

Abstract
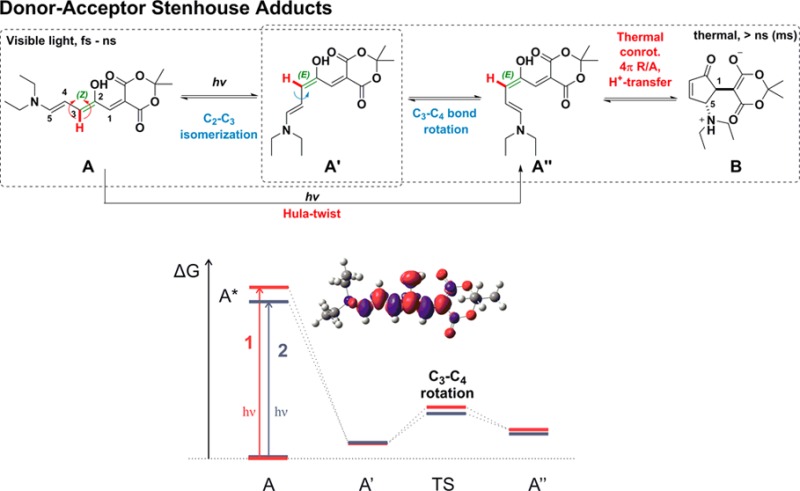
Donor–acceptor Stenhouse adducts (DASAs) are negative photochromes that hold great promise for a variety of applications. Key to optimizing their switching properties is a detailed understanding of the photoswitching mechanism, which, as yet, is absent. Here we characterize the actinic step of DASA-photoswitching and its key intermediate, which was studied using a combination of ultrafast visible and IR pump–probe spectroscopies and TD-DFT calculations. Comparison of the time-resolved IR spectra with DFT computations allowed to unambiguously identify the structure of the intermediate, confirming that light absorption induces a sequential reaction path in which a Z–E photoisomerization of C2–C3 is followed by a rotation around C3–C4 and a subsequent thermal cyclization step. First and second-generation DASAs share a common photoisomerization mechanism in chlorinated solvents with notable differences in kinetics and lifetimes of the excited states. The photogenerated intermediate of the second-generation DASA was photo-accumulated at low temperature and probed with time-resolved spectroscopy, demonstrating the photoreversibility of the isomerization process. Taken together, these results provide a detailed picture of the DASA isomerization pathway on a molecular level.
Molecular photoswitches1 allow reversible optical control in a plethora of applications.2−7 Each photoswitch has its own favorable and adverse properties that ultimately determine how well it is suited for photoregulating a particular responsive system. Highly desirable properties include switching with visible light8,9 and negative photochromism.10
Donor–acceptor Stenhouse adducts (Figure 1a) are an emerging class of photoswitches.11,12 They are particularly attractive due to their modular nature and rapid synthesis and undergo a large structural change upon photoswitching with visible light. Theoretical studies have provided a first effort to rationalize their photoswitching characteristics.13,14 Structural improvements of these adducts have led to a second generation of DASAs.15,16 Successful applications of DASAs have already emerged for smart materials,17−22 sensors,23,24 catalysis,25 and drug-carriers.11,26 Although such applications are promising and highlight the potential of DASAs, the understanding of their photoswitching behavior is far from complete.
Figure 1.
