

Abstract
Classical galactosemia is detected through newborn screening by measuring galactose-1-phosphate uridylyltransferase (GALT) in the USA primarily via the Beutler spot assay. We report on an 18-month-old patient with glucose-6-phosphate dehydrogenase (G6PD) deficiency that was originally diagnosed with classical galactosemia. The patient presented with elevated liver function enzymes and bilirubinemia and was immediately treated with soy-based formula. Confirmatory tests revealed deficiency of the GALT enzyme, however, full-sequencing of GALT was normal, suggestive of a different ideology. The Beutler spot assay uses three other enzymatic steps in addition to GALT. A deficiency in either of these enzymes can result in suspected decreased GALT activity when using the Beutler assay. Congenital Disorders of Glycosylation screening for phosphoglucomutase-1 deficiency was negative. Quantitative analysis of G6PD enzyme in red blood cells showed a severe deficiency and a deletion in G6PD. Soy-formula, the standard treatment for galactosemia, has been reported to trigger hemolysis in G6PD deficient patients. G6PD and phosphoglucomutase-1 deficiencies should be considered when confirmatory tests are negative for pathogenic variants in GALT and galactose-1-phosphate level is normal.
Introduction
Newborn screening (NBS) for classical galactosemia (MIM 230400) in the USA has identified over 2,500 infants with this potentially lethal metabolic disorder (Pyhtila et al. 2014a). Classical galactosemia is inherited by autosomal recessive pattern and affects the metabolism of galactose as a result of a deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.12). Symptoms can manifest within a few days after birth if dietary restriction of galactose is not initiated. These include feeding difficulties, hypotonia, jaundice, failure to thrive, hepatosplenomegaly, sepsis, cataract, intellectual disability, and eventually death (Bosch 2006).
Multiple fluorescent and radioactive enzyme assays have been developed to screen for classical galactosemia (Li et al. 2010). However, the semiquantitative fluorescent Beutler spot assay is commonly used for diagnosis and mass screening in the USA (Fig. 1) (Fujimoto et al. 2000). This test, in addition to GALT, relies on phosphoglucomutase-1 (PGM1), glucose-6-phosphate dehydrogenase (G6PD), and 6-phosphogluconate dehydrogenase (6PGD) in stepwise process to reduce NADP+ to NADPH. The fluorescence of NADPH is measured to determine if an infant is presumptive positive for classical galactosemia. Therefore, the Beutler spot assay is influenced by the enzyme activity of PGM1, G6PD, and 6PGD to accurately quantify GALT (Fujimoto et al. 2000). Considering there are several enzymes in this pathway that can cause disorders when deficiencies are present, newborn screening using the Beutler method may lead to misdiagnosis for a select number of newborns with 6PGD.
Fig. 1.
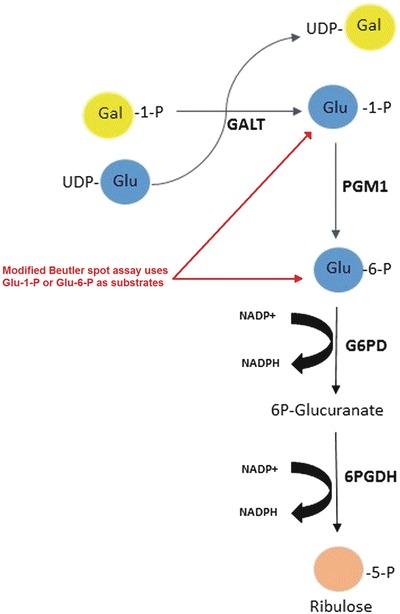
In the Beutler spot assay, the substrates Gal-1-P and uridine diphosphate glucose (UDP-glucose) are part of the test reagent and breakdown into glucose-1-phosphate in the presence of GALT. Glucose-1-phosphate is then further metabolized, stepwise, to ribulose-5-phosphate by PGM1, G6PD, and 6PGD. The last two reactions are in conjunction with the reduction of NADP+ to NADPH. The fluorescence of NADPH is measured to determine GALT deficiency. The Beutler spot assay can be modified to measure G6PD enzyme activity instead of GALT. This was achieved in our laboratory by adding glucose-6-phosphate as the main substrate. Glucose-6-phosphate dehydrogenase (G6PDH) catalyzes the conversion of glucose-6-phosphate to 6-phosphogluconate. In presence of NADP, glucose-6-phosphate is oxidized by G6PD to generate 6-phosphogluconate. This reaction also generates NADPH by the reduction of NADP. The formation of NADPH produces fluorescence, which magnitude is proportional to the G6PD enzyme activity. By using glucose-6-phosphate, the preceding GALT and PGM1 enzymes present/involved in the pathway are avoided and G6PD enzyme is directly measured. This technique has been described previously (Beutler 1994)
Case Report
A Caucasian male born term, in the state of Louisiana, was reported as presumptive positive for galactosemia on his newborn screen. A routine newborn screen was completed on the second day of life. He had a GALT enzyme activity less than 2.5 U/g Hb (reference: >3.5 U/g Hb), and was referred to a biochemical geneticist in New Orleans. There was no known family history of jaundice, liver failure, sepsis, or metabolic disorders, and his parents were nonconsanguineous. His birth history was unremarkable. He was breastfed from birth until 6 days of age when he was switched to a galactose free diet (soy-based formula) upon the report of his presumptive positive galactosemia newborn screen. His family was counseled on the biochemical background of galactosemia, the role of galactose-free diet, outcomes of galactosemia, and inheritance.
