6.0 | NDh | NDh |
| LysoFosb | rmin c | rmax c | Dr slow (107 s−1)d | Dr fast (107 s−1)d | Rh max (nm)e | ΔRh (nm)f |
| OmpG | 0.229 ± 0.006 | 0.307 ± 0.001 | 1.2 ± 0.1 | 3.0 ± 0.2 | 2.4 | 0.61 ± 0.04 |
| FhuA ΔC/Δ5L | 0.223 ± 0.007 | 0.330 ± 0.001 | 0.84 ± 0.01 | 3.1 ± 0.2 | 2.7 | 0.95 ± 0.05 |
| FhuA ΔC/Δ5L_25N | 0.177 ± 0.001 | 0.313 ± 0.001 | 1.1 ± 0.02 | 5.0 ± 0.1 | 2.4 | 0.96 ± 0.02 |
| FhuA ΔC/Δ7L_30N | 0.184 ± 0.003 | 0.294 ± 0.005 | 1.4 ± 0.1 | 4.7 ± 0.1 | 2.2 | 0.73 ± 0.06 |
| FhuA ΔC/Δ5L in Gdm-HClg | ~0.16 | ~0.16 | ~6.0 | ~6.0 | 1.4 | NAi |
To reach low detergent concentrations below CMC, the Gdm-HCl-solubilized protein was refolded at various detergent concentrations above the CMC.
Full names of the detergents are provided in Experimental and computational methods.
rmin was extrapolated for the lowest detergent concentration in the well. rmax was determined at detergent concentrations above the CMC.
Dr slow and Dr fast indicate the rotational diffusion coefficients under solvation and desolvation conditions, respectively.
Rh max is the maximum hydrodynamic radius of the proteomicelle.
ΔRh is the decrease in the hydrodynamic radius, Rh, as a result of the detergent desolvation.
Anisotropy was determined in 6 M Gdm-HCl.
Not determined.
Not applicable.
Table 4. Summary of the fitting results of the two-state, concentration-dependent anisotropy curves of the endpoints of the detergent desolvation phase with neutral detergentsa,b.
This was determined with three FhuA derivatives and OmpG as well as a panel of five neutral detergents of varying hydrophobic chain and hydrophilic head group. The FP measurements were carried in 200 mM NaCl, 50 mM HEPES, pH 7.4 and at a temperature of 24°C. All data were derived as averages ± SDs of three independent data acquisitions.
| DMc | pd | Kde (mM) | qf (mM−1) | ΔGg (kcal/mol) | Balanceh |
| OmpG | 4.1 ± 1.2 | 1.8 ± 0.4 | 0.064 | −3.7 ± 0.1 | Fadh ≅ Fcoh |
| FhuA ΔC/Δ5L | 3.5 ± 0.5 | 1.7 ± 0.1 | 0.072 | −3.8 ± 0.1 | Fadh ≅ Fcoh |
| FhuA ΔC/Δ5L_25N | 27 ± 3 | 0.9 ± 0.1 | 1.30 | −4.1 ± 0.1 | Fadh ≫ Fcoh |
| FhuA ΔC/Δ7L_30N | 27 ± 6 | 0.9 ± 0.1 | 1.07 | −4.1 ± 0.1 | Fadh ≫ Fcoh |
| CYMAL-4c | pd | Kde(mM) | qf (mM−1) | ΔGg (kcal/mol) | Balanceh |
| OmpG | 6.7 ± 0.1 | 4.6 ± 0.1 | 0.25 | −3.2 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L | 3.7 ± 0.1 | 5.3 ± 0.1 | 0.38 | −3.1 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L_25N | 5.2 ± 0.3 | 5.7 ± 0.1 | 0.28 | −3.1 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ7L_30N | 2.3 ± 0.9 | 4.5 ± 1.1 | 0.17 | −3.2 ± 0.1 | Fadh > Fcoh |
| OGc | pd | Kd e (mM) | qf (mM−1) | ΔGg (kcal/mol) | Balanceh |
| OmpG | 4.9 ± 0.5 | 11 ± 1 | 0.017 | −2.7 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L | 5.3 ± 0.9 | 13 ± 1 | 0.013 | −2.5 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L_25N | NDi | NDi | NDi | NDi | Fadh ≪ Fcoh |
| FhuA ΔC/Δ7L_30N | NDi | NDi | NDi | NDi | Fadh ≪ Fcoh |
| CHAPSc | pd | Kde (mM) | qf (mM−1) | ΔGg (kcal/mol) | Balanceh |
| OmpG | ~1.6 | < 0.6 | NDi | ~−7.0 | Fadh ≫ Fcoh |
| FhuA ΔC/Δ5L | 1.7 ± 0.6 | 3.3 ± 1.8 | 0.015 | −3.4 ± 0.5 | Fadh > Fcoh |
| FhuA ΔC/Δ5L_25N | NDi | NDi | NDi | NDi | Fadh ≪ Fcoh |
| FhuA ΔC/Δ7L_30N | NDi | NDi | NDi | NDi | Fadh ≪ Fcoh |
| LysoFosc | pd | Kde(mM) | qf (mM−1) | ΔGg (kcal/mol) | Balanceh |
| OmpG | 5.6 ± 1.5 | 0.26 ± 0.03 | 0.41 | −4.9 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L | 9.1 ± 6.2 | 0.47 ± 0.04 | 0.51 | −4.5 ± 0.1 | Fadh > Fcoh |
| FhuA ΔC/Δ5L_25N | 4.5 ± 0.5 | 0.71 ± 0.03 | 0.22 | −4.3 ± 0.1 | Fadh ≅ Fcoh |
| FhuA ΔC/Δ7L_30N | 3.5 ± 0.7 | 0.73 ± 0.04 | 0.13 | −4.3 ± 0.1 | Fadh ≅ Fcoh |
To reach low detergent concentrations below the CMC, the Gdm-HCl-solubilized (FhuA derivatives) or urea-solubilized (OmpG) proteins were refolded at various detergent concentrations above the CMC.