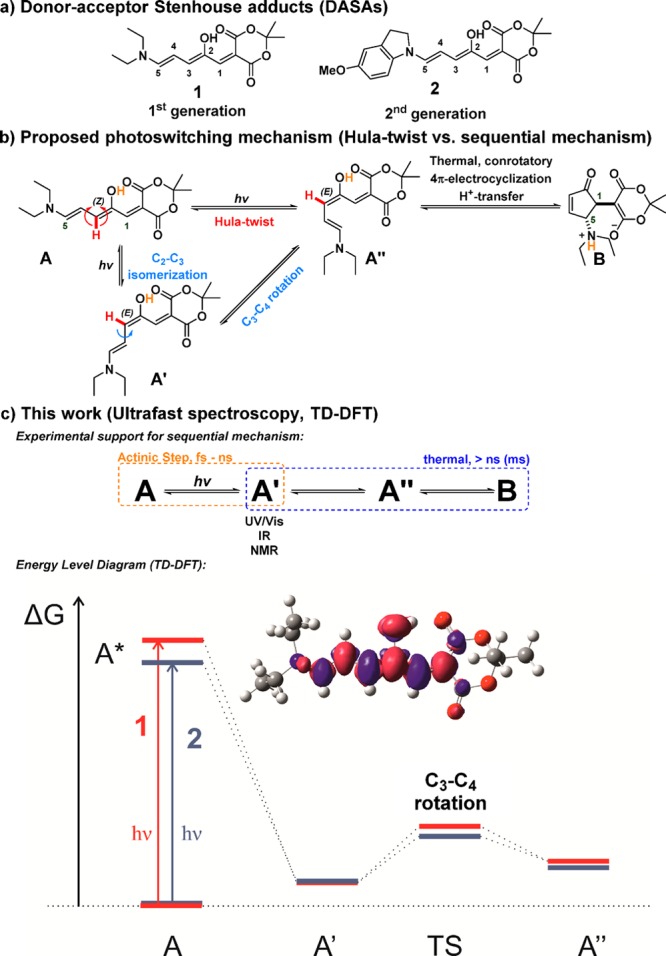
Photoswitching of DASAs: (a) photoswitches used in this study; (b) refined mechanistic proposal; and (c) findings of this work. The red (blue) relative energy levels correspond to DASA 1 (2) (see SI sections 6.1 and 6.3). In the electron density differences plot, the blue (red) regions correspond to decrease (increase) in electron density upon electronic transition for DASA 1.
Initial insights into the DASA photoswitching mechanism stem from the observation of an intermediate, presenting a bathochromically shifted transient absorption band in the UV/vis spectrum with respect to the main absorption band upon light irradiation.27 Time- and temperature-dependent studies revealed a strong dependence on temperature and light intensity, and suggested that this intermediate results from a photoinduced Z–E isomerization followed by a thermal conrotatory 4π-electrocyclization (Figure 1b).27 This observation suggests the separation of the mechanism into an actinic step and a thermal step. Intermediate A′′ (Figure 1b) is needed for successful cyclization, but a simple Z–E isomerization leads only to intermediate A′. Intermediate A′′ could result from a photoisomerization around C2–C3 followed by a rotation around C3–C4 or from a concerted Hula-twist movement as commonly observed in confined spaces such as protein cavities.28
Time-resolved spectroscopy in both the UV/vis and IR regions has proven very successful in elucidating structural changes occurring on the femto-/picosecond time scale29,30 and in characterizing the electronic properties of short-lived reaction intermediates. Here we report in-depth studies on the photoswitching mechanism of DASAs using ultrafast visible and mid-IR spectroscopy, elucidating the time scale of the photoinduced isomerization process and the structure of the intermediate resulting from the actinic step. This intermediate was trapped and manipulated at low temperature and could be structurally confirmed to be A′. Comparison between the experimental transient infrared and DFT-computed spectra supports a sequential photoswitching mechanism (Figure 1b,c), showing that in the analyzed solvents (chloroform/dichloromethane) isomerization between the elongated triene form A and the twisted intermediate form A′′ does not occur through a hula-twist mechanism. Furthermore, we provide evidence that—even though the same intermediate is formed for both first- and second-generation DASAs—the second generation isomerizes at least 10 times slower.
Figure 2 reports the evolution-associated difference spectra (EADS) obtained by global analysis31 of visible pump–probe data recorded for DASA 1 and 2 in chloroform (SI section 4). DASA 1 does not cyclize in chlorinated solvents, while 2 undergoes a reversible cyclization reaction to form a neutral cyclic form reminiscent of B (SI section 3).15,16 Similar spectra with comparable EADS lifetimes are obtained for 1 in toluene, where reversible cyclization occurs (SI Figure S4.1).
Figure 2.
