

Abstract
“Cross-correction,” the transfer of soluble lysosomal enzymes between neighboring cells, forms the foundation for therapeutics of lysosomal storage disorders (LSDs). However, “cross-correction” poses a significant barrier to studying the role of specific cell types in LSD pathogenesis. By expressing the native enzyme in only one cell type, neighboring cell types are invariably corrected. In this study, we present a strategy to limit “cross-correction” of palmitoyl-protein thioesterase-1(PPT1), a lysosomal hydrolase deficient in Infantile Neuronal Ceroid Lipofuscinosis (INCL, Infantile Batten disease) to the lysosomal membrane via the C-terminus of lysosomal associated membrane protein-1 (LAMP1). Tethering PPT1 to the lysosomal membrane prevented “cross-correction” while allowing PPT1 to retain its enzymatic function and localization in vitro. A transgenic line harboring the lysosomal membrane-tethered PPT1 was then generated. We show that expression of lysosome-restricted PPT1 in vivo largely rescues the INCL biochemical, histological, and functional phenotype. These data suggest that lysosomal tethering of PPT1 via the C-terminus of LAMP1 is a viable strategy and that this general approach can be used to study the role of specific cell types in INCL pathogenesis, as well as other LSDs. Ultimately, understanding the role of specific cell types in the disease progression of LSDs will help guide the development of more targeted therapeutics.
One Sentence Synopsis: Tethering PPT1 to the lysosomal membrane is a viable strategy to prevent “cross-correction” and will allow for the study of specific cellular contributions in INCL pathogenesis.
Keywords: Infantile Batten disease, Lysosomal storage disorders, Neuronal ceroid lipofuscinosis, Palmitoyl-protein thioesterase-1, Soluble lysosomal enzymes
Introduction
Lysosomal storage diseases are monogenic diseases that affect many cell types and present with a complex clinical phenotype. Understanding the role of individual cell types in the disease process will allow for the rational development of effective therapies. However, “cross-correction” interferes with the ability to address basic biological questions in a complex setting.
Inter-cellular trafficking of lysosomal enzymes, “cross-correction,” forms the foundation of therapeutic approaches to treat lysosomal storage disorders (Neufeld and Fratantoni 1970; Sands and Davidson 2006). Soluble lysosomal enzymes are glycosylated and phosphorylated, then transported to the lysosome via the mannose-6-phophate receptor (M6P/IGFIIr) (Neufeld et al. 1977; Dahms et al. 1989). A small proportion of lysosomal enzymes escapes the canonical mannose-6-phosphate pathway and is secreted into the intercellular space. The secreted enzymes can be endocytosed by adjacent cells via the plasma membrane-localized M6P/IGFIIr and trafficked to the lysosome (Neufeld 1980; Dahms et al. 1989).
Deficiency of the lysosomal hydrolase palmitoyl-protein thiosesterase-1 (PPT1) results in infantile neuronal ceroid lipofuscinosis (INCL) (Vesa et al. 1995; Gupta et al. 2001; Verkruyse and Hofmann 1996). Human INCL and PPT1-deficient mice have progressive deficits in vision, motor function, and cognition (Macauley et al., 2009; Griffey et al., 2005; Santavuori et al. 1973; Dearborn et al. 2015). INCL histology reveals autofluorescent storage material (AFSM), neuronal death, glial activation, and neuroinflammation, implicating a variety of cell lineages in the pathogenesis (Haltia et al. 1973; Bible et al. 2004; Kielar et al. 2007; Macauley et al. 2009, 2011). Proteomic and metabolomic studies in INCL mice also demonstrated that PPT1 deficiency affects a variety of cell types (Woloszynek et al. 2007; Khaibullina et al. 2012; Tikka et al. 2016). To understand the relative contribution of each cell type to the disease process, it is necessary to prevent “cross-correction.”
In this study, we tethered PPT1 to the C-terminus of lysosome-associated membrane protein-1 (LAMP1). The C-terminus of LAMP1 is necessary and sufficient for lysosomal targeting (Guarnieri et al. 1993; Rohrer et al. 1996). To target specific lineages, we employed the Cre-lox system. We report that membrane-tethered PPT1 retains enzyme activity in vitro and prevents the biochemical, histological, and behavioral phenotype of murine INCL. Unfortunately, cell specificity was lost due to rearrangement of the transgene. Nonetheless, this study demonstrates a proof-of-principle that this general methodology is a viable approach to address questions regarding the role of specific cell types in the progression of INCL.