The dose-response equilibrium curves were fitted by the four-parameter Hill equation.
This column indicates the names of the detergents and proteins used in this work.
p is the Hill coefficient.
The apparent dissociation constant, Kd, was determined as the midpoint of the dose-dependent dissociation phase.13
The slope factor or transition steepness was calculated at the midpoint of the dissociation phase.
Free energies were determined using the standard thermodynamic relationship ΔG = RT ln Kd.
The quantitative balance between the adhesive protein-detergent (Fadh) and cohesive detergent-detergent interactions (Fcoh) of the proteomicelles.
Not determined.
We were able to refold the acidic β-barrel proteins in glucoside-containing neutral detergent (OG) and noted a detergent desolvation-induced transition with maximum and minimum anisotropy values of ~0.30 and ~0.16, respectively (Table 3, Table 4). In contrast, the experiments with the basic β-barrel proteins revealed very low anisotropy values of ~0.17, near r1, which corresponded to the most rotationally diffusive FhuA ΔC/Δ5L, indicating poor solubility features under these experimental conditions. Because the two-state detergent desolvation-induced transition was only observed with acidic, but not basic β barrels, it is conceivable that the anisotropy value, r, is strongly dependent on the characteristics of the PDC, even if there is some mobility restriction of the fluorophore by the detergent coat. Therefore, these examples illustrate how extensive changes in the charge distribution across the solvent-accessible surface of the β barrel proteins produced dramatic alterations in the magnitude of adhesive interactions.
Zwitterionic detergents
We extended these studies to zwitterionic detergents. Interestingly, we were able to refold the acidic β barrels in CHAPS, but not the basic β barrels (Fig. 4). This situation resembles that found with OG. Indeed, the time-dependent changes in the FP anisotropy revealed a fast dissociation of CHAPS from FhuA ΔC/Δ5L at a detergent concentration of 2 mM, which is ~3-fold lower than its CMC (Table 1, Supporting Information, Fig. S4). In contrast, we found a strong binding interaction between CHAPS and OmpG, with a Kd < 0.6 mM (Table 4). n-dodecyl-N,N-dimethylglycine (LD), another zwitterionic detergent, showed a closely similar signature, encompassing adhesive interactions with the acidic β barrels, but weak interactions with the basic β barrels (Supporting Information, Fig. S5). In excellent accord with the outcomes pertaining to the above-mentioned zwitterionic detergents, LysoFos exhibited stronger adhesive interactions with the acidic β-barrels than those interactions with the basic β barrels (Table 3 and Table 4). On the other hand, the Kd values noted with the interaction of LysoFos with the basic β barrels matched the CMC under similar experimental conditions, indicating no significant difference between adhesive and cohesive interactions (Table 1 and Table 4). Therefore, LysoFos was found as a satisfactorily solubilizing detergent for both the acidic and basic β barrels.
Figure 4. Dose-response changes in fluorescence anisotropy recorded with zwitterionic detergents and proteins of varying isoelectric point pI.

This panel shows a desorption isotherms recorded with 3-[(3-Cholamidopropyl) dimethylammonio]-1-Propanesulfonate (CHAPS). Vertical bars represent the magnitudes of the CMC and Kd of the PDCs of the proteins of varying isoelectric point. The horizontal dashed bar represents the minimum anisotropy value, r0 = ~0.16, obtained with FhuA ΔC/5L in 6 M Gdm-HCl (Table 3). All the other experimental conditions were the same as in Fig. 3.
Does pH alter the interfacial interactions of the PDC with neutral detergents?
Here, we asked whether pH alters the balance between the adhesive and cohesive interactions. It is worth mentioning that Texas Red is a pH insensitive fluorophore.52 Because pH modifications affect the protein electrostatics, but not the cohesive interactions within proteomicelles formed by a neutral detergent, we examined the PDC interfacial interactions mediated by DM (Fig. 5; Supporting Information, Table S3 and Table S4). The rationale of this choice resided in the fact that at physiological pH DM showed substantially increased adhesive interactions with the basic β barrels (Kd ~ 0.9 mM), as compared with the acidic β barrels (Kd ~ 1.8 mM), although it is a neutral detergent (Fig. 3C). At acidic pH values, no significant distinctions between Kd and CMC were observed, despite a broad pI range among the four β barrels. In contrast to all FhuA derivatives, DM-refolded OmpG showed no significant pH-dependent alterations in the balance between adhesive and cohesive interactions when examined in the pH range 4.6 – 8.2 (Supporting Information, Table S4; last column), likely due to very strong hydrophobic interactions at the PDC interface.
Figure 5. Dose-response changes in fluorescence anisotropy acquired with DM under acidic conditions.


(A) pH 4.6; (B) pH 5.6; (C) pH 6.8; (D) pH 8.2; and (E) pH 10.0. The buffer was either 50 mM HEPES (pH 6.8), or 50 mM NaOAc (pH 4.6, pH 5.6). The salt concentration was 200 mM NaCl. Vertical bars represent the magnitudes of the CMC and Kd of the PDCs of varying isoelectric point of the proteins. The horizontal dashed bar represents the minimum anisotropy value, r0 = ~0.16, obtained with FhuA ΔC/5L in either 40 mM SDS or 6 M Gdm-HCl (Table 3). All the other experimental conditions were the same as in Fig. 3.
