

Abstract
Ongoing speciation in the most important African malaria vectors gives rise to cryptic populations, which differ remarkably in their behavior, ecology, and capacity to vector malaria parasites. Understanding the population structure and the drivers of genetic differentiation among mosquitoes is crucial for effective disease control because heterogeneity within vector species contributes to variability in malaria cases and allow fractions of populations to escape control efforts. To examine population structure and the potential impacts of recent large‐scale control interventions, we have investigated the genomic patterns of differentiation in mosquitoes belonging to the Anopheles nili group—a large taxonomic group that diverged ~3 Myr ago. Using 4,343 single nucleotide polymorphisms (SNPs), we detected strong population structure characterized by high‐F ST values between multiple divergent populations adapted to different habitats within the Central African rainforest. Delineating the cryptic species within the Anopheles nili group is challenging due to incongruence between morphology, ribosomal DNA, and SNP markers consistent with incomplete lineage sorting and/or interspecific gene flow. A very high proportion of loci are fixed (F ST = 1) within the genome of putative species, which suggests that ecological and/or reproductive barriers are maintained by strong selection on a substantial number of genes.
Keywords: Anopheles nili, divergent selection, high‐FST regions, speciation
1. INTRODUCTION
One of the principal goals of population genetics is to summarize the genetic similarities and differences between populations (Wright, 1984). This task can be relatively straightforward for some taxa, but the genetic relationship among populations can also be difficult to summarize, especially for species whose evolutionary history is complex and reticulate. The best known mosquito species of the genus Anopheles—which includes all vectors of human malaria parasites—exhibit very complex rangewide population structure due to the combined effects of cryptic speciation, adaptive flexibility and ongoing gene flow across strong but incomplete reproductive barriers (Harbach, 2013; Krzywinski & Besansky, 2003). For example, almost all major malaria vectors of the Afrotropical region belong to large taxonomic groups encompassing multiple incipient species relatively isolated reproductively and geographically from one another (reviewed by Sinka et al., 2010; Antonio‐Nkondjio & Simard, 2013; Coetzee & Koekemoer, 2013; Dia, Guelbeogo, & Ayala, 2013; Lanzaro & Lee, 2013). These characteristics make them promising model systems to study speciation and the processes which contribute to reproductive barriers (e.g., Turner, Hahn, & Nuzhdin, 2005; Lawniczak et al., 2010; Neafsey et al., 2010; Fontaine et al., 2015; Weng, Yu, Hahn, & Nakhleh, 2016), but can also have far‐reaching practical consequences. Both spatial and temporal variabilities in malaria cases and the effectiveness of vector control measures are greatly impacted by heterogeneity within vector species (Molineaux & Gramiccia, 1980; Van Bortel et al., 2001). For these reasons, research on the genetic structure among the major African malaria vector mosquitoes has intensified over the last few decades (Antonio‐Nkondjio & Simard, 2013; Coetzee & Koekemoer, 2013; Dia et al., 2013; Lanzaro & Lee, 2013).
The recent scaling up of insecticide‐treated nets usage and indoor insecticide spraying to a lesser extent have led to a dramatic reduction in malaria morbidity and mortality across the continent (WHO, 2016). However, other consequences of these large‐scale interventions include increased insecticide resistance (reviewed by Hemingway et al., 2016; Ranson & Lissenden, 2016), range shift (e.g., Bøgh, Pedersen, Mukoko, & Ouma, 1998; Derua et al., 2012; Mwangangi et al., 2013) and profound evolutionary changes among vector populations. In contrast to insecticide resistance and range shift, which have been extensively studied, the recent adaptive changes among mosquito populations have yet to be addressed significantly. These changes—which involve local adaptation and genetic differentiation, introgressive hybridization, and selective sweeps across loci conferring resistance to xenobiotics—are particularly evident in the most anthropophilic species (Barnes et al., 2017; Clarkson et al., 2014; Kamdem, Fouet, Gamez, & White, 2017; Norris et al., 2015).
The ecology, taxonomic complexity, geographic distribution, role in transmission, and evolutionary potential of each vector species are unique. Consequently, further research is needed to specifically resolve population structure and the genomic targets of natural selection at a fine scale in all of the important taxa including currently understudied species. The present work focused on a group of malaria vector species representing a large taxonomic unit named Anopheles nili group. Despite the significant role some of its species play in sustaining high malaria transmission, this group has received little attention. To date, four species that occur in forested areas of Central and West Africa and are distinguishable by slight morphological variations are known within the An. nili group: An. nili sensu stricto (hereafter An. nili), An. ovengensis, An. carnevalei, and An. somalicus (Awono‐Ambene, Kengne, Simard, Antonio‐Nkondjio, & Fontenille, 2004; Gillies & Coetzee, 1987; Gillies & De Meillon, 1968). These species are characterized by reticulate evolution and complex phylogenies that have been challenging to resolve so far (Awono‐Ambene et al., 2004, 2006; Kengne, Awono‐Ambene, Antonio‐Nkondjio, Simard, & Fontenille, 2003; Ndo et al., 2010, 2013; Peery et al., 2011; Sharakhova et al., 2013). Populations of An. nili and An. ovengensis are very anthropophilic and efficient vectors of Plasmodium in rural areas where malaria prevalence is particularly high (Antonio‐Nkondjio et al., 2006).
To delineate genomic patterns of differentiation, we sampled mosquito populations throughout the range of species of the An. nili group in Cameroon and used reduced representation sequencing to develop genomewide SNP markers that we genotyped in 145 individuals. We discovered previously unknown subpopulations characterized by high pairwise differentiation within An. ovengensis and An. nili. We further explored the genetic differentiation across the genome and revealed the presence of a very high number of outlier loci that are targets of selection among locally adapted subpopulations. These findings provide significant baseline data on the genetic underpinnings of adaptive divergence and pave the way for further genomic studies in this important group of mosquitoes. Notably, a complete reference genome will enable us to conduct in‐depth studies in order to decipher the functional and phenotypic characteristics of the numerous differentiated loci as well as the contribution of recent selective events in ongoing adaptation.
2. MATERIALS AND METHODS
2.1. Mosquito species
We surveyed 28 locations within the geographic ranges of species of the An. nili group previously described in Cameroon (Figure 1) (Antonio‐Nkondjio et al., 2009; Awono‐Ambene et al., 2004, 2006; Ndo et al., 2010, 2013). The genetic structure of Anopheles species is most often based on macrogeographic or regional subdivisions of gene pools, but can also involve more subtle divergence between larvae and adults, or between adult populations found in or around human dwellings (e.g., Riehle et al., 2011). To effectively estimate the genetic diversity and identify potential cryptic populations within species, we collected larvae and adult mosquitoes within and around human dwellings using several sampling techniques (Service, 1993) in September–October 2013 (Table S1). To identify the four currently known members of the An. nili. group, we used morphological keys and a diagnostic PCR, which discriminates species based on point mutations of the ribosomal DNA (Awono‐Ambene et al., 2004; Gillies & Coetzee, 1987; Gillies & De Meillon, 1968; Kengne et al., 2003).
Figure 1.