Materials and Methods
PPT1-LAMP1 Transgenic Mouse and Husbandry
Transgenic animals were generated by microinjection of the transgene directly into C57Bl/6 embryos (Transgenic Knockout Microinjection Core, Washington University). Transgenic animals were crossed onto the PPT1−/− background (also on the congenic C57Bl/6 background) until PPT1−/− homozygosity. Congenic C57BL/6 wild-type and PPT1−/− mice were used as controls (Griffey et al. 2004). All procedures were carried out in accordance with an approved IACUC protocol from Washington University School of Medicine.
Chimeric PPT1-LAMP1 and Lentiviral Expression
The chimeric PPT1-LAMP1 expression plasmid includes the promoter and first intron of the chicken β-actin gene, the human PPT1 cDNA, a six-glycine linker, the 120 bp sequence encoding the transmembrane domain and lysosomal localization sequence of LAMP1 followed by a rabbit β-globin polyadenylation signal (Fig. 1d). The transgene is identical to the expression plasmid except that it contains a loxP-STOP-loxP sequence (L-S-L) from pBS302 (Addgene, Cambridge, MA).
Fig. 1.
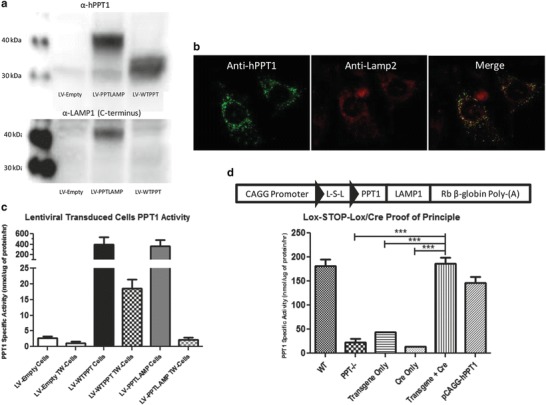
In vitro characterization of PPT1-LAMP1 and generation of a Cre responsive transgene. (a) Expression of PPT1-LAMP1 in vitro. PPT1−/− cells were transduced with LV-Empty, LV-PPTLAMP, or LV-WTPPT. The top panel is probed for anti-hPPT1. hPPT1 has a predicted molecular weight of ~32 and 36 kDa, and hPPT1-LAMP1 is predicted at ~40 kDa. The bottom panel was probed for the C-terminus of LAMP-1. The left lanes show the markers for 30 and 40 kDa. A ~32 and ~36 kDa PPT1 signal was detected in the LV-WTPPT lane. PPT1 signal was detected at ~40 kDa in the LV-PPTLAMP lane. LAMP-1 signal was detected in the LV-PPTLAMP lane. No PPT1 or LAMP1 signal was detected in LV-Empty lane. (b) Immunofluorescence of PPT1 (green) and LAMP-2 (red). (c) Transwell experiment for “cross-correction.” Cells were transduced with LV-Empty, LV-PPTLAMP, or LV-WTPPT. There was a significant increase in enzyme activity in the LV-WTPPT cells (394.7 nmol/μg/h) compared to LV-Empty (2.7 nmol/μg/h). LV-WTPPT cells and LV-PPTLAMP cells (367.9 nmol/μg/h) had similar levels of activity. There was a significant (p < 0.05) increase in PPT1 activity in the transwell inserts (TW-cells) in the LV-WTPPT TW cells (18.5 nmol/μg/h) compared to LV-Empty TW cells (1.1 nmol/μg/h) and LV-PPTLAMP TW cells (2.2 nmol/μg/h). (d) In vitro testing of cell-specific transgene. Schematic of PPT1LAMP1 transgene is above the graph. The transgene consists of the CAGG Promoter (Chicken β-actin first exon and intron), human palmitoyl-protein thioesterase-1 (PPT1) cDNA, lysosomal-associated membrane protein (LAMP-1) cDNA, and the rabbit β-globin polyadenylation terminator (Rb β-globin Poly-(A)). A loxp-STOP-loxp (L-S-L) cassette was added to the transgene (loxp sites: black triangles). There was not a significant increase in PPT1 activity in the PPT1−/− cells transfected with the transgene alone (43.9 nmol/μg/h) or the Cre-containing plasmid alone (14.0 nmol/μg/h) compared to PPT1−/− cells (22.4 nmol/μg/h). There was an increase in PPT1 activity when the transgene and the Cre-containing plasmid were transfected together (186.4 nmol/μg/h) to WT levels (181.5 nmol/μg/h). pCAGG-hPPT1 plasmid was used as a transfection control
Third-generation SIN lentiviral vectors were created for in vitro experiments (Dull et al. 1998; Zufferey et al. 1998). Briefly, the lentivirus contained the phosphoglycerate kinase (PGK) promoter, the target gene, an SV40 polyadenylation site, and a puromycin resistant cassette. The target gene contained wild type hPPT1 (LV-WT) or hPPT1-LAMP1 (LV-PPTLAMP). The empty lentiviral vector did not contain a target gene (LV-Empty).