MD simulations of the interactions between DDM and the β-barrel proteins
To gain insights into the PDC interactions at the submicroscopic level, we simulated spontaneous aggregation of DDM detergents around OmpG, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N, using the MD method of Bond and colleagues (2004).53 Each simulation system contained one copy of the protein, 21 or 105 DDM molecules, which translates into 20 or 100 mM DDM concentration, respectively, and 200 mM NaCl electrolyte (Fig. 6A). Two independent simulations were performed for each system differing by the initial arrangements of the DDM molecules (Methods). Starting from a disperse configuration, DDM molecules were seen to aggregate at the surface of the proteins, reaching a dynamic equilibrium after ~100 ns (Fig. 6B; Supporting Information, Fig. S6A). In all simulations, all DDM molecules were observed to eventually form a complex with the protein (Supporting Information, Fig. S6B–D). All proteins maintained their structural integrity at our simulation timescale. Reflecting the progress of the aggregation process, the radius of gyration of the DDM-protein complex, Rg, reached steady-state values of ~2.2, ~2.9, ~2.7, and ~2.6 nm for proteomicelles with OmpG, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N, respectively (Supporting Information, Fig. S6B, C). Interestingly, increasing DDM concentration by 5-fold produced a rather modest (~0.6 nm) increase of Rg (Supporting Information, Fig. S6D).
Figure 6. MD simulations of DDM binding to the β-barrel proteins.

(A) Initial setup of a typical MD simulation. FhuA ΔC/Δ5L is shown using a cartoon representation (yellow); the head and the tail regions of the DDM molecules are shown as orange spheres and cyan lines, respectively. The magenta and green spheres indicate the sodium and chloride ions, respectively; the semitransparent surface indicate the volume occupied by the electrolyte; (B) A sequence of microscopic configurations realized in a typical MD simulation. Images in the top and bottom rows depict the same system from two different viewpoints; (C) The number of the DDM molecules bound to FhuA ΔC/Δ5L with their tail (top) or head (bottom) parts versus simulation time. The simulation system contained 20 mM DDM initially placed on a cubic lattice around the protein. To count as a binding event, any atom of a DDM must reside within 4 Å of any atom of the protein. The traces show 0.48 ns block average of 2.4 ps-sampled data. The inset image shows a zoomed-in view of a 10-ns fragment of the binding trace. The standard deviation of the number of bound DDM molecules, σ, is used as an effective measure of the molecules’ binding affinity: smaller deviation indicates stronger binding; (D) The mean equilibrium number of DDM molecules bound to the proteins (left) and the mean equilibrium standard deviation (right) of the number of DDM molecules bound to the proteins. In each figure, the left two columns characterize binding of the tail or head groups of DDM to the proteins; the right three columns characterize binding of entire DDM molecules to the hydrophobic, hydrophilic and charged residues of the proteins. The data were averaged over the steady-state (last ~70 ns) parts of two independent MD trajectories for each protein and then over the four protein systems; (E) Four β-barrel proteins colored according to their local propensity for forming an interface with DDM molecules. For each residue, the contact probability was calculated as the fraction of the time it was bound to a DDM molecule within the last ~70 ns of the equilibration simulation; (F) The average fraction of the hydrophobic, hydrophilic, positively and negatively charged residues in the four β-barrel proteins (open bars) and the fraction of those residues that bind DDM (filled bars) during the steady-state (last ~70 ns) parts of the MD trajectories. The data were averaged over the two independent MD trajectories for each protein and then over the four protein systems. In panels D and F, error bars represent standard deviations among the eight simulations.
The steady-state parts of the trajectories were used to extract information about DDM-protein interactions. Fig. 6C shows two typical traces characterizing binding of DDM’s hydrophobic (tail) and hydrophilic (head) parts to FhuA ΔC/Δ5L. The tail parts of all DDM molecules bind to the protein surface, which is not the case for the head groups of which only ~80% have atoms that are in contact with the protein surface (Fig. 6D). The most dramatic difference, however, is seen in the magnitude of the steady-state fluctuations, σ, which we use as an effective measure of binding affinity. Indeed, assuming that the binding of a detergent to a protein can be described by a harmonic potential and that the conditions of the equipartition theorem are met, the spring constant of the harmonic potential should be inversely proportional to the square of the standard deviation. According to this argument, the head group of DDM binds to the protein ~8 times less strongly than the tail part (Fig. 6D). Similarly, we find that the binding of DDM to hydrophobic residues to be ~2.6 times stronger than to hydrophilic residues and ~8 times stronger than to charged residues (Fig. 6D; Supporting Information, Fig. S7A). Here and anywhere else in the paper, hydrophilic residues include polar and charged residues. Analyzing the binding of head groups and tails of DDM separately, we find only the tail domain to exhibit considerable dependence of binding strength on the residue type (Supporting Information, Fig. S7B–C). Further analysis found no significant correlation between the DDM binding affinity and the sign of the charged residues (Supporting Information, Fig. S7D–F).
Fig. 6E shows the structure of the four proteins colored by the local probability of binding DDM molecules. Interestingly, DDM molecules did not uniformly cover the hydrophobic belt of the protein and tended to form half-micelle like aggregates at the junction of the loops and the β barrel. One possible explanation for such an arrangement is that detergent molecules seek such configurations where both their hydrophobic and hydrophilic parts are placed in the most favorable environment. At the same time, the pattern of DDM binding (Fig. 6E) is very similar to the pattern of hydrophobic and hydrophilic residues in the protein structures (Supporting Information, Fig. S8A). Note that increasing the number of DDM molecules does not lead to formation of half-micelles at the hydrophobic belt of the protein (Supporting Information, Fig. S6A). Unfortunately, statistical sampling of binding events was not sufficient in our simulations to elucidate the effect of point mutations on DDM binding. Nevertheless, we could infer this information by evaluating the effect that a residue type has on its probability to bind DDM. Fig. 6F plots the fraction of hydrophobic, hydrophilic, as well as positively and negatively charged residues in the respective protein structures, which was averaged over the four proteins. Fig. S8A (Supporting Information) show the same data for individual proteins. If the binding of DDM molecules to a protein were completely random, the fraction of residues that would bind detergent would be the same as the fraction of the residues in the protein. Analysis of MD simulations, however, does not support this conjecture. DDM molecules are found to bind hydrophobic residues 50% more likely than suggested by their abundance in the structure whereas binding hydrophilic residues was 30% less likely (Fig. 6F). Interestingly, the hydrophobic residues of OmpG bind DDM considerably stronger than those of FhuA variants (Supporting Information, Fig. S8B), in agreement with the proteins’ grand average of hydropathicity indices (Table 2). Substantial reduction of DDM binding is also observed in the case of positively and negatively charged residues.