The initial confirmatory tests ordered included serum GALT enzyme, serum galactose-1-phosphate (Gal-1-P), galactosemia mutation panel for Q188R and N314D, and a liver profile. The GALT enzyme activity was 0.4 μmol/hr/ml blood (reference: 4.0–12.0 μmol/hr/ml blood) at less than 10% of residual activity. The initial Gal-1-P level was 0.3 mg/dl (reference: <1.0 mg/dl), but the child was on a lactose-free diet for 1 day prior to obtaining sample. The Gal-1-P level was undetectable after the child was on lactose-free diet for 5 days. The galactosemia mutation panel was negative for Q188R and N314D in the GALT gene. The liver profile revealed an elevated total bilirubin of 17.5 mg/dl (reference: <1.1 mg/dl) and a direct bilirubin of 0.4 mg/dl (reference: <0.4 mg/dl). Gal-1-P was measured again after 4 days on the soy-based formula and the level was undetectable.
The patient was scheduled for a follow-up visit 4 months later, but due to a miscommunication, was not seen until the following year. The patient’s hemoglobin level was measured during a pediatric well-visit at 9 months of age at 8.3 g/dl (reference: 11.5–13.5 g/dl). At approximately 1 year of age, he was evaluated by genetics for a follow-up visit. His mom reported he had maintained a galactose-restricted diet and was still on a soy-based formula. The family was further counseled on implications of galactosemia and inheritance, and further genetic testing was recommended. Full-sequencing of the GALT gene was completed and the results showed no sequence pathogenic variants. These results triggered further testing to rule out possible PGM1 or G6PD deficiency. The tests included the modified Beutler assay, using specific substrates to target PGM1 and G6PD enzymes (Fig. 1), Congenital Disorders of Glycosylation (CDG) screening using transferrin isoelectric focusing, and quantitative measurement of G6PD. The modified Beutler assay measured PGM1 enzyme activity at <1 μmol/hr/ml (reference: 8–14 μmol/hr/ml) and G6PD enzyme activity at 2.5 μmol/hr/ml (reference: 4–16 μmol/hr/ml). Transferrin isoelectric focusing detected no abnormalities, ruling out PGM1 deficiency. Quantification of G6PD enzyme via kinetic assay was 13 IU/trillion RBC (reference: 146–376 IU/trillion RBC) at <5% of residual enzyme activity. Decreased G6PD enzyme activity and clinical features coinciding with G6PD deficiency warranted genetic testing for G6PD sequencing and deletion/duplication. Results suggested the patient was hemizygous in the G6PD gene for an in-frame duplication, c.1301–1318dup18 (predicted pathogenic variant). Lastly, sequencing in the G6PD gene was completed in the mother showing hemizygosity for the same unknown variant found in her son. The mother was clinically unaffected. At 19 months of age the patient had a fully normal development, but persistent elevated total bilirubin of 2.3 mg/dl, mild jaundice of the sclera, and slightly elevated liver function tests (AST: 99 (10–50 U/L), ALT: 227 (5–45 U/L). We counseled the parents on avoidance of oxidative drugs and fava beans and recommended removing soy products and switching to regular milk. Four months after removing soy from the patient’s diet, a liver profile and complete blood count (CBC) revealed an improved total bilirubin of 1.7 mg/dl (reference: 0.1–2.0 mg/dl, direct bilirubin 0.5 mg/dl (reference: 0.1–0.4 mg/dl), and hemoglobin of 11.4 g/dl (11.5–13.6 g/dl).