Cross-Correction Experiments
PPT1−/− murine fibroblasts were plated, transduced, and then selected with puromycin. Untransduced PPT1−/− murine cells were plated on transwell inserts (Corning, Corning, NY) in a separate dish prior to transfer into the experimental plate. Three days post co-incubation, PPT1 enzyme activity was measured. “Cross-correction” experiments using primary dermal fibroblasts from F42 mice were performed similarly.
Genomic DNA Isolation and Sequencing
Genomic DNA from brain and liver was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). The recombined transgene was sequenced using primers flanking the loxP-STOP-loxP sequence (PNACL, Washington University). Copy number was determined by qPCR on genomic DNA using primers flanking the human PPT1 and LAMP1 cDNA junction and calculated as previously described (Joshi et al. 2008).
SDS-PAGE/Western Blot
Western blots were performed on homogenates from transduced cells or primary fibroblast as previously described (Benitez et al. 2015). Membranes were probed for human PPT1 (Abcam 89022; 1:1,000) or the cytoplasmic domain of LAMP1 (Sigma L1418; 1:1,000).
PPT1 Enzyme Activity and Secondary Enzyme Elevations
PPT1 assays were performed using the 4-MU-palmitate fluorometric assay and normalized to total protein (Griffey et al. 2004). Whole blood was collected slowly by cardiac puncture through a 23G needle in an attempt to avoid cell lysis. The blood was allowed to clot then spun at 3,000 rpm. Serum was collected and flash frozen for subsequent PPT1 assays. β-glucuronidase assays were performed using 4-MU-β-D-glucuronide fluorometric assay and normalized to total protein as previously described (Roberts et al. 2012). Significance was determined using a two-way ANOVA followed by a Bonferroni post-hoc analysis.
Autofluorescent Storage Material
Autofluorescent storage material (AFSM) was imaged and quantified as previously described (Griffey et al. 2004). Twelve-micrometer sections were imaged using confocal microscopy.
Lifespan, Rotarod, and Electroretinography
Lifespan was determined by death or euthanasia out to 1 year (n = 10–11 mice/group). A Kaplan-Meier lifespan curve was used to measure survival and significant differences were determined using a log-rank analysis (p < 0.05).
Mice (n = 10/group) were tested on a constant-speed rotarod (3 rpm) at 7 months as previously described (Dearborn et al. 2015). Statistical significance was determined using one-way ANOVA followed by a Bonferroni post hoc test.
Electroretinography (ERG) was performed under dark or light conditioning as previously described (Vision Core, Washington University) (Griffey et al. 2005). b-wave amplitudes (microvolts) were recorded (UTAS-E-3000 LKC system, LKC Technologies, Gaithersburg, MD).
Results
PPT1-LAMP1 Function In Vitro
PPT1−/− fibroblasts were transduced in vitro with LV-Empty, LV-PPTLAMP, or LV-WT. Western blot analysis showed an increased molecular weight of the hPPT1-LAMP1 (~40 kDa) compared to the wild type human PPT1(~32–36 kDa). This was consistent with the addition of the LAMP1 transmembrane domain and six-glycine linker (~5 kDa). Endogenous LAMP1 (~120 kDa) and a signal at ~40 kDa were detected when the same membrane was probed with an anti-LAMP1 antibody (Fig. 1a). Immunofluorescent staining for LAMP2 and PPT1 in PPT1−/− fibroblasts transduced with LV-PPTLAMP demonstrates that PPT1 enzyme is localized to the lysosome (Fig. 1b).