Do α-helical transmembrane proteins undergo a two-state detergent desolvation-induced transition?
One question is whether we can extend this FP-based approach to other membrane proteins, which are different in structure from β barrels. Therefore, we inspected SELENOS and SELENOK, two small, human membrane proteins that are not related in structure and homology with either OmpG or FhuA. SELENOS and SELENOK are single-pass polypeptides with a short luminal segment, a single transmembrane helix, and a cytoplasmic domain housing a selenocysteine (Sec) residue.54 The cytoplasmic regions contain an unstructured segment rich in glycine, proline, and polar residues (Fig. 7A). Both SELENOS34 and SELENOK32 are homodimers (Fig 7B). In this work, we explored SELENOK U92C and SELENOS U188S, in which the selenocysteine was mutated either to cysteine (SELENOK) or serine (SELENOS), leaving a sole cysteine in the protein for fluorescent labeling (Supporting Information, Table S5). We also noted that these proteins underwent a two-state DDM desolvation transition in proteomicelles, but between significantly lower rmax and rmin values than those determined with the FhuA derivatives (Supporting Information, Table S6). For both α-helical proteins, the apparent Kd values were greater than the CMC, suggesting that the cohesive interactions were greater than the adhesive interactions (Fig. 7C; Supporting Information, Table S7; last column).
Figure 7. Dose-response in fluorescence anisotropy acquired with SELENOK U92C and SELENOS U188S, two short single α-helical transmembrane proteins solubilized in DDM.

(A) Cartoon presenting the transmembrane topography of the SELENOK U92C and SELENOS U188S proteins; (B) Domain organization and the position of relevant Cys and Sec residues of SELENOS and SELENOK. TM stands for the transmembrane region of these proteins; (C) The protein concentration in the well was 200 nM. The initial DDM concentration was 1.3 mM. The FP measurements were carried out using a solution that contained 200 mM NaCl, 50 mM HEPES, pH 7.4 at a temperature of 24°C. Vertical bars represent the magnitudes of the CMC and Kd. All the other experimental conditions were the same as in Fig. 3.
DISCUSSION
In this work, we inspected the interfacial interactions between detergents and water-insoluble membrane proteins. The detergent desolvation of insoluble membrane proteins is closely related to protein unfolding. Recently, using temperature-dependent circular dichroism (CD) spectroscopy and chemical denaturant-induced protein unfolding, we showed that rmax and rmin correspond to the folded and unfolded states, respectively.35 This finding implies that the unfolding transition of these β barrel proteins in aqueous phase occurs in between these states. However, the observed changes in the FP anisotropy directly reflected adhesion-dissociation of detergent monomers from the protein, not protein folding-unfolding. The two-state transition of detergent desolvation was due to detergent depletion in the proximity of a hydrophobic membrane protein or weak adhesive PDC interactions. The steady-state FP anisotropy values on these plots represent the endpoints of the desolvation reaction; thereby, the signal resulting from more fluorophores has no impact on the endpoints of the desolvation reaction. For example, we show the ability of obtaining the two-state Langmuir-Hill dissociation curves using two dimeric selenoproteins of unknown structure. Moreover, three distinct protein instances (e.g., FhuA, OmpG, and selenoproteins) indicate the effective labeling of the membrane proteins within the aqueous phase-exposed domains for quantitative FP studies.
In general, the very acidic OmpG exhibited stronger adhesive interactions with both neutral and zwitterionic detergents than the other FhuA protein mutants, likely due to strong hydrophobic PDC contacts. The all-atom MD simulations confirmed a stronger binding interaction of DDM to OmpG than to other FhuA derivatives (Fig. 3; Table S2; Supporting Information, Fig. S8B). At physiological conditions, the adhesive interactions were greater than the cohesive interactions in the case of acidic β barrels solubilized by neutral, short-hydrophobic tail detergents CYMAL-4 and OG, as well as by zwitterionic detergents CHAPS, LD, and LysoFos. In contrast, the basic β barrels could not be folded in OG, CHAPS and LD, but showed comparable adhesive and cohesive interactions when incubated in LysoFos. These findings imply that for the zwitterionic detergents the electrical dipoles of the monomers are attracted by the dominant negative charges of the acidic β barrels, but repelled by the dominant positive charges of the basic β proteins. Another clear distinction between acidic and basic barrels was noted with DM at physiological pH. Closely similar adhesive and cohesive interactions were apparent for the acidic β barrels, but strong adhesive interactions were found for the basic β barrels. These few examples illuminate the entanglement and importance of the hydrophobic and electrostatic interactions in mediating the PDC interface.
rmin and rmax, defining the two sub-states of the desolvation transition, were significantly smaller for the shorter polypeptides, which is in accord with a greater rotational mobility of a lower-molecular mass proteomicelle. For example, the 102-residue SELENOK U92C and 190-residue SELENOS U188S showed rmin values of 0.095 ± 0.002 and 0.103 ± 0.002, respectively, when they were solubilized in 1.3 mM DDM. These values correspond to rotational diffusion coefficients, Drfast, of ~1.3×108 and ~1.1×108 s−1 (Supporting Information, Table S6), respectively, giving rotational correlation times, θ, in the range 1.3 –1.5 ns. This time interval compares well with the rotational correlation time θ = 14.2 ns, as calculated for Stam2 VHS-domain (VHS), a 17.7 kDa protein, at 20°C.55 Another interesting aspect of the hydrodynamics of DDM-containing proteomicelles is that the average radius, Rh, determined with β barrel proteins, ranged a narrow interval between 2.5 and 2.8 nm, whereas that calculated for the shorter helical polypeptides was ~2.1 nm. The MD trajectories of the DDM-mediated proteomicellizations with all four β barrels indicated a gyration radius covering a range between 2.2 nm and 2.9 nm. Our full-atomistic MD computational studies also indicated that a substantial increase in the DDM concentration did not produce a significant change in the PDC gyration radius. This finding is in accord with the FP anisotropy measurements, which did not reveal alterations in the FP anisotropy at detergent concentrations much greater than the CMC (Supporting Information, Fig. S2).