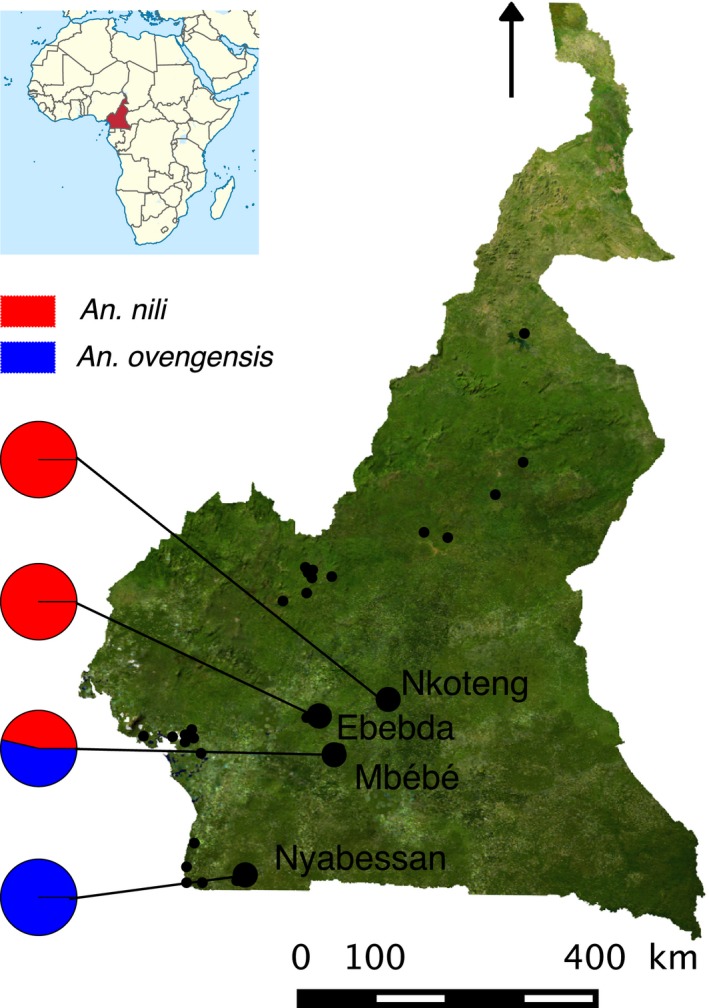
Map showing the sampling locations and the relative frequencies of the morphologically defined species An. nili and An. ovengensis in Cameroon. Small and large black dots indicate, respectively, the 28 locations surveyed and the four sampling sites where mosquitoes were collected
2.2. Library preparation, sequencing, and SNP discovery
We created double‐digest restriction site‐associated DNA (ddRAD) libraries as described in Kamdem et al. (2017) using a modified version of the protocol designed by Peterson, Weber, Kay, Fisher, and Hoekstra (2012). Briefly, genomic DNA of mosquitoes was extracted using the DNeasy Blood and Tissue kit (Qiagen) and the Zymo Research MinPrep kit for larvae and adult samples, respectively. Approximately 50 ng (10 μl) of DNA of each mosquito was digested simultaneously with MluC1 and NlaIII restriction enzymes. Digested products were ligated to adapter and barcode sequences enabling identification of individuals. Samples were pooled, purified, and 400‐bp fragments selected. The resulting libraries were amplified via PCR and purified, and fragment size distribution was checked using the BioAnalyzer. PCR products were quantified, diluted and single‐end sequenced to 100 base reads on Illumina HiSeq2000.
2.3. SNP discovery and genotyping
The process_radtags program of the Stacks v 1.35 pipeline (Catchen, Hohenlohe, Bassham, Amores, & Cresko, 2013; Catchen, Amores, Hohenlohe, Cresko, & Postlethwait, 2011) was used to demultiplex and clean Illumina sequences. Reads that passed quality filters were aligned to the An. nili Dinderesso draft genome assembly (Giraldo‐Calderon et al., 2015) made up of 51,048 short contigs (~200–30,512 bp long) using Gsnap (Wu & Nacu, 2010). To identify and call SNPs within consensus RAD loci, we utilized the ref_map.pl program of Stacks. We set the minimum number of reads required to form a stack to three and allowed two mismatches during catalogue creation. We generated SNP files in different formats for further downstream analyses using the populations program of Stacks and Plink v1.09 (Purcell et al., 2007).
2.4. Population genomics analyses
We analyzed the genetic structure of An. nili sensu lato (s.l.) populations using a principal component analysis (PCA) and an unrooted Neighbor‐Joining tree (NJ). We also examined ancestry proportions and admixtures between populations in Admixture v1.23 (Alexander, Novembre, & Lange, 2009) and Structure v2.3.4 (Pritchard, Stephens, & Donnelly, 2000). We used the package adegenet (Jombart, 2008) to implement the PCA in R (R Development Core Team 2016). The individual‐based NJ network was generated from SNP allele frequencies via a matrix of Euclidian distance using the R package ape (Paradis, Claude, & Strimmer, 2004). We ran Admixture with 10‐fold cross‐validation for values of k from 1 through 8. Similarly, we analyzed patterns of ancestry from k ancestral populations in Structure, testing five replicates of k = 1–8. We used 200,000 iterations and discarded the first 50,000 iterations as burn‐in for each Structure run. Clumpp v1.1.2 (Jakobsson & Rosenberg, 2007) was used to summarize assignment results across independent runs. To identify the optimal number of genetic clusters in our sample, we applied simultaneously the lowest cross‐validation error in Admixture, the ad hoc statistic deltaK (Earl & VonHoldt, 2012; Evanno, Goudet, & Regnaut, 2005) and the discriminant analysis of principal component (DAPC) method implemented in adegenet. To examine the level of genomic divergence among populations, we assessed genetic differentiation (F ST) across SNPs using the populations program of the Stacks pipeline. Mean F ST values were also used to quantify pairwise divergence between populations. To infer the demographic history of different populations, we used the diffusion approximation method implemented in the package ∂a∂i v 1.6.3 (Gutenkunst, Hernandez, Williamson, & Bustamante, 2009). Single‐population models were fitted to allele frequency spectra, and the best model was selected using the lowest likelihood and Akaike information criterion as well as visual inspections of residuals.
3. RESULTS
3.1. SNP genotyping
We collected mosquitoes from four locations out of 28 sampling sites (Figure 1, Table S1) and sequenced 145 individuals belonging, according to morphological criteria and diagnostic PCRs, to two species (An. nili [n = 24] and An. ovengensis [n = 121]). We assembled 197,724 RAD loci that mapped to unique positions throughout the reference genome. After applying stringent filtering rules, 408 loci present in all populations and in at least 50% of individuals in each population were retained. Within these loci, we identified 4,343 high‐quality biallelic markers that were used to analyze population structure and genetic differentiation.