Transwell experiments confirmed that PPT1-LAMP1 retained enzyme activity and was not secreted (Fig. 1c). Following LV-WT, PPT1 activity (~400 nmol/μg/h) was detected in the transduced cells and in the transwell cells (~20 nmol/μg/h). Nearly identical PPT1 activity to LV-WT was detected following LV-PPTLAMP transduction but no activity was detected in the transwell cells.
To achieve cell specificity for the expression of the transgene, a lox-STOP-lox sequence was included within the transgene (Fig. 1d). In vitro transfection of PPT1−/− cells with a Cre-recombinase plasmid or the transgene-containing plasmid alone resulted in no increase in PPT1 activity. Upon co-transfection of the plasmids containing Cre recombinase and the lox-STOP-lox PPTLAMP transgene, a significant increase in PPT1 activity to near WT-PPT1 activity levels was observed (Fig. 1d).
Generation of a PPT1-LAMP1 Transgenic Mouse
To study the role of specific cell types in INCL in vivo, transgenic animals were generated harboring the PPT1-LAMP1 transgene. Eight founders were identified and five demonstrated germ-line transmission. Only two founders (F38 and F42) were able to generate colonies. F38 did not express PPT1-LAMP1 in the presence or absence of Cre-recombinase. Surprisingly, a PPT1 assay of F42 detected supraphysiological levels of PPT1 activity in the absence of Cre recombinase.
Founder #42 Analyses
Genomic transgene characterization of F42 mice identified a ~1.5 Kb band consistent with the intact transgene and a ~260 bp band consistent with recombination of the loxP sites (Fig. 2a). Sequencing confirmed Cre-independent recombination. Approximately 11 transgene copies per genome were calculated using quantitative PCR.
Fig. 2.
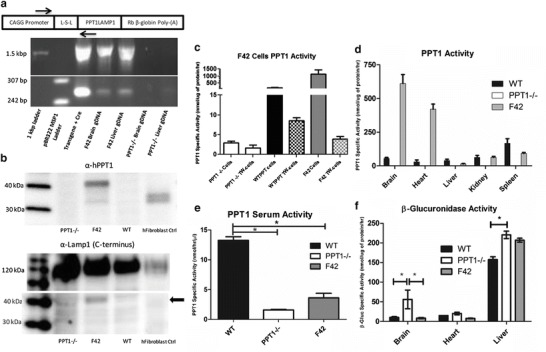
Biochemical and molecular characterization of F42 mice. (a) Schematic of the transgene with the position of PCR and sequencing primers. PCR gel identifying spontaneous recombination in Founder #42. Intact loxp-STOP-loxp cassette is ~1.5 kbp and the rearranged cassette is ~260 bp. Lane 1: 1 kbp ladder, Lane 2: pBR322-MSPI digest ladder, Lane 3: in vitro Cre recombinase and transgene plasmid, Lane 4: genomic DNA isolated from F42 brain, Lane 5: genomic DNA isolated from F42 Liver, Lane 6: genomic DNA isolated from PPT1−/− brain, Lane 7: genomic DNA isolated from PPT1−/− liver, Lane 8: negative control. (b) Western blot for PPT1-LAMP1 expression. Expression of PPT1-LAMP1 was confirmed in primary dermal fibroblasts from F42. The top panel was probed for anti-human PPT1. A strong signal was detected at ~40 kDa in the F42 lane, and at the predicted ~32 (unglycosylated) and ~36 kDA (glycosylated) in the human control fibroblasts. The bottom panel was probed for the C-terminus of LAMP-1. There was a signal at ~120 kDa (endogenous LAMP1) in all lanes and ~40 kDa (PPT1-LAMP1) signal in the F42 lane (black arrow). (c) Transwell experiment for “cross-correction.” Primary dermal fibroblast cells were used. There was a significant increase in enzyme activity in the WT-PPT1 (184.0 nmol/μg/h) compared to PPT1−/− cells (2.9 nmol/μg/h). F42 cells (1135.1 nmol/μg/h) have ~6-fold greater levels of PPT1 activity compared to WT cells. There was an increase in PPT1 activity in the transwell inserts in the WT-PPT1 transwell (8.5 nmol/μg/h) compared to PPT1−/− (1.5 