In some cases (e.g., all desorption isotherms in Fig. 3B), the rmin values acquired with the β-barrel proteins were greater than the value corresponding to most rotationally diffusive FhuA ΔC/Δ5L protein (r1 = ~0.16), which was acquired under denaturing conditions by excess of Gdm-HCl. At least two possibilities can explain these slightly elevated rmin values. First, there might be a small residual amount of yet-bound detergent monomers at the lowest detergent concentrations used in this work, thus contributing to a decreased rotational mobility of the desolvated protein. Second, there are effects of the soluble local aggregation, again decreasing the tumbling rate of the desolvated protein. It should be noted that soluble aggregates of proteins would increase the anisotropy due to the size increase from monomers. Because the proteins examined in this work are hydrophobic, we do also see, as expectedly, insoluble aggregation at detergent concentrations much smaller than the CMC, resulting in a decrease in raw polarization signal.
We used Texas Red, a bright fluorophore,36 enabling a low concentration of the inspected protein. This is a very important asset of this approach, given the limited expression and purification yields of water-insoluble membrane proteins. Previous FP methods also involved time-resolved anisotropy measurements that require a very fast detector.56 This latter FP method facilitates the determination of anisotropy decays of proteins exposed to excitation light pulses shorter than the decay time constant of the sample. In this way, time-resolved anisotropy studies can reveal details lost in the averaging process, such as molecular shape, conformational sub-states, and local flexibility. Because of the need for sophisticated equipment, these time-resolved anisotropy measurements cannot be expanded to a multiplexed format for inspecting a large sample number, which is a critical requirement in the HTS area of the PDC interactions. One immediate question is whether this semi-quantitative FP-based approach can be expanded by employing intrinsic tryptophan fluorescence. This is because in general membrane proteins have multiple tryptophan residues exposed to their hydrophobic interface.24 We judge that it is not very convenient to use intrinsic tryptophan fluorescence for the FP-based spectroscopy studies for a number of reasons. They include complex contributions of individual-residue tryptophan spectra to the overall FP spectrum of the protein as well as rapid tryptophan quenching, because the indole nucleus is prone to electron donation during the excitation state.57 Moreover, the presence of multiple tryptophan residues in any given membrane protein requires their mutagenesis with non-fluorescent side chains.14 Therefore, many applications of the FP spectroscopy rely on covalently attached intense fluorophores, such as Texas Red from this work. It should be mentioned that this approach cannot be coupled with large fluorophores, such as green fluorescence protein (GFP) and its derivatives, because they can potentially impact the local tumbling rate, flexibility, and even conformation of the inspected protein.
There are various ways to identify contributions (if any significant) of light scattering to the FP anisotropy signal. We think that the light scattering has negligible effects to our acquired fluorescence anisotropy signal for the following independent reasons: (i) the Spectramax i3 plate reader that we used is equipped with emission filters for rhodamine derivatives (Texas Red is one of them). These filters are designed for excitation at 535 nm and emission at 595 nm. This rather large separation between excitation and emission (~ 60 nm) ensures that scattering is minimal in our data; (ii) the large wavelength of emission was strategically used to avoid Raman and Rayleigh scattering effects. This is because the light intensities of both scattering contributions are proportional to λ−4, where λ is the wavelength;58 (iii) in our very preliminary stage of these studies, we have increased the concentration of labeled proteins up to a level, in which the signal was independent on protein concentration;59 (iv) we conducted control experiments with proteins of closely similar molecular mass, but that exhibit a broad range of detergent solubilization properties under identical micellization conditions. The basal fluorescence anisotropy of the unfolded FhuA variants under excess of Gdm-HCl was ~0.16. We demonstrated that acidic FhuA proteins were refolded in OG, showing an anisotropy signal of ~0.3. On the contrary, the basic FhuA variants were not refolded in OG and aggregated in solution in the presence of OG-induced micelles, exhibiting a fluorescence anisotropy of ~0.16. This control experiment demonstrated that both the light scattering contributions and protein aggregation did not affect the FP anisotropy signal under OG-induced micellization conditions. Such a control experiment was also recapitulated with other detergent micelles (e.g., CHAPS in Table 3, Fig. 4). These experimental outcomes indicates that the fluorophore directly probed whether the protein is in a detergent solvated or desolvated state.35 Overall, we think that the light scattering of the incident excitation light into the emission pathway does not affect the anisotropy values reported here.