3.2. Morphologically defined species do not correspond to genetic clusters
The PCA and the NJ tree show that the genetic variation across 4,343 SNPs is best explained by more than two clusters, implying subdivisions within An. nili and An. ovengensis (Figure 2). Three subgroups are apparent within An. nili while two distinct clusters segregate in An. ovengensis (hereafter referred to as An. nili group 1, An. nili group 2, An. nili group 3, An. ovengensis group 1 and An. ovengensis group 2). These five subpopulations are strongly correlated with the different sampling sites suggesting local adaptation of divergent populations. Importantly, Structure and Admixture analyses reveal that, at k = 2, one population identified by morphology and the diagnostic PCR as An. nili has almost the same ancestry pattern as the largest An. ovengensis cluster (Figure 3). Such discrepancies between morphology‐based and molecular taxonomies can be due to a variety of processes including phenotypic plasticity, introgressive hybridization, or incomplete lineage sorting (i.e., when independent loci have different genealogies by chance) (Arnold, 1997; Combosch & Vollmer, 2015; Fontaine et al., 2015; Weng et al., 2016). At k = 2 and k = 3, some populations also exhibit half ancestry from each morphological species suggestive of gene flow. We found a conflicting number of genetic clusters in our samples likely reflecting the complex history of subdivisions and admixtures among populations (Figure 4). The Evanno et al. (2005) method, which highlights the early stages of divergence between An. nili and An. ovengensis, indicates two probable ancestors. DAPC and the Admixture cross‐validation error, which are more sensitive to recent hierarchical population subdivisions, show five or more distinct clusters as revealed by the PCA and the NJ tree (Figure 4).
Figure 2.
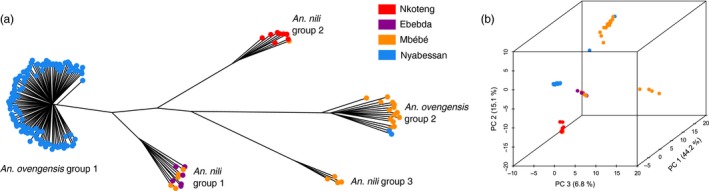
Population genetic structure inferred from 4,343 SNPs using a PCA (a) and a neighbor‐joining tree (b). The percentage of variance explained is indicated on each PCA axis. Note the strong association between the five genetic clusters and the different sampling locations
Figure 3.
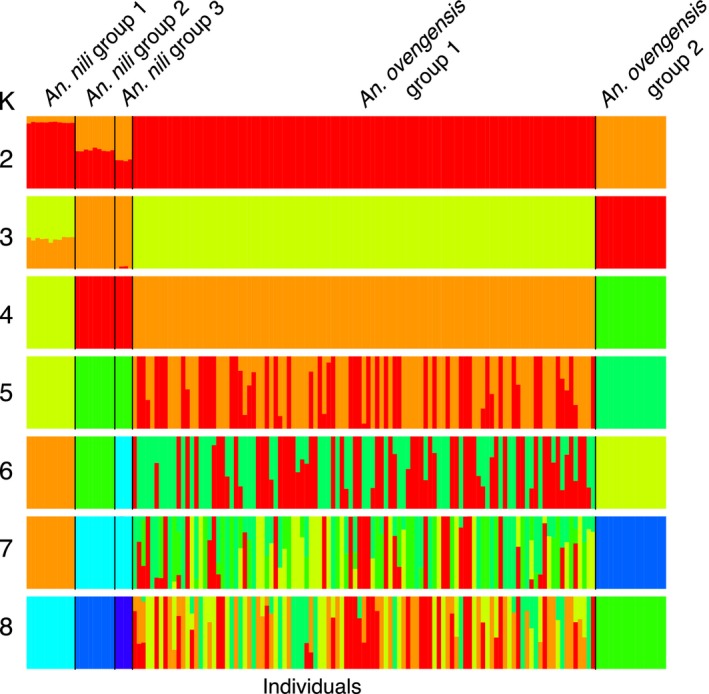
Ancestry proportions inferred in Admixture with k = 2–8
Figure 4.
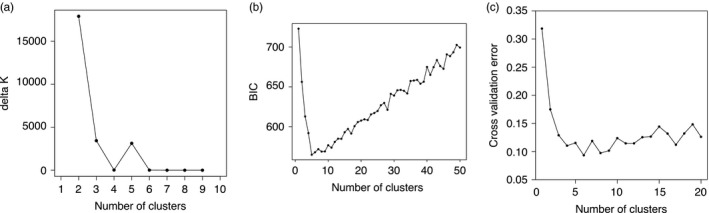
Identification of the optimal number of genetic clusters using the delta k method of Evanno et al. (2005) (a), DAPC (b) and 10‐fold cross‐validation in Admixture (c). The lowest Bayesian information criterion (BIC) and cross‐validation error and the highest delta k indicate the most probable number of clusters
As suggested by the long internal branches, which connect subpopulations on the NJ tree, there is strong differentiation between and within morphological species characterized by globally high‐F ST values (Table 1). Relatively lower F ST values observed between certain clusters may be due to greater interpopulation migration and intermixing or more recent divergence. The F ST values do not reflect the morphological delimitation of species. Indeed, the level of genetic differentiation is higher between some subpopulations within the same morphological species. Overall, patterns of genetic structure and differentiation reveal a group of populations whose phylogenies and species status are likely confounded by hybridization and/or incomplete lineage sorting. We argue that the current taxonomy based on morphology and ribosomal DNA does not capture the optimal reproductive units among populations of this group of mosquitoes.
Table 1.
Pairwise F ST between divergent subpopulations of An. nili s.l
| F ST | An. nili group 1 | An. nili group 2 | An. nili group 3 | An. ovengensis group 1 | An. ovengensis group 2 |
|---|---|---|---|---|---|
| An. nili group 1 | – | ||||
| An. nili group 2 | 0.374 | – | |||
| An. nili group 3 | 0.506 | 0.552 | – | ||
| An. ovengensis group 1 | 0.135 | 0.275 | 0.364 | – | |
| An. ovengensis group 2 | 0.432 | 0.458 | 0.492 | 0.349 | – |
3.3. Genomic signatures of divergent selection and demographic history
We analyzed patterns of genetic differentiation across SNP loci throughout the genome. Pairwise comparisons are based on filtered variants that satisfy all criteria to be present in both populations, which explains the discrepancy in the number of SNPs observed between specific paired comparisons (Figure 5). The distribution of locus‐specific F ST values between the five subpopulations revealed a U‐shape characterized by two peaks around 0 and 1. The large majority of SNPs have low‐to‐moderate divergence, but a substantial number of variants are extremely differentiated between populations. The maximum F ST among SNPs is 1, and the proportion of loci with F ST = 1 varies from 6.52% between the populations we termed An. nili group 1 and An. ovengensis group 1 to 44.74% between the subgroups called An. nili group 2 and An. nili group 3 (Figure 5). This pattern of genomewide divergence suggests that a very high number of sites with abrupt differentiation—which likely contain genes that contribute to divergent selection and/or reproductive isolation—coexist with regions of weak divergence that can be freely exchanged between species. As is the case with the overall genetic differentiation, morphology is not a reliable predictor of locus‐specific divergence. Precisely, the lowest percentage of fixed SNPs is found between An. ovengensis from Nyabessan and An. nili collected from Mbébé and Ebebda (Figures 1 and 5). In contrast, the largest proportion of fixed loci is observed between locally adapted subgroups within the same morphological species: An. nili. The draft reference genome made up of short contigs did not enable us to test hypotheses about the genomic distribution of differentiated loci. For example, it remains unknown whether the numerous SNPs that are fixed among populations are spread throughout the entire genome or clustered within genomic regions of low recombination including chromosomal inversions and chromosome centers (Nosil & Feder, 2012; Roesti, Hendry, Salzburger, & Berner, 2012).