nmol/μg/h) and F42 transwells (3.9 nmol/μg/h). (d) PPT1 enzyme activity tissue survey. Supraphysiological levels of PPT1 activity were detected in the brain and heart. Near normal levels were detected in the kidney, and ~50% activity in the spleen. The liver had ~5% normal levels of PPT1 activity. (e) PPT1 enzyme activity in the serum. There is a significant (p < 0.05) decrease in PPT1 activity in PPT1−/− serum compared to WT serum (1.6 vs 13.3 nmol/μg/h). PPT1 activity was significantly decreased in serum from F42 mice (3.6 nmol/μg/h, p < 0.05) compared to WT mice. There is no significant difference in PPT1 activity between F42 and PPT1−/− serum. (f) β-glucuronidase activity in the brain, heart, and liver from F42 and controls. There was a significant (p < 0.05) increase in β-glucuronidase activity in the brain (56.2 vs 10.2 nmol/μg/h) and liver (220.4 vs 157.4 nmol/μg/h) of PPT1−/− animals compared to WT (p < 0.05). β-glucuronidase activity was normalized to WT levels in the F42 brain (8.5 nmol/μg/h), however, the β-glucuronidase activity was not reduced in F42 liver (206.7 nmol/μg/h). The heart showed a small increase in β-glucuronidase activity in PPT1−/− animals compared to WT animals (20.2 vs 15.0 nmol/μg/h) but this increase was not statistically significant. There appeared to be decreased β-glucuronidase activity in the F42 heart (7.8 nmol/μg/h)
Western blot analysis was performed on dermal fibroblasts from F42. A PPT1-specific signal was detected in human fibroblasts corresponding to differentially glycosylated PPT1 but was not detected in PPT1−/− or WT murine fibroblasts (Fig. 2b). A protein of ~40 kDa was detected in homogenates from F42 fibroblasts probed with an anti-hPPT1 antibody. Similarly, a protein of ~40 kDa was detected with an anti-LAMP1 antibody along with endogenous LAMP1 (~120 kDa). A transwell experiment showed that PPT1-LAMP1 from F42 fibroblasts does not “cross-correct” (Fig. 2c). Transwell insert cells exposed to WT cell media had increased PPT1 activity of ~10% of WT levels. In contrast, little to no PPT1 activity was detected in the transwell cells exposed to F42 cell media even though F42 fibroblasts had ~6-fold greater PPT1 activity compared to WT cells.
PPT1-LAMP1 Expression In Vivo Is Ubiquitous and Prevents AFSM Accumulation
Supraphysiological levels of PPT1 activity were detected in the brain and heart, and near normal levels in the kidney and spleen of F42 mice compared to wild type (Fig. 2d). PPT1 activity was not significantly increased in the serum of F42 mice compared to PPT1−/− mice (Fig. 2e). Surprisingly, very low PPT1 activity was detected in the liver of F42 mice. There is an increase in secondary lysosomal enzyme activity in the murine model of INCL (Fig. 2f) (Griffey et al. 2004). The β-glucuronidase activity levels in the brain and heart of F42 animals were restored to WT levels. However, β-glucuronidase activity in the F42 liver was not significantly reduced.
PPT1-LAMP1 Prevents the Pathological and Clinical Defects Associated with Murine INCL
There was widespread AFSM throughout the PPT1−/− brain and liver and none detected in WT animals (Fig. 3a). Accumulation of AFSM was not detected in the cortex, cerebellum or any other brain region in F42 mice. There was abundant AFSM throughout the liver of F42 mice (Fig. 3a).
Fig. 3.