CONCLUSIONS
In summary, we report a comparative study of the detergent desolvation-induced transitions of membrane proteins of varying biophysical and structural fingerprint. This approach for deriving the energetics of detergent desolvation was used for four robust β-barrels, but extended to two α-helical ones, whose X-ray crystal structure is not yet available. These membrane proteins were expressed, solubilized, purified, and refolded under very distinctive protocols, reinforcing the impact of this approach on other membrane proteins to examine their interfacial PDC interactions. Therefore, this method may be applied to diverse mixtures of detergents with complementary interfacial features. For example, such measurements might be expanded to mechanistic studies of the PDC interactions of newly developed detergent-like compounds, such as steroid-based facial amphiphiles,60 lipopeptides61 and amphipols.62
Supplementary Material
Acknowledgments
We thank Motahareh Larimi and Avinash Thakur valuable feedback and stimulating discussions. This study was supported by the US National Institutes of Health grants GM113299 (A.R.B.), GM115442 (M.C.), GM115762 (S.N.L.), and GM088403 (L.M.), as well as National Science Foundation grant MCB-1616178 (S.R.). The Delaware COBRE program supported this research with the US National Institutes of Health grants P20 GM104316 and P30 GM110758-02. The computational studies in this article were supported by a grant from the National Institutes of Health P41-RR005969. The authors acknowledge supercomputer time provided through the XSEDE Allocation Grant MCA05S028 and the Blue Waters petascale supercomputer system at the University of Illinois at Urbana-Champaign. W.S. acknowledges financial support from the China Scholarship Council (CSC201506090040) and the National Natural Science Foundation of China (Grant No. 51435003).
Footnotes
Supporting Information. (i) Characterization of selenoproteins prior to and following labeling with Texas Red; (ii) Example of steady-state FP traces illustrating no time-dependent alterations in the anisotropy readout at detergent concentrations much greater than the CMC; (iii) Example of steady-state FP traces showing no time-dependent alterations in the anisotropy readout after 24 hours; (iv) Hydrodynamic changes of the proteomicelles during the transition of detergent desolvation; (v) Table that summarizes the recorded minima and maxima of the anisotropy with maltoside-containing detergents; (vi) Summary of the fitting results of the two-state, concentration-dependent anisotropy curves acquired with maltoside-containing detergents; (vii) Time-dependent changes in the FP anisotropy acquired with CHAPS; (viii) Time-dependent FP anisotropy acquired with LD; (ix) Table that summarizes the recorded minima and maxima of the anisotropy readout with DM at various pH values; (x) Summary of the fitting results of the two-state desolvation curves acquired with DM at various pH values; (xi) MD simulations of DDM molecules binding to β-barrel proteins; (xii) Differential affinity of DDM molecules to residues of β-barrels; (xiii) DDM binding versus residue type; (xiv) Biophysical properties of the selenoproteins; (xv) Table that summarizes the recorded minima and maxima of the anisotropy readout with the α-helical membrane proteins solubilized in DDM; (xvi) Summary of the fitting results of the two-state, concentration-dependent anisotropy curves acquired with α-helical transmembrane proteins. These materials are available free of charge via the Internet at http://pubs.acs.org
References
- 1.Stangl M, Veerappan A, Kroeger A, Vogel P, Schneider D. Detergent properties influence the stability of the glycophorin A transmembrane helix dimer in lysophosphatidylcholine micelles. Biophys J. 2012;103(12):2455–64. doi: 10.1016/j.bpj.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Z, Wang C, Zhou Q, An J, Hildebrandt E, Aleksandrov LA, Kappes JC, DeLucas LJ, Riordan JR, Urbatsch IL, et al. Membrane protein stability can be compromised by detergent interactions with the extramembranous soluble domains. Protein Sci. 2014;23(6):769–89. doi: 10.1002/pro.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roy A. Membrane preparation and solubilization. Methods Enzymol. 2015;557:45–56. doi: 10.1016/bs.mie.2014.11.044. [DOI] [PubMed] [Google Scholar]
- 4.Sadaf A, Cho KH, Byrne B, Chae PS. Amphipathic agents for membrane protein study. Methods Enzymol. 2015;557:57–94. doi: 10.1016/bs.mie.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Brouillette CG. A guide to differential scanning calorimetry of membrane and soluble proteins in detergents. Methods Enzymol. 2016;567:319–58. doi: 10.1016/bs.mie.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Miles AJ, Wallace BA. Circular dichroism spectroscopy of membrane proteins. Chem Soc Rev. 2016;45(18):4859–72. doi: 10.1039/c5cs00084j. [DOI] [PubMed] [Google Scholar]
- 7.Raschle T, Rios Flores P, Opitz C, Muller DJ, Hiller S. Monitoring backbone hydrogen-bond formation in beta-barrel membrane protein folding. Angew Chem Int Ed Engl. 2016;55(20):5952–5. doi: 10.1002/anie.201509910. [DOI] [PubMed] [Google Scholar]
- 8.Borysik AJ, Hewitt DJ, Robinson CV. Detergent release prolongs the lifetime of native-like membrane protein conformations in the gas-phase. J Am Chem Soc. 2013;135(16):6078–83. doi: 10.1021/ja401736v. [DOI] [PubMed] [Google Scholar]
- 9.Jahnke N, Krylova OO, Hoomann T, Vargas C, Fiedler S, Pohl P, Keller S. Real-time monitoring of membrane-protein reconstitution by isothermal titration calorimetry. Anal Chem. 2014;86(1):920–927. doi: 10.1021/ac403723t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Textor M, Keller S. Automated analysis of calorimetric demicellization titrations. Anal Biochem. 2015;485:119–21. doi: 10.1016/j.ab.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Qin X, Liu M, Yang D, Zhang X. Concentration-dependent aggregation of CHAPS investigated by NMR spectroscopy. J Phys Chem B. 2010;114(11):3863–8. doi: 10.1021/jp911720w. [DOI] [PubMed] [Google Scholar]