Figure 5.
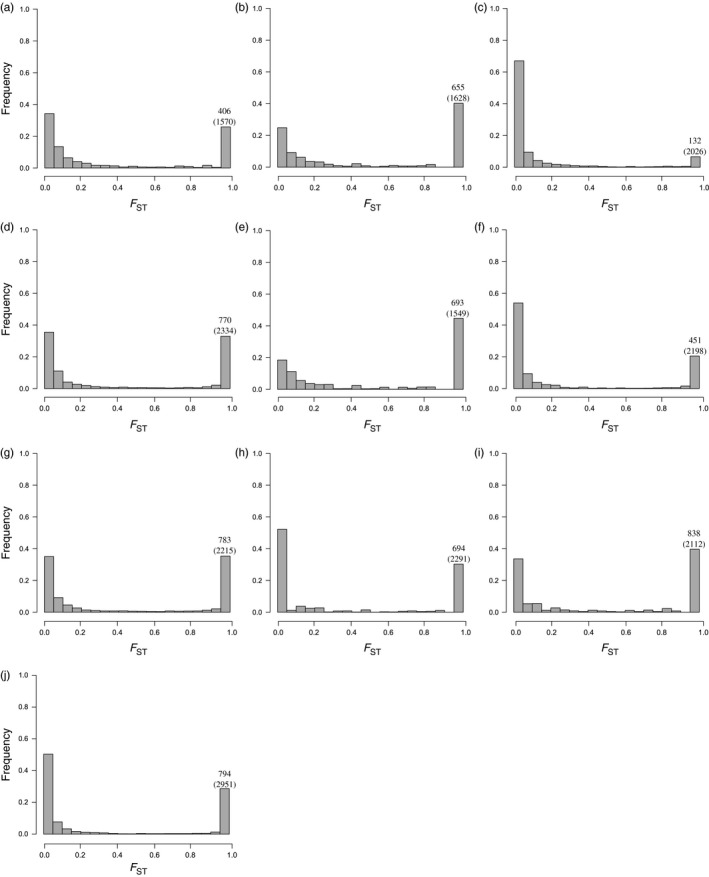
Distribution of F ST values throughout the genome between An. nili group 1 and An. nili group 2 (a); An. nili group 1 and An. nili group 3 (b); An. nili group 1 and An. ovengensis group 1 (c); An. nili group 1 and An. ovengensis group 2 (d); An. nili group 2 and An. nili group 3 (e); An. nili group 2 and An. ovengensis group 1 (f); An. nili group 2 and An. ovengensis group 2 (g); An. nili group 3 and An. ovengensis group 1 (h); An. nili group 3 and An. ovengensis group 2 (i); An. ovengensis group 1 and An. ovengensis group 2 (j). The number of SNPs with F ST = 1 is indicated in each pairwise comparison as well as the total number of SNPs in parenthesis
Models of population demography indicate that all subgroups have experienced an increase in effective size in a more or less recent past (Table 2). Nevertheless, confidence intervals of population parameters are high in some populations, and our results should be interpreted with the necessary precautions. The population growth is less significant in An. nili group 1.
Table 2.
Demographic models of different subgroups of An. nili s.l
| Population | Best model | Log‐likelihood | Final population sizea (95% CI) | Timeb (95% CI) |
|---|---|---|---|---|
| An. nili group 1 | Growth | −18.42 | 6.41 (5.326–20.71) | 3.70 (1.11–13.31) |
| An. nili group 2 | Two‐epoch | −19.97 | 17.87 (9.33–35.50) | 11.27 (4.93–19.64) |
| An. ovengensis group 1 | Growth | −112.18 | 13.04 (12.15–17.26) | 0.70 (0.58–1.08) |
| An. Ovengensis group 2 | Growth | −22.98 | 19.95 (14.45–45.70) | 5.11 (2.33–15.13) |
Relative to ancestral population size.
Expressed in units 2Ne generations from start of growth to present.
4. DISCUSSION
4.1. Genetic differentiation
Advances in sequencing and analytical approaches have opened new avenues for the study of genomes of disease vectors. We have focused on malaria mosquitoes of the An. nili group, whose taxonomy and population structure have been challenging to resolve with low‐resolution markers. We analyzed genetic structure using genomewide SNPs and found strong differentiation and local adaption among populations belonging to the two morphologically defined species An. nili and An. ovengensis. The exact number of subpopulations remains contentious, with the suggested number of divergent clusters varying from two to five. Significant population structure at eight microsatellite loci has been described among An. nili populations from Cameroon, with F ST values as high as 0.48 between samples from the rainforest area (Ndo et al., 2013). By contrast, An. ovengensis was discovered recently and the genetic structure of this vector remains understudied. This species was initially considered as a sibling of An. nili (Awono‐Ambene et al., 2004, 2006; Kengne et al., 2003), but more recent studies have started to challenge the assumed relatedness between the two species due to the high divergence revealed by polytene chromosomes (Sharakhova et al., 2013). Our findings call for a careful review of the current taxonomy within this group of species, which is a necessary first step for accurately delineating the role played by the different subpopulations in malaria transmission.
Our samples were collected from locations characterized by a more or less degraded forest within the rainforest area of Cameroon. In these habitats, larvae of An. nili s.l. exploit relatively similar breeding sites consisting of slow‐moving rivers (Antonio‐Nkondjio et al., 2009). The ecological drivers of genetic differentiation remain unknown, and will be difficult to infer from our data given the apparent similarity of habitats among the divergent populations we described. Further study is needed to clearly address the environmental variables that may be correlated with ongoing adaptive divergence at adult and larval stages. One of the most expected outcomes of current large‐scale malaria control measures that are underway in sub‐Saharan African countries concerns the effects of increased insecticide exposure on the genetic diversity and population demography of vectors. A substantial population decline that may considerably affect the adaptive potential of vector species has been occasionally reported following a major insecticide‐treated bed net distribution campaign and/or indoor residual house spraying (e.g., Athrey et al., 2012). The inferred demographic history of the different subpopulations within the An. nili group does not reveal signatures of bottlenecks that can be potentially correlated with increased usage of insecticides and insecticide‐treated nets. This result is consistent with the demography of several other important malaria vectors of the Afrotropical region, including An. gambiae, An. coluzzii, An. funestus and An. moucheti, which reveals a substantial population increase suggesting that intense insecticide exposure has yet to leave deep or detectable impacts on patterns of genetic variation among mosquito populations (Fouet, Kamdem, Gamez, & White, 2017; Kamdem et al., 2017; O'Loughlin et al., 2014).