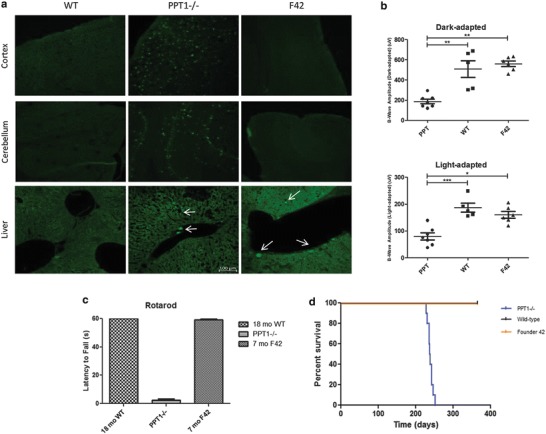
Histological and clinical parameters of F42. (a) Representative images show AFSM accumulation in PPT1−/− brains (cortex and cerebellum). In contrast, AFSM was not detected in the cortex or cerebellum of WT or F42 animals. Representative images show similar levels and distribution of AFSM in the livers of PPT1−/− and F42 animals. No AFSM was detected in WT livers. All images were taken at the same magnification of 20x (100 μm). (b) Electroretinography of F42 and control eyes at 7 months. There was a significant decrease in b-wave amplitudes in the light-adapted (WT p < 0.0001, F42 p < 0.01) and dark-adapted (WT p < 0.001, F42 p < 0.001) PPT1−/− mice compared to WT and F42 mice. There was no significant difference between F42 and WT b-wave amplitudes in light-adapted or dark-adapted conditions. (c) Rotarod analysis of F42 mice showed no deficit in motor function at 7 months of age. PPT1−/− animals could not stay on the rotarod at 7 months of age. However, 7-month-old F42 mice were able to stay on the rotarod for 60 s. Similarly, the WT mice were also able to stay on for the duration of the study. (d) The lifespan of F42 mice (orange) was not reduced compared to WT animals (black) out to 1 year. In contrast, PPT1−/− animals had a median lifespan of 238 days (blue)
PPT1−/− mice showed a significant decline in ERG amplitudes and motor function by 7 months of age, and are terminal by ~8.5 months. F42 mice showed no significant decline in either dark-adapted or light-adapted ERG amplitudes (Fig. 3b). At 7 months, F42 mice remained on the rotarod for 60 s and were indistinguishable from wild type mice (Fig. 3c). Lastly, the F42 mice lived to at least 1 year without any obvious clinical signs of disease (Fig. 3d).
Discussion
The first goal of this study was to generate PPT1 that was sequestered within the lysosome. We successfully generated a lysosomal membrane-tethered form of human PPT1 that was properly trafficked, enzymatically active, and retained within the lysosomal compartment in vitro. This demonstrates proof-of-principle that PPT1-LAMP1 can be used as a tool to limit enzyme expression to select cells. The second goal was to utilize the Cre-lox system and generate a mouse model where PPT1-LAMP1 was limited to specific cell types, thereby creating novel models of cell-specific enzyme deficiencies. We succeeded in generating a mouse model of PPT1-LAMP1 using a loxP-STOP-loxP cassette. However, a spontaneous rearrangement resulted in near-ubiquitous expression of PPT1-LAMP1 throughout the mouse. Therefore, we characterized the mouse model with widespread expression of the transgene to determine whether PPT1-LAMP1 could correct INCL pathology. Since native PPT1 is a soluble protein, it was critical to determine if membrane-tethered PPT1 could access its substrates.
We demonstrated that tethering PPT1 to the lysosomal membrane eliminates “cross-correction” of neighboring cells in vitro and in vivo. Near-ubiquitous expression of PPT1-LAMP1 in vivo was sufficient to prevent the hallmark INCL pathology of AFSM accumulation in most tissues indicating its retention in the lysosome. AFSM did accumulate in the liver, which is consistent with the low level of PPT1-LAMP1 expression, possibly resulting from transgene integration into a closed genomic domain within hepatic cells. These data, combined with the very low level of PPT1 activity in the serum and high level of expression in other tissues, strongly support our conclusion that PPT1-LAMP1 is not secreted nor capable of “cross-correction.” The apparent small increase in serum PPT1 activity in the F42 mice was not significantly greater than that measured in PPT1−/− mice but could be due to cell lysis during blood collection. Finally, we found that restricting PPT1 expression to the lysosome is sufficient to prevent several functional deficits observed in INCL mice, including loss of vision and motor deficits.