- 12.Turman DL, Nathanson JT, Stockbridge RB, Street TO, Miller C. Two-sided block of a dual-topology F- channel. Proc Natl Acad Sci US A. 2015;112(18):5697–701. doi: 10.1073/pnas.1505301112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoddart LA, White CW, Nguyen K, Hill SJ, Pfleger KD. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br J Pharmacol. 2016;173(20):3028–3037. doi: 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jutila A, Zhu K, Patkar SA, Vind J, Svendsen A, Kinnunen PK. Detergent-induced conformational changes of Humicola lanuginosa lipase studied by fluorescence spectroscopy. Biophys J. 2000;78(3):1634–42. doi: 10.1016/S0006-3495(00)76715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen KK, Oliveira CL, Larsen KL, Poulsen FM, Callisen TH, Westh P, Pedersen JS, Otzen D. The role of decorated SDS micelles in sub-CMC protein denaturation and association. J Mol Biol. 2009;391(1):207–26. doi: 10.1016/j.jmb.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Naidu KT, Prabhu NP. Protein-surfactant interaction: sodium dodecyl sulfate-induced unfolding of ribonuclease A. J Phys Chem B. 2011;115(49):14760–7. doi: 10.1021/jp2062496. [DOI] [PubMed] [Google Scholar]
- 17.Fano M, van de Weert M, Moeller EH, Kruse NA, Frokjaer S. Ionic strength-dependent denaturation of Thermomyces lanuginosus lipase induced by SDS. Arch Biochem Biophys. 2011;506(1):92–8. doi: 10.1016/j.abb.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Qiu XJ. Quantification of membrane protein self-association with a high-throughput compatible fluorescence assay. Biochemistry. 2017;56(14):1951–1954. doi: 10.1021/acs.biochem.7b00009. [DOI] [PubMed] [Google Scholar]
- 19.Kubler D, Bergmann A, Weger L, Ingenbosch KN, Hoffmann-Jacobsen K. Kinetics of detergent-induced activation and inhibition of a minimal lipase. J Phys Chem B. 2017;121(6):1248–1257. doi: 10.1021/acs.jpcb.6b11037. [DOI] [PubMed] [Google Scholar]
- 20.Khao J, Arce-Lopera J, Sturgis JN, Duneau JP. Structure of a protein-detergent complex: the balance between detergent cohesion and binding. Eur Biophys J. 2011;40(10):1143–55. doi: 10.1007/s00249-011-0745-9. [DOI] [PubMed] [Google Scholar]
- 21.Yildiz O, Vinothkumar KR, Goswami P, Kuhlbrandt W. Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. EMBO J. 2006;25(15):3702–3713. doi: 10.1038/sj.emboj.7601237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locher KP, Rees B, Koebnik R, Mitschler A, Moulinier L, Rosenbusch JP, Moras D. Transmembrane signaling across the ligand-gated FhuA receptor: crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell. 1998;95(6):771–778. doi: 10.1016/s0092-8674(00)81700-6. [DOI] [PubMed] [Google Scholar]
- 23.Mohammad MM, Howard KR, Movileanu L. Redesign of a plugged beta-barrel membrane protein. J Biol Chem. 2011;286(10):8000–8013. doi: 10.1074/jbc.M110.197723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfe AJ, Mohammad MM, Thakur AK, Movileanu L. Global redesign of a native beta-barrel scaffold. Biochim Biophys Acta. 2016;1858(1):19–29. doi: 10.1016/j.bbamem.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niedzwiecki DJ, Mohammad MM, Movileanu L. Inspection of the engineered FhuA deltaC/delta4L protein nanopore by polymer exclusion. Biophys J. 2012;103(10):2115–2124. doi: 10.1016/j.bpj.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thakur AK, Larimi MG, Gooden K, Movileanu L. Aberrantly large single-channel conductance of polyhistidine arm-containing protein nanopores. Biochemistry. 2017;56(36):4895–4905. doi: 10.1021/acs.biochem.7b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammad MM, Iyer R, Howard KR, McPike MP, Borer PN, Movileanu L. Engineering a rigid protein tunnel for biomolecular detection. J Am Chem Soc. 2012;134(22):9521–9531. doi: 10.1021/ja3043646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomita N, Mohammad MM, Niedzwiecki DJ, Ohta M, Movileanu L. Does the lipid environment impact the open-state conductance of an engineered beta-barrel protein nanopore? BiochimBiophysActa -Biomembr. 2013;1828(3):1057–1065. doi: 10.1016/j.bbamem.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linke D. Detergents: an overview. Methods Enzymol. 2009;463:603–617. doi: 10.1016/S0076-6879(09)63034-2. [DOI] [PubMed] [Google Scholar]
- 30.Fahie M, Chisholm C, Chen M. Resolved single-molecule detection of individual species within a mixture of anti-biotin antibodies using an engineered monomeric nanopore. ACS nano. 2015;9(2):1089–1098. doi: 10.1021/nn506606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M, Khalid S, Sansom MS, Bayley H. Outer membrane protein G: engineering a quiet pore for biosensing. Proc Natl Acad Sci US A. 2008;105(17):6272–6277. doi: 10.1073/pnas.0711561105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Zhang Z, Rozovsky S. Selenoprotein K form an intermolecular diselenide bond with unusually high redox potential. FEBS Lett. 2014;588(18):3311–21. doi: 10.1016/j.febslet.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Srinivasan P, Pham DN, Rozovsky S. Expression and purification of the membrane enzyme selenoprotein K. Protein Expr Purif. 2012;86(1):27–34. doi: 10.1016/j.pep.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Li F, Rozovsky S. The intrinsically disordered membrane protein selenoprotein S is a reductase in vitro. Biochemistry. 2013;52(18):3051–61. doi: 10.1021/bi4001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfe AJ, Hsueh YC, Blanden AR, Mohammad MM, Pham B, Thakur AK, Loh SN, Chen M, Movileanu L. Interrogating detergent desolvation of nanopore-forming proteins by fluorescence polarization spectroscopy. Analytical chemistry. 2017;89(15):8013–8020. doi: 10.1021/acs.analchem.7b01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gradinaru CC, Marushchak DO, Samim M, Krull UJ. Fluorescence anisotropy: from single molecules to live cells. Analyst. 2010;135(3):452–9. doi: 10.1039/b920242k. [DOI] [PubMed] [Google Scholar]