4.2. Genomic architecture of geographic and reproductive isolation
Understanding the genomic architecture of reproductive isolation may reveal crucial information on the sequence of events that occur from the initial stages of divergence among populations to the onset of strong reproductive barriers between species (e.g., Turner et al., 2005; Harr, 2006; Nadeau et al., 2012; Ellegren et al., 2012; Carneiro et al., 2014; Burri et al., 2015). One influential concept of speciation coined the “genic view of species” proposes that boundaries between species are properties of individual genes or genome regions and not of whole organisms or lineages (Barton & Hewitt, 1985; Harrison & Larson, 2014; Harrison, 1990; Key, 1968; Nosil & Feder, 2012; Rieseberg, Whitton, & Gardner, 1999; Wu, 2001). We have discovered a substantially high number of SNPs that are strongly differentiated between populations and often fixed within subgroups of An. nili s.l. Interpreting this intriguing pattern of genomic differentiation is not straightforward due to the complex interactions between numerous forces—including positive or negative selection, recombination, introgressive hybridization and incomplete lineage sorting—that can affect the level of divergence among SNPs (Begun & Aquadro, 1992; Cutter & Payseur, 2013; Harrison & Larson, 2016; Nachman & Payseur, 2012; Roesti et al., 2012). Some of these variants exhibiting high divergence among populations certainly contain markers of ecological and/or reproductive isolation. However, as far as reproductive barriers are concerned, recent studies have indicated a complex relationship between the degree of genetic differentiation and gene flow at the genome level (e.g., Gompert et al., 2012; Hamilton, Lexer, & Aitken, 2013a,b; Larson, Andrés, Bogdanowicz, & Harrison, 2013; Larson, White, Ross, & Harrison, 2014; Parchman et al., 2013; Taylor, Curry, White, Ferretti, & Lovette, 2014). Highly divergent genomic regions do not necessarily coincide with regions of reduced gene flow among established or emerging species. Several alternative interpretations exist for the numerous high‐F ST regions we detected in all pairwise comparisons (Cruickshank & Hahn, 2014; Delmore et al., 2015; Nachman & Payseur, 2012; Noor & Bennett, 2009). Nevertheless, careful examination of these outliers of differentiation may reveal significant insights into the wide range of genes and traits that contribute to ecological divergence and/or reproductive isolation between subgroups of An. nili s.l. A complete genome assembly will be necessary to better delineate specific regions of the genome under natural selection, and therefore clarify the genomic basis of phenotypic fitness differences between divergent populations. This will also help understand the extent to which recent selection associated with human interventions contribute to local adaptation and genetic differentiation as observed in An. gambiae and An. coluzzii (Kamdem et al., 2017).
Signals consistent with gene flow between An. nili and An. ovengensis are apparent in our data although it has been proposed that the two morphological species diverged ~3 Myr ago (Ndo et al., 2013). Some individuals display almost half ancestry from each morphological species. The disagreement between morphology/PCR and molecular taxonomies observed in Structure and Admixture analyses also suggests that incongruent genealogies may be widespread along chromosomes due to hybridization. However, hybridization can be difficult to detect because other factors such as incomplete lineage sorting or technical artifacts can leave signatures that are similar to those of interspecific gene flow (Liu et al., 2014; Patterson et al., 2012). A complete reference genome is also needed to analyze the detailed distribution of genealogies across small genomic windows and to disentangle the relative contribution of processes that generate the putative admixtures and species confusion observed among divergent populations (Fontaine et al., 2015; Martin et al., 2013; Weng et al., 2016).
5. CONCLUSIONS AND IMPLICATIONS
Delineating the fine‐scale population structure of mosquito populations is crucial for understanding their epidemiological significance and their potential response to vector control measures. Moreover, recent malaria control efforts affect interspecific gene flow, genetic differentiation, population demography and natural selection in mosquitoes (Athrey et al., 2012; Barnes et al., 2017; Clarkson et al., 2014; Kamdem et al., 2017; Norris et al., 2015). Deciphering the signatures of these processes across mosquito genomes is important to minimize their negative impacts on vector control. Our findings shed some light on the complex evolutionary history and provide a framework for future investigations into the genetic basis of ecological and reproductive barriers among species of the An. nili group.
DATA ARCHIVING STATEMENT
Raw data (fastq files) for 145 Anopheles nili individuals are available at: https://doi.org/10.5061/dryad.5d5b3
AUTHOR CONTRIBUTIONS
CF, CK, and BJW conceived and designed the experiments. CF, CK, SG, and BJW performed the experiments. CF and CK analyzed the data. CF, CK, and BJW wrote the manuscript.
Supporting information
ACKNOWLEDGEMENTS
Funding for this project was provided by the University of California Riverside and NIH grants 1R01AI113248 and 1R21AI115271 to BJW. We thank inhabitants and administrative authorities of the sampling sites included in this study for their collaboration.
Fouet C, Kamdem C, Gamez S, White BJ. Genomic insights into adaptive divergence and speciation among malaria vectors of the Anopheles nili group. Evol Appl. 2017;10:897–906. https://doi.org/10.1111/eva.12492
Contributor Information
Caroline Fouet, Email: caroline.fouet@ucr.edu.