A previous study limited the secretion of sphingomyelinase (SMase), another soluble lysosomal hydrolase, by tethering SMase to the lysosomal membrane using the C-terminus of LAMP-1 (Marathe et al. 2000). The original goal of their study was to eliminate the secreted form ubiquitously. The transgenic/knockout hybrid model of Neimann-Pick A/B had modest lysosomal SMase activity in the brain (~20% WT) with little or no neurological disease. However, there was lower activity in systemic tissues (~1–14% WT) with widespread visceral disease. The data in the INCL mouse are similar with rescue of CNS pathology while liver pathology persisted. Although these represent two different disease models, taken together, the data suggest that secreted enzymes might be of greater importance to visceral organs compared to the CNS. Alternatively, these data suggest that secreted PPT1 serves no vital function since there is essentially no disease pathology in any tissue examined with the exception of the liver which has very low activity.
Our original model utilized a loxP-STOP-loxP system that was to be combined with cell-specific Cre-driver lines. However, a spontaneous rearrangement of the loxP sites led to loss of Cre-responsiveness. We detected ~11 transgene copies/genome in F42 mice which is consistent with previous studies demonstrating multiple integrants using a traditional transgenic approach (Haruyama et al. 2009). It is possible that the direct repeat sequences in the concatamers made the transgene prone to recombination and led to deletion of the loxP-STOP-loxP sequence (Bill and Nickoloff 2001; Hendricks et al. 2003; Wurtele et al. 2005).
While the current transgenic model may not allow for the study of cell autonomous expression of PPT1-LAMP1 in vivo, the model provides evidence that this basic approach is sound. In addition, expressing PPT1-LAMP1 in cultured cells will allow for in vitro experiments without the confounding issues of “cross-correction.”
Acknowledgments
We thank J. Michael White (Transgenic Knockout Micro-Injection Core, WUSTL) for his help with generating the transgenic founders. We thank Dr. Anne Hennig (Vision Research Core, WUSTL) for her help with the electroretinography. In addition, we thank Dr. Bruno Benitez for his advice and technical assistance.
Contributions
C.S. and M.S.S. wrote the paper. C.S., S.L.M., and M.S.S. designed research. C.S., S.L.M. and J.T.D. performed research. C.S. and J.T.D. analyzed the data. M.S.S. will serve as guarantor.
Compliance with Ethical Standards
Conflict of Interests: No conflicts of interest to report.
Animal Rights: IACUC approval #20130254.
Funding
This work was funded by a grant from the NIH NINDS 043205.
Contributor Information
Mark S. Sands, Email: mssands@wustl.edu
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Benitez BA, Cairns NJ, Schmidt RE, et al. Clinically early-stage CSPalpha mutation carrier exhibits remarkable terminal stage neuronal pathology with minimal evidence of synaptic loss. Acta Neuropathol Commun. 2015;3:73. doi: 10.1186/s40478-015-0256-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:346–359. doi: 10.1016/j.nbd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Bill CA, Nickoloff JA. Spontaneous and ultraviolet light-induced direct repeat recombination in mammalian cells frequently results in repeat deletion. Mutat Res. 2001;487:41–50. doi: 10.1016/S0921-8777(01)00101-X. [DOI] [PubMed] [Google Scholar]
- Dahms NM, Lobel P, Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem. 1989;264:12115–12118. [PubMed] [Google Scholar]
- Dearborn JT, Harmon SK, Fowler SC, et al. Comprehensive functional characterization of murine infantile Batten disease including Parkinson-like behavior and dopaminergic markers. Sci Rep. 2015;5:12752. doi: 10.1038/srep12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffey M, Bible E, Vogler C, et al. Adeno-associated virus 2-mediated gene therapy decreases autofluorescent storage material and increases brain mass in a murine model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:360–369. doi: 10.1016/j.nbd.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Griffey M, Macauley SL, Ogilvie JM, Sands MS. AAV2-mediated ocular gene therapy for infantile neuronal ceroid lipofuscinosis. Mol Ther. 2005;12:413–421. doi: 10.1016/j.ymthe.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Guarnieri FG, Arterburn LM, Penno MB, Cha Y, August JT. The motif Tyr-X-X-hydrophobic residue mediates lysosomal membrane targeting of lysosome-associated membrane protein 1. J Biol Chem. 1993;268:1941–1946. [PubMed] [Google Scholar]