- 37.Titus JA, Haugland R, Sharrow SO, Segal DM. Texas Red, a hydrophilic, red-emitting fluorophore for use with fluorescein in dual parameter flow microfluorometric and fluorescence microscopic studies. J Immunol Methods. 1982;50(2):193–204. doi: 10.1016/0022-1759(82)90225-3. [DOI] [PubMed] [Google Scholar]
- 38.Jameson DM, Ross JA. Fluorescence polarization/anisotropy in diagnostics and imaging. Chem Rev. 2010;110(5):2685–708. doi: 10.1021/cr900267p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prinz H. Hill coefficients, dose-response curves and allosteric mechanisms. J Chem Biol. 2010;3(1):37–44. doi: 10.1007/s12154-009-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31(4):671–90. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoo JJ, Aksimentiev A. Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J Phys Chem Lett. 2012;3(1):45–50. [Google Scholar]
- 43.Yoo J, Aksimentiev A. Improved parameterization of amine-carboxylate and amine-phosphate interactions for molecular dynamics simulations using the CHARMM and AMBER force fields. J Chem Theory Comput. 2016;12(1):430–43. doi: 10.1021/acs.jctc.5b00967. [DOI] [PubMed] [Google Scholar]
- 44.Andersen HC. RATTLE - A velocity version of the shake algorithm for molecular-dynamics calculations. J Comput Phys. 1983;52(1):24–34. [Google Scholar]
- 45.Miyamoto S, Kollman PA. SETTLE - An analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13(8):952–962. [Google Scholar]
- 46.Darden T, York D, Pedersen L. Particle mesh Ewald - An N.LOG(N) method for the Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 47.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177–4189. [Google Scholar]
- 48.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-pressure molecular dynamics simulation - The langevin piston method. J Chem Phys. 1995;103(11):4613–4621. [Google Scholar]
- 49.Brunger AT. X-PLOR, Version 3.1: A System for X-ray Crystallography and NMR. The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry, Yale University; New Haven: CT: 1992. [Google Scholar]
- 50.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 51.Grosse W, Psakis G, Mertins B, Reiss P, Windisch D, Brademann F, Burck J, Ulrich A, Koert U, Essen LO. Structure-based engineering of a minimal porin reveals loop-independent channel closure. Biochemistry. 2014;53(29):4826–38. doi: 10.1021/bi500660q. [DOI] [PubMed] [Google Scholar]
- 52.Sandoval RM, Kennedy MD, Low PS, Molitoris BA. Uptake and trafficking of fluorescent conjugates of folic acid in intact kidney determined using intravital two-photon microscopy. Am J Physiol Cell Physiol. 2004;287(2):C517–26. doi: 10.1152/ajpcell.00006.2004. [DOI] [PubMed] [Google Scholar]
- 53.Bond PJ, Cuthbertson JM, Deol SS, Sansom MS. MD simulations of spontaneous membrane protein/detergent micelle formation. J Am Chem Soc. 2004;126(49):15948–9. doi: 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- 54.Shchedrina VA, Novoselov SV, Malinouski MY, Gladyshev VN. Identification and characterization of a selenoprotein family containing a diselenide bond in a redox motif. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(35):13919–24. doi: 10.1073/pnas.0703448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen PC, Hologne M, Walker O. Computing the rotational diffusion of biomolecules via molecular dynamics simulation and quaternion orientations. J Phys Chem B. 2017;121(8):1812–1823. doi: 10.1021/acs.jpcb.6b11703. [DOI] [PubMed] [Google Scholar]
- 56.Lakowicz JR. Principles of fluorescence microscopy. 2. Springer; New York: 2006. [Google Scholar]
- 57.Mann TL, Krull UJ. Fluorescence polarization spectroscopy in protein analysis. Analyst. 2003;128(4):313–7. doi: 10.1039/b300873h. [DOI] [PubMed] [Google Scholar]
- 58.Splinter RH, BA . An introduction to biomedical optics. Taylor & Francis; New York: 2007. p. 602. [Google Scholar]
- 59.Zhang H, Wu Q, Berezin MY. Fluorescence anisotropy (polarization): from drug screening to precision medicine. Expert Opin Drug Discov. 2015;10(11):1145–61. doi: 10.1517/17460441.2015.1075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, Stout CD, Yeager MJ, Zhang Q. Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc Natl Acad Sci U S A. 2013;110(13):E1203–11. doi: 10.1073/pnas.1221442110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prive GG. Lipopeptide detergents for membrane protein studies. Curr Opin Struct Biol. 2009;19(4):379–85. doi: 10.1016/j.sbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Kleinschmidt JH, Popot JL. Folding and stability of integral membrane proteins in amphipols. Arch Biochem Biophys. 2014;564:327–43. doi: 10.1016/j.abb.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 63.le Maire M, Champeil P, Moller JV. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim Biophys Acta. 2000;1508(1–2):86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 64.Stafford RE, Fanni T, Dennis EA. Interfacial properties and critical micelle concentration of lysophospholipids. Biochemistry. 1989;28(12):5113–20. doi: 10.1021/bi00438a031. [DOI] [PubMed] [Google Scholar]
- 65.Neugebauer JM. Detergents: an overview. Methods Enzymol. 1990;182:239–53. doi: 10.1016/0076-6879(90)82020-3. [DOI] [PubMed] [Google Scholar]
- 66.Chattopadhyay A, Harikumar KG. Dependence of critical micelle concentration of a zwitterionic detergent on ionic strength: implications in receptor solubilization. FEBS letters. 1996;391(1–2):199–202. doi: 10.1016/0014-5793(96)00733-8. [DOI] [PubMed] [Google Scholar]
- 67.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Apel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: JMW, editor. Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 68.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 69.Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88(6):1895–1898. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.