Bradley J. White, Email: bwhite@ucr.edu
REFERENCES
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonio‐Nkondjio, C. , Kerah, C. H. , Simard, F. , Awono‐Ambene, P. , Chouaibou, M. , Tchuinkam, T. , & Fontenille, D. (2006). Complexity of the malaria vectorial system in Cameroon: Contribution of secondary vectors to malaria transmission. Journal of Medical Entomology, 43, 1215–1221. https://doi.org/10.1603/0022-2585 (2006)43[1215:COTMVS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Antonio‐Nkondjio, C. , Ndo, C. , Costantini, C. , Awono‐Ambene, P. , Fontenille, D. , & Simard, F. (2009). Distribution and larval habitat characterization of Anopheles moucheti, Anopheles nili, and other malaria vectors in river networks of southern Cameroon. Acta Tropica, 112(3), 270–276. https://doi.org/10.1016/j.actatropica.2009.08.009 [DOI] [PubMed] [Google Scholar]
- Antonio‐Nkondjio, C. , & Simard, F . (2013). Highlights on Anopheles nili and Anopheles moucheti, Malaria Vectors in Africa In Manguin S. (Ed.), Anopheles mosquitoes ‐ New insights into malaria vectors (INTECH) (pp. 828). Croatia, European Union: InTech. ISBN 978‐953‐51‐1188‐7. [PubMed] [Google Scholar]
- Arnold, M. L. (1997). Natural hybridization and evolution. Oxford: Oxford University Press. [Google Scholar]
- Athrey, G. , Hodges, T. K. , Reddy, M. R. , Overgaard, H. J. , Matias, A. , & Ridl, F. C , … Slotman, M. A . (2012). The effective population size of malaria mosquitoes: Large impact of vector control. PLoS Genetics, 8(12), e1003097 https://doi.org/10.1371/journal.pgen.1003097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awono‐Ambene, H. P. , Kengne, P. , Simard, F. , Antonio‐Nkondjio, C. , & Fontenille, D. (2004). Description and bionomics of Anopheles (Cellia) ovengensis (Diptera: Culicidae), a new malaria vector species of the Anopheles nili group from south Cameroon. Journal of Medical Entomology, 41, 561–568. https://doi.org/10.1603/0022-2585-41.4.561 [DOI] [PubMed] [Google Scholar]
- Awono‐Ambene, H. P. , Simard, F. , Antonio‐Nkondjio, C. , Cohuet, A. , Kengne, P. , & Fontenille, D. (2006). Multilocus enzyme electrophoresis supports speciation within the Anopheles nili group of malaria vectors in Cameroon. The American Journal of Tropical Medicine and Hygiene, 75, 656–658. https://doi.org/75/4/656 [pii] [PubMed] [Google Scholar]
- Barnes, K. G. , Weedall, G. D. , Ndula, M. , Irving, H. , Mzihalowa, T. , Hemingway, J. , & Wondji, C. S. (2017). Genomic Footprints of Selective Sweeps from Metabolic Resistance to Pyrethroids in African Malaria Vectors Are Driven by Scale up of Insecticide‐Based Vector Control. PLoS Genetics, 13(2), e10 1–22. https://doi.org/10.1371/journal.pgen.1006539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. , & Hewitt, G. (1985). Analysis of hybrid zones. Annual Review of Ecology and Systematics, 16, 113–148. [Google Scholar]
- Begun, D. , & Aquadro, C. (1992). Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster . Nature, 356, 519–520. [DOI] [PubMed] [Google Scholar]
- Bøgh, C. , Pedersen, E. M. , Mukoko, D. A , & Ouma, J. H. (1998). Permethrin‐impregnated bednet effects on resting and feeding behaviour of lymphatic filariasis vector mosquitoes in Kenya. Medical and Veterinary Entomology, 12(1), 52–59. [DOI] [PubMed] [Google Scholar]
- Burri, R. , Nater, A. , Kawakami, T. , Mugal, C. F. , Olason, P. I. , & Smeds, L , … Ellegren, H . (2015). Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Research, 25(11), 1656–1665. https://doi.org/10.1101/gr.196485.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro, M. , Albert, F. W. , Afonso, S. , Pereira, R. J. , Burbano, H. , & Campos, R , … Ferrand, N . (2014). The genomic architecture of population divergence between subspecies of the European Rabbit. PLoS Genetics, 10(8), e1003519 https://doi.org/10.1371/journal.pgen.1003519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. , Cresko, W. , & Postlethwait, J. H. (2011). Stacks: Building and genotyping Loci de novo from short‐read sequences. G3 (Bethesda, Md.), 1(3), 171–182. https://doi.org/10.1534/g3.111.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. a. , Bassham, S. , Amores, A. , & Cresko, W. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22, 3124–3140. https://doi.org/10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson, C. S. , Weetman, D. , Essandoh, J. , Yawson, A. E. , Maslen, G. , & Manske, M , … Donnelly, M. J . (2014). Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nature Communications, 5, 4248 https://doi.org/10.1038/ncomms5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee, M. , & Koekemoer, L. (2013). Molecular Systematics and Insecticide Resistance in the Major African Malaria Vector Anopheles funestus. Annual Review of Entomology, 58, 393–412. [DOI] [PubMed] [Google Scholar]
- Combosch, D. J. , & Vollmer, S. V. (2015). Trans‐Pacific RAD‐Seq population genomics confirms introgressive hybridization in Eastern Pacific Pocillopora corals. Molecular Phylogenetics and Evolution, 88, 154–162. https://doi.org/10.1016/j.ympev.2015.03.022 [DOI] [PubMed] [Google Scholar]
- Cruickshank, T. E. , & Hahn, M. W. (2014). Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Molecular Ecology, 23(13), 3133–3157. https://doi.org/10.1111/mec.12796 [DOI] [PubMed] [Google Scholar]
- Cutter, A. D. , & Payseur, B. A. (2013). Genomic signatures of selection at linked sites: Unifying the disparity among species. Nature Reviews. Genetics, 14(4), 262–274. https://doi.org/10.1038/nrg3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore, K. E. , Hübner, S. , Kane, N. C. , Schuster, R. , Andrew, R. L. , & Câmara, F , … Irwin, D. E . (2015). Genomic analysis of a migratory divide reveals candidate genes for migration and implicates selective sweeps in generating islands of differentiation. Molecular Ecology, 24(8), 1873–1888. [DOI] [PubMed] [Google Scholar]
- Derua, Y. a , Alifrangis, M. , Hosea, K. M. , Meyrowitsch, D. W. , Magesa, S. M. , Pedersen, E. M. , & Simonsen, P. E. (2012). Change in composition of the Anopheles gambiae complex and its possible implications for the transmission of malaria and lymphatic filariasis in north‐eastern Tanzania. Malaria Journal, 11(1), 188 https://doi.org/10.1186/1475-2875-11-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dia, I. , Guelbeogo, M. , & Ayala, D . (2013). Advances and Perspectives in the Study of the Malaria Mosquito Anopheles funestus In Manguin S. (Ed.), Anopheles mosquitoes ‐ New insights into malaria vectors (INTECH) (pp. 828). Croatia, European Union: InTech. [Google Scholar]
- Earl, D. A. , & VonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(359–361). [Google Scholar]
- Ellegren, H. , Smeds, L. , Burri, R. , Olason, P. I. , Backström, N. , & Kawakami, T , … Wolf, J. B. W . (2012). The genomic landscape of species divergence in Ficedula flycatchers. Nature, 491(7426), 756–760. https://doi.org/10.1038/nature11584 [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Goudet, J. , & Regnaut, S. (2005). Detecting the number of clusters of individuals using the software structure: A simulation study. Molecular Ecology, 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Fontaine, M. C. , Pease, J. B. , Steele, a. , Waterhouse, R. M. , Neafsey, D. E. , & Sharakhov, I. V , … Besansky, N. J . (2015). Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science, 347(6217), 1258524 https://doi.org/10.1126/science.1258524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouet, C. , Kamdem, C. , Gamez, S. , & White, B. J. (2017). Extensive genetic diversity among populations of the malaria mosquito Anopheles moucheti revealed by population genomics. Infection, Genetics and Evolution, 48, 27–33. https://doi.org/10.1016/j.meegid.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Gillies, M. T. , & Coetzee, M . (1987). A supplement to the Anophelinae of Africa south of the Sahara. Johannesburg: The South African Institute for Medical Research. [Google Scholar]