- Gupta P, Soyombo AA, Atashband A, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci U S A. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia M, Rapola J, Santavuori P, Keranen A. Infantile type of so-called neuronal ceroid-lipofuscinosis. 2. Morphological and biochemical studies. J Neurol Sci. 1973;18:269–285. doi: 10.1016/0022-510X(73)90076-2. [DOI] [PubMed] [Google Scholar]
- Haruyama N, Cho A, Kulkarni AB (2009) Overview: engineering transgenic constructs and mice. Curr Protoc Cell Biol. Chapter 19: Unit 19.10. [DOI] [PMC free article] [PubMed]
- Hendricks CA, Almeida KH, Stitt MS, et al. Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:6325–6330. doi: 10.1073/pnas.1232231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M, Keith Pittman H, Haisch C, Verbanac K. Real-time PCR to determine transgene copy number and to quantitate the biolocalization of adoptively transferred cells from EGFP-transgenic mice. BioTechniques. 2008;45:247–258. doi: 10.2144/000112913. [DOI] [PubMed] [Google Scholar]
- Khaibullina A, Kenyon N, Guptill V, et al. In a model of Batten disease, palmitoyl protein thioesterase-1 deficiency is associated with brown adipose tissue and thermoregulation abnormalities. PLoS One. 2012;7:e48733. doi: 10.1371/journal.pone.0048733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielar C, Maddox L, Bible E, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2007;25:150–162. doi: 10.1016/j.nbd.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, Wozniak DF, Kielar C, Tan Y, Cooper JD, Sands MS. Cerebellar pathology and motor deficits in the palmitoyl protein thioesterase 1-deficient mouse. Exp Neurol. 2009;217:124–135. doi: 10.1016/j.expneurol.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, Pekny M, Sands MS. The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J Neurosci. 2011;31:15575–15585. doi: 10.1523/JNEUROSCI.3579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marathe S, Miranda SR, Devlin C, et al. Creation of a mouse model for non-neurological (type B) Niemann-Pick disease by stable, low level expression of lysosomal sphingomyelinase in the absence of secretory sphingomyelinase: relationship between brain intra-lysosomal enzyme activity and central nervous system function. Hum Mol Genet. 2000;9:1967–1976. doi: 10.1093/hmg/9.13.1967. [DOI] [PubMed] [Google Scholar]
- Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16:77–84. [PubMed] [Google Scholar]
- Neufeld EF, Fratantoni JC. Inborn errors of mucopolysaccharide metabolism. Science. 1970;169:141–146. doi: 10.1126/science.169.3941.141. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Sando GN, Garvin AJ, Rome LH. The transport of lysosomal enzymes. J Supramol Struct. 1977;6:95–101. doi: 10.1002/jss.400060108. [DOI] [PubMed] [Google Scholar]
- Roberts MS, Macauley SL, Wong AM, et al. Combination small molecule PPT1 mimetic and CNS-directed gene therapy as a treatment for infantile neuronal ceroid lipofuscinosis. J Inherit Metab Dis. 2012;35:847–857. doi: 10.1007/s10545-011-9446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J, Schweizer A, Russell D, Kornfeld S. The targeting of Lamp1 to lysosomes is dependent on the spacing of its cytoplasmic tail tyrosine sorting motif relative to the membrane. J Cell Biol. 1996;132:565–576. doi: 10.1083/jcb.132.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Santavuori P, Haltia M, Rapola J, Raitta C. Infantile type of so-called neuronal ceroid-lipofuscinosis. 1. A clinical study of 15 patients. J Neurol Sci. 1973;18:257–267. doi: 10.1016/0022-510X(73)90075-0. [DOI] [PubMed] [Google Scholar]
- Tikka S, Monogioudi E, Gotsopoulos A, et al. Proteomic profiling in the brain of CLN1 disease model reveals affected functional modules. Neruomol Med. 2016;18:109–133. doi: 10.1007/s12017-015-8382-6. [DOI] [PubMed] [Google Scholar]
- Verkruyse LA, Hofmann SL. Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem. 1996;271:15831–15836. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- Vesa J, Hellsten E, Verkruyse LA, et al. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- Woloszynek JC, Coleman T, Semenkovich CF, Sands MS. Lysosomal dysfunction results in altered energy balance. J Biol Chem. 2007;282:35765–35771. doi: 10.1074/jbc.M705124200. [DOI] [PubMed] [Google Scholar]
- Wurtele H, Gusew N, Lussier R, Chartrand P. Characterization of in vivo recombination activities in the mouse embryo. Mol Genet Genomics. 2005;273:252–263. doi: 10.1007/s00438-005-1112-2. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]