Discussion
The Beutler spot assay is an inexpensive, efficient method for detecting classical galactosemia in mass newborn screening. Although there are other screening methods for galactosemia, predominantly measurement of total galactose (galactose and galactose-1-phosphate) in blood spots, there is a higher frequency of false-positives (Ohlsson et al. 2011). Several states use both measurements to screen for classical galactosemia. A study by Pyhtila et al. looked at the testing strategies of state labs in the USA in 2011–2012 (Pyhtila et al. 2014b). Of the 19 states that responded, 40% exclusively used GALT, 40% used GALT plus total galactose, and 20% only used total galactose if the GALT was low. In the state of Louisiana, newborn screening for classical galactosemia is conducted using the Perkin Elmer Neonatal GALT kit based on the Beutler spot assay. Presumptive positive newborn screens are based solely on GALT enzyme activity. The GALT cut-off is set at <3.5 U/g Hb. The current reporting is <2.5 U/g Hb is the lower limit of detection, between 2.5 and 3.0 U/g Hb is reported as presumptive positive, and 3.1–≤3.5 U/g Hb is reported as borderline and >3.5 IU is considered normal enzyme activity. Values were determined using the Perkin Elmer package insert and Louisiana in-state data. The Beutler spot assay measures fluorescent NADPH as the final product in a stepwise reaction (Fig. 1). Although GALT is the rate-limiting enzyme, the Beutler spot assay requires normal enzyme activity of PGM1, G6PD, and 6PGD to avoid false-positive newborn screens for galactosemia (Fujimoto et al. 2000). Deficiency in any of these enzymes can result in a misdiagnosis of galactosemia when using the Beutler spot assay. PGM1 deficiency is a type of congenital disorders of glycosylation (CDG) caused by inborn errors of glycan metabolism (Scott et al. 2014). Only a few cases of 6PGD deficiency have been described (Caprari et al. 2001). Reported signs include hemolytic anemia and scleral jaundice.
The guidelines set by the American College of Medical Genetics recommend molecular testing for common pathogenic variants in the GALT gene in congruence with a quantitative GALT assay (American College of Medical Genetics 2001). Additional tests should be conducted if initial confirmatory testing reveals decreased GALT activity using the Beutler spot assay in combination with no detection of common GALT gene variants and a normal Gal-1-P. Additional testing should include full-sequencing of the GALT gene, transferrin glycoform analysis (CDG screening), and measure of G6PD. Interestingly, the Beutler spot assay can be modified to bypass the GALT and PGM1 enzymes, and therefore, measure G6PD enzyme activity (Fig. 1).
The USA currently does not recommend newborn screening for G6PD deficiency, an X-linked condition. Individuals with severe G6PD deficiency may be identified through newborn screening via the Beutler spot assay resulting in a false positive newborn screen for galactosemia (Frazier and Summer 1974). Deficiency in the G6PD enzyme occurs in at least 400 million people worldwide, making it the most common human enzyme deficiency. Over 400 variants in the G6PD gene have been identified. Variants associated with G6PD deficiency are divided into four classes based on the level of residual activity and the severity of phenotype ranging from severe (Class I) to mild (Class IV) (Frank and Maj 2005). The patient in discussion, although having a variant of unknown significance, had a residual G6PD activity of less than 10%. Despite G6PD being a very common disease, a severe deficiency in G6PD enzyme is required to trigger an abnormal NBS for galactosemia when using the Beutler spot assay. This underlines the possibility of identifying individuals with severe G6PD deficiency through newborn screening of classic galactosemia using the Beutler spot assay.
Treatment of galactosemia includes strict dietary avoidance of galactose. During infancy, this involves substituting breast milk or milk-based formula with a soy-based formula or other galactose-free formula. Treatment of G6DP deficiency includes avoidance of exogenous agents such as oxidative drugs and fava beans (Frank and Maj 2005). Fava beans are recognized to cause hemolysis in G6PD-deficient patients by an unknown oxidative compound, possibly vicine, convicine, isouramil, or divicine (Frank and Maj 2005; Chevion et al. 1982). In addition, other food items such as soy and legumes have been reported by the G6PD Deficiency Association to cause hemolysis based on increased hemoglobin levels. Although there are no scientific studies on the effect of soy and legumes on hemolysis in severe G6PD deficiency, it may be clinically relevant to remove these items from the individual’s diet. If a severely G6PD deficient patient is reported as presumptive positive for galactosemia on their newborn screen, and, placed on soy-based formula, it may do more harm than good.
In conclusion, severe enzyme deficiency of G6PD can result in a presumptive positive newborn screen for classical galactosemia when using the Beutler spot assay. G6PD should be included in the differential diagnosis when confirmatory testing is negative for galactosemia variants and Gal-1-P level is normal. Under these circumstances, second tier tests such as a comprehensive metabolic panel, quantitative G6PD assay, and transferrin glycoform analysis should be conducted to rule out possible G6PD or PGM1 deficiencies.