- Gillies, M. T. , & De Meillon, B. (1968). The Anophelinae of Africa South of the Sahara, 2nd edn Johannesburg: Publications of the South African Institute for Medical Research. [Google Scholar]
- Giraldo‐Calderon, G. I. , Emrich, S. J. , MacCallum, R. M. , Maslen, G. , Dialynas, E. , & Topalis, P , … Lawson, D . (2015). VectorBase: An updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Research, 43(D1), D707–D713. https://doi.org/10.1093/nar/gku1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompert, Z. , Lucas, L. K. , Nice, C. C. , Fordyce, J. A. , Forister, M. L. , & Buerkle, C. A. (2012). Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution, 66(7), 2167–2181. https://doi.org/10.5061/dryad.f0b2f083 [DOI] [PubMed] [Google Scholar]
- Gutenkunst, R. N. , Hernandez, R. D. , Williamson, S. H. , & Bustamante, C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5(10), e1000695 https://doi.org/10.1371/journal.pgen.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, J. a. , Lexer, C. , & Aitken, S. N. (2013a). Differential introgression reveals candidate genes for selection across a spruce (Picea sitchensis × P. glauca) hybrid zone. New Phytologist, 197(3), 927–938. https://doi.org/10.1111/nph.12055 [DOI] [PubMed] [Google Scholar]
- Hamilton, J. A. , Lexer, C. , & Aitken, S. N. (2013b). Genomic and phenotypic architecture of a spruce hybrid zone (Picea sitchensis × P. glauca). Molecular Ecology, 22(3), 827–841. https://doi.org/10.1111/mec.12007 [DOI] [PubMed] [Google Scholar]
- Harbach, R. E . (2013). The Phylogeny and Classification of Anopheles In Manguin S. (Ed.), Anopheles mosquitoes ‐ New insights into malaria vectors (INTECH) (pp. 828) Croatia, European Union: InTech. ISBN 978‐953‐51‐1188‐7. [Google Scholar]
- Harr, B . (2006). Genomic islands of differentiation between house mouse subspecies, 730–737. https://doi.org/10.1101/gr.5045006.entiation [DOI] [PMC free article] [PubMed]
- Harrison, R. G . (1990). Hybrid zones: Windows on evolutionary process. Oxford Surveys in Evolutionary Biology, 7, 69–128. [Google Scholar]
- Harrison, R. G. , & Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. Journal of Heredity, 105(S1), 795–809. https://doi.org/10.1093/jhered/esu033 [DOI] [PubMed] [Google Scholar]
- Harrison, R. G. , & Larson, E. L. (2016). Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Molecular Ecology, 25, 2454–2466. https://doi.org/10.1111/mec.13582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemingway, J. , Ranson, H. , Magill, A. , Kolaczinski, J. , Fornadel, C. , & Gimnig, J , … Hamon, N . (2016). Averting a malaria disaster: Will insecticide resistance derail malaria control? The Lancet, 387(10029), 1785–1788. https://doi.org/10.1016/s0140-6736(15)00417-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. (2007). CLUMPP: A cluster matching and permutation program for dealing with multimodality in analysis of population structure. Bioinformatics, 23, 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24, 1403–1405. [DOI] [PubMed] [Google Scholar]
- Kamdem, C. , Fouet, C. , Gamez, S. , & White, B. J. (2017). Pollutants and insecticides drive local adaptation in African malaria mosquitoes. Molecular Biology and Evolution, 34(5), 1261–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kengne, P. , Awono‐Ambene, H. P. , Antonio‐Nkondjio, C. , Simard, F. , & Fontenille, D. (2003). Molecular identification of the Anopheles nili group African malaria vectors. Medical and Veterinary Entomology, 17, 67–74. [DOI] [PubMed] [Google Scholar]
- Key, K. H. L. (1968). The concept of stasipatric speciation. Systematic Zoology, 17, 14–22. [Google Scholar]
- Krzywinski, J. , & Besansky, N. J. (2003). Molecular systematics of Anopheles : From subgenera to subpopulations. Annual Review of Entomology, 48(1), 111–139. https://doi.org/10.1146/annurev.ento.48.091801.112647 [DOI] [PubMed] [Google Scholar]
- Lanzaro, G. C. , & Lee, Y . (2013). Speciation in Anopheles gambiae — The Distribution of Genetic Polymorphism and Patterns of Reproductive Isolation Among Natural Populations In Manguin S. (Ed.), Anopheles mosquitoes ‐ New insights into malaria vectors (INTECH). (pp. 828). Croatia, European Union: InTech. [Google Scholar]
- Larson, E. L. , Andrés, J. A. , Bogdanowicz, S. M. , & Harrison, R. G. (2013). Differential introgression in a mosaic hybrid zone reveals candidate barrier genes. Evolution, 67(12), 3653–3661. https://doi.org/10.1111/evo.12205 [DOI] [PubMed] [Google Scholar]
- Larson, E. L. , White, T. A. , Ross, C. L. , & Harrison, R. G. (2014). Gene flow and the maintenance of species boundaries. Molecular Ecology, 23(7), 1668–1678. https://doi.org/10.1111/mec.12601 [DOI] [PubMed] [Google Scholar]
- Lawniczak, M. K. N. , Emrich, S. J. , Holloway, A. K. , Regier, A. P. , Olson, M. , & White, B , … Besansky, N. J . (2010). Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science (New York, N.Y.), 330(6003), 512–514. https://doi.org/10.1126/science.1195755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, K. J. , Dai, J. , Truong, K. , Song, Y. , Kohn, M. H. , & Nakhleh, L. (2014). An HMM‐Based Comparative Genomic Framework for Detecting Introgression in Eukaryotes. PLoS Computational Biology, 10(6), e1003649 https://doi.org/10.1371/journal.pcbi.1003649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S. H. , Dasmahapatra, K. K. , Nadeau, N. J. , Salazar, C. , Walters, J. R. , Simpson, F. , … Jiggins, C. D. (2013). Genome‐wide evidence for speciation with gene flow in Heliconius butterflies. Genome Research, 23(11), 1817–1828. https://doi.org/10.1101/gr.159426.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineaux, L. , & Gramiccia, G . (1980). The Garki Project. Research on the epidemiology and control of malaria in the sudan savanna of West Africa. Geneva: World Health Organization, 311 pp. ISBN: 9241560614, 9789241560610. [Google Scholar]