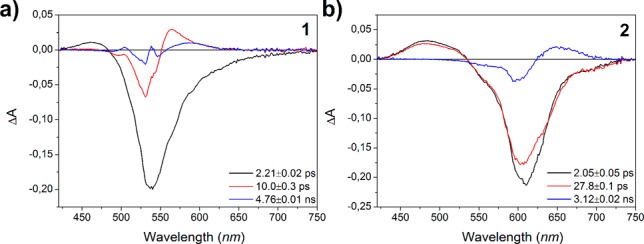
Evolution-associated difference spectra obtained from global analysis31 of time-resolved visible data recorded for 1 (a) and 2 (b) in chloroform.
Upon photoexcitation, bleaching of the ground-state absorption (Figure 2, black lines) is observed as a negative signal peaking at 537 nm for 1 and 610 nm for 2. Importantly, this short living component exhibits a blue-shifted excited-state absorption, peaking at 460 and 482 nm for 1 and 2, respectively. At long pump–probe delays, the appearance of a positive band (Figure 2, blue lines), red-shifted compared to the bleaching signal, identifies the formation of a photogenerated intermediate corresponding to the transient absorption band previously identified for 1.27 For sample 1, the A → A′ photoreaction happens on a 2 ps time scale as indicated by the appearance of a positive band peaking at 567 nm in the second EADS (red EADS in Figure 2a), while both the ground-state bleaching of A and its excited-state absorption band, peaked at 460 nm, significantly decrease. On the following 10 ps time scale, the induced absorption band associated with the photoproduct is subject to a spectral evolution due to a vibrational cooling process in the ground state of the non-isomerized form A (vide infra). In case of sample 2 (Figure 2b) the 2 ps component is mostly associated with an excited-state relaxation of A, while the appearance of the intermediate band (peaked at 649 nm) occurs on a longer 28 ps time scale (red to blue curve evolution in Figure 2b). The quantum yields for the intermediate formation estimated from the residual bleaching signal in transient absorption spectra are ∼10% for 1 and ∼17% for 2.
In order to get detailed insight into the conformation of the intermediate, visible-pump/Mid-IR probe spectra were measured for both samples in the 1100–1750 cm–1 spectral range. Figure 3 shows the EADS obtained by global analysis31 of the time-resolved infrared (TRIR) spectra for sample 1 and 2, together with their steady-state FTIR spectra, which identify the vibrational bands of the elongated form. The time constants obtained from the analysis of TRIR spectra match those obtained from the visible pump–probe experiments except for the long time scale. This is due to the larger error on the long time constant in the TRIR measurements, for which a smaller acquisition time window was used.
Figure 3.
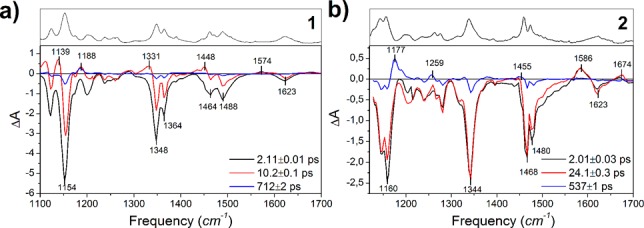
EADS obtained from global analysis31 of the TRIR spectra of 1 (a) and 2 (b) in chloroform. The black spectrum at the top of each panel is the FTIR spectrum of the linear A form.
The long-living component (Figure 3, blue lines) allows to identify the structural changes that occur during the formation of the intermediate. For both samples, major spectral changes occur in the 1150 and 1450 cm–1 region, mainly associated with triene chain C—C/C=C stretching and C—H rocking/scissoring vibrations according to our DFT calculations. In particular, for the long-lived spectral component (Figure 3, blue lines), negative/positive bands are present at 1154/1188 cm–1 for 1 and at 1160/1170 cm–1 for 2. A differential band pattern in the same regions has been previously observed in different molecules undergoing cis/trans photoisomerization such as rhodopsin32,33 and the photoactive yellow protein.34 This pattern strongly suggests that a similar process occurs in the present compounds. We have compared the long living experimental TRIR spectral component with computed IR difference spectra obtained at the B3LYP/6-31++G(d,p)/SMD level35−38 (SI section 6) for isomerizations