Abbreviations
- 6PGD
6-Phosphogluconate dehydrogenase
- ALT
Alanine transaminase
- AST
Aspartate aminotransferase
- CBC
Complete blood count
- CDG
Congenital disorders of glycosylation
- dl
Deciliter
- G6PD
Glucose-6-phosphate dehydrogenase
- Gal-1-P
Galacose-1-phosphate
- GALT
Galactose-1-phosphate uridylyltransferase
- Hb
Hemoglobin
- hr
Hour
- IU
International unit
- μmol
Micromole
- mg
Milligram
- ml
Milliliter
- NADP+
Nicotinamide adenine dinucleotide phosphate
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NBS
Newborn screening
- PGM1
Phosphoglucomutase-1
- U/g
Units per gram
- UDP-glucose
Uridine diphosphate glucose
Synopsis
Decreased GALT enzyme by Beutler spot assay can mask PGM1 or G6PD deficiencies and warrants further metabolic analysis in the absence of pathogenic variants in GALT. Soy-based formula intake can trigger hemolysis in G6PD deficiency.
Compliance with Ethics Guidelines
Conflict of Interest
Grace Stuhrman, Stefanie J. Perez Juanazo, Kea Crivelly, Jennifer Smith, Dr. Hans Andersson and Dr. Eva Morava declare that they have no conflicts of interest.
Informed Consent/Animal Rights
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). This article does not contain any studies with animal subjects performed by the any of the authors.
Details of the Contributions of Individual Authors
Grace Stuhrman drafted the initial manuscript and revised the manuscript.
Stefanie J. Perez Juanazo contributed to drafting the initial manuscript and conducted biochemical assays.
Jennifer Smith conducted biochemical assays.
Kea Crivelly, Eva Morava-Kozicz, and Hans Andersson critically reviewed and revised the manuscript.
All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Contributor Information
Grace Stuhrman, Email: gstuhrma@tulane.edu.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- American College of Medical Genetics (2001) ACMG ACT sheets and confirmatory algorithms. Bethesda. http://www.ncbi.nlm.nih.gov/books/NBK55832/. Accessed 14 Nov 2016
- Beutler E. G6PD deficiency. Blood. 1994;84(11):3613–3636. [PubMed] [Google Scholar]
- Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29(4):516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- Caprari P, Caforio MP, Cianciulli P, et al. 6-Phosphogluconate dehydrogenase deficiency in an Italian family. Ann Hematol. 2001;80(1):41–44. doi: 10.1007/s002770000233. [DOI] [PubMed] [Google Scholar]
- Chevion M, Navok T, Glaser G, Mager J. The chemistry of favism-inducing compounds. The properties of isouramil and divicine and their reaction with glutathione. Eur J Biochem. 1982;127(2):405–409. doi: 10.1111/j.1432-1033.1982.tb06886.x. [DOI] [PubMed] [Google Scholar]
- Frank JF, Maj MC. Diagnosis and management of G6PD deficiency. Am Fam Physician. 2005;72(7):1277–1282. [PubMed] [Google Scholar]
- Frazier PDM, Summer GK. Automated fluorometric micromethod for detection of transferase-deficiency galactosemia. J Lab Clin Med. 1974;83(2):334–338. [PubMed] [Google Scholar]
- Fujimoto A, Okano Y, Miyagi T, Isshiki G, Oura T. Quantitative Beutler test for newborn mass screening of galactosemia using a fluorometric microplate reader. Clin Chem. 2000;46(6):806–810. [PubMed] [Google Scholar]
- Li Y, Ptolemy AS, Harmonay L, Kellogg M, Berry GT. Quantification of galactose-1-phosphate uridyltransferase enzyme activity by liquid chromatography–tandem mass spectrometry. Clin Chem. 2010;56(5):772–780. doi: 10.1373/clinchem.2009.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson A, Guthenberg C, von Döbeln U. Galactosemia screening with low false-positive recall rate: the Swedish experience. JIMD Rep. 2011;2:113–117. doi: 10.1007/8904_2011_59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyhtila BM, Shaw KA, Neumann SE, Fridovich-Keil JL. A brief overview of galactosemia newborn screening in the United States. J Inherit Metab Dis. 2014;37(4):649–650. doi: 10.1007/s10545-014-9694-7. [DOI] [PubMed] [Google Scholar]
- Pyhtila BM, Shaw KA, Neumann SE, Fridovich-Keil JL. Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep. 2014;15:79–93. doi: 10.1007/8904_2014_302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K, Gadomski T, Kozicz T, Morava E. Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis. 2014;37(4):609–617. doi: 10.1007/s10545-014-9720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]