- Mwangangi, J. M. , Mbogo, C. M. , Orindi, B. O. , Muturi, E. J. , Midega, J. T. , Nzovu, J. , … Beier, J. C. (2013). Shifts in malaria vector species composition and transmission dynamics along the Kenyan coast over the past 20 years. Malaria Journal, 12, 13 https://doi.org/10.1186/1475-2875-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman, M. W. , & Payseur, B. A. (2012). Recombination rate variation and speciation : Theoretical predictions and empirical results from rabbits and mice. Philosophical Transactions of the Royal Society B: Biological Sciences, 367, 409–421. https://doi.org/10.1098/rstb.2011.0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau, N. J. , Whibley, A. , Jones, R. T. , Davey, J. W. , Dasmahapatra, K. K. , & Baxter, S. W , … Jiggins, C. D. . (2012). Genomic islands of divergence in hybridizing Heliconius butterflies identified by large‐scale targeted sequencing, Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 367(1587), 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndo, C. , Antonio‐Nkondjio, C. , Cohuet, A. , Ayala, D. , Kengne, P. , Morlais, I. , … Simard, F. (2010). Population genetic structure of the malaria vector Anopheles nili in sub‐Saharan Africa. Malaria Journal, 9, 161 https://doi.org/10.1186/1475-2875-9-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndo, C. , Simard, F. , Kengne, P. , Awono‐Ambene, P. , Morlais, I. , Sharakhov, I. , … Antonio‐Nkondjio, C. (2013). Cryptic Genetic Diversity within the Anopheles nili group of Malaria Vectors in the Equatorial Forest Area of Cameroon (Central Africa). PLoS One, 8(3), 1–12. https://doi.org/10.1371/journal.pone.0058862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey, D. E. , Lawniczak, M. K. N. , Park, D. J. , Redmond, S. N. , Coulibaly, M. B. , Traoré, S. F. , … Muskavitch, M. A. T. (2010). SNP genotyping defines complex gene‐flow boundaries among African malaria vector mosquitoes. Science (New York, N.Y.), 330(6003), 514–517. https://doi.org/10.1126/science.1193036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor, M. A. F. , & Bennett, S. M. (2009). Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species Heredity, 103(6), 439–444. https://doi.org/10.1038/hdy.2010.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, L. C. , Main, B. J. , Lee, Y. , Collier, T. C. , Fofana, A. , Cornel, A. J. , & Lanzaro, G. C. (2015). Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide‐treated bed nets. Proceedings of the National Academy of Sciences, 2014, 18892 https://doi.org/10.1073/pnas.1418892112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil, P. , & Feder, J. L. (2012). Widespread yet heterogeneous genomic divergence. Molecular Ecology, 21(12), 2829–2832. https://doi.org/10.1111/j.1365-294x.2012.05580.x [DOI] [PubMed] [Google Scholar]
- O'Loughlin, S. M. , Magesa, S. , Mbogo, C. , Mosha, F. , Midega, J. , Lomas, S. , & Burt, A. (2014). Genomic analyses of three malaria vectors reveals extensive shared polymorphism but contrasting population histories. Molecular Biology and Evolution, 1, 14 https://doi.org/10.1093/molbev/msu040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis, E. , Claude, J. , & Strimmer, K. (2004). Analyses of Phylogenetics and Evolution in R language. Bioinformatics, 20(2), 289–290. [DOI] [PubMed] [Google Scholar]
- Parchman, T. L. , Gompert, Z. , Braun, M. J. , Brumfield, R. T. , McDonald, D. B. , Uy, J. A. C. , … Buerkle, C. A. (2013). The genomic consequences of adaptive divergence and reproductive isolation between species of manakins. Molecular Ecology, 22(12), 3304–3317. https://doi.org/10.1111/mec.12201 [DOI] [PubMed] [Google Scholar]
- Patterson, N. , Moorjani, P. , Luo, Y. , Mallick, S. , Rohland, N. , Zhan, Y. , … Reich, D. (2012). Ancient admixture in human history. Genetics, 192(3), 1065–1093. https://doi.org/10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peery, A. , Sharakhova, M. V , Antonio‐Nkondjio, C. , Ndo, C. , Weill, M. , Simard, F. , & Sharakhov, I. V. (2011). Improving the population genetics toolbox for the study of the African malaria vector Anopheles nili: Microsatellite mapping to chromosomes. Parasites & Vectors, 4(1), 202 https://doi.org/10.1186/1756-3305-4-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non‐Model Species. PLoS One, 7(5), e37135.https://doi.org/10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. , Bender, D. , … Sham, P. (2007). PLINK: A toolset for whole‐genome association and population‐based linkage analysis. American Journal of Human Genetics, 81(3), 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2016). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ranson, H. , & Lissenden, N. (2016). Insecticide Resistance in African Anopheles Mosquitoes: A Worsening Situation that Needs Urgent Action to Maintain Malaria Control. Trends in Parasitology, 32(3), 187–196. https://doi.org/10.1016/j.pt.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Riehle, M. M. , Guelbeogo, W. M. , Gneme, A. , Eiglmeier, K. , Holm, I. , Bischoff, E. , … Vernick, K. D. (2011). A cryptic subgroup of Anopheles gambiae is highly susceptible to human malaria parasites. Science (New York, N.Y.), 331(6017), 596–598. https://doi.org/10.1126/science.1196759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg, L. H. , Whitton, J. , & Gardner, K . (1999). Hybrid Zones and the Genetic Architecture of a Barrier to Gene Flow Between Two Sunflower Species. [DOI] [PMC free article] [PubMed]
- Roesti, M. , Hendry, A. P. , Salzburger, W. , & Berner, D. (2012). Genome divergence during evolutionary diversification as revealed in replicate lake‐stream stickleback population pairs. Molecular Ecology, 21(12), 2852–2862. https://doi.org/10.1111/j.1365-294x.2012.05509.x [DOI] [PubMed] [Google Scholar]
- Service, M. W . (1993). Mosquito ecology: Field sampling methods. London: UK Elsevier Applied Science, Ed. [Google Scholar]
- Sharakhova, M. V. , Peery, A. , Antonio‐Nkondjio, C. , Xia, A. , Ndo, C. , Awono‐Ambene, P. , … Sharakhov, I. V. (2013). Cytogenetic analysis of Anopheles ovengensis revealed high structural divergence of chromosomes in the Anopheles nili group. Infection, Genetics and Evolution, 16, 341–348. https://doi.org/10.1016/j.meegid.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinka, M. E. , Bangs, M. J. , Manguin, S. , Coetzee, M. , Mbogo, C. M. , Hemingway, J. , … Hay, S. I. (2010). The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: Occurrence data, distribution maps and bionomic précis. Parasites & Vectors, 3(1), 117 https://doi.org/10.1186/1756-3305-3-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S. A. , Curry, R. L. , White, T. A. , Ferretti, V. , & Lovette, I. (2014). Spatiotemporally consistent genomic signatures of reproductive isolation in a moving hybrid zone. Evolution, 68(11), 3066–3081. https://doi.org/10.1111/evo.12510 [DOI] [PubMed] [Google Scholar]
- Turner, T. L. , Hahn, M. W. , & Nuzhdin, S. V. (2005). Genomic islands of speciation in Anopheles gambiae. PLoS Biology, 3(9), 1572–1578. https://doi.org/10.1371/journal.pbio.0030285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortel, W. , Harbach, R. E. , Trung, H. D. , Roelants, P. , Backeljau, T. , & Coosemans, M. (2001). Confirmation of Anopheles varuna in vietnam, previously misidentified and mistargeted as the malaria vector Anopheles minimus, The American Journal of Tropical Medicine and Hygiene, 65(6), 729–732. [DOI] [PubMed] [Google Scholar]
- Weng, D. , Yu, Y. , Hahn, M. W. , & Nakhleh, L. (2016). Reticulate evolutionary history and extensive introgression in mosquito species revealed by phylogenetic network analysis. Molecular Ecology, 25(11), 2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . (2016). World malaria report 2016. Geneva: WHO; [Google Scholar]
- Wright, S . (1984). Evolution and the genetics of populations: Genetics and biometric foundations v. 1. Chicago, IL: University of Chicago; 480 pp. ISBN‐13: 978‐0226910383. [Google Scholar]
- Wu, C. (2001). The genic view of the process of speciation. Journal of Evolutionary Biology, 14, 851–865. [Google Scholar]
- Wu, T. D. , & Nacu, S. (2010). Fast and SNP‐tolerant detection of complex variants and splicing in short reads. Bioinformatics, 26(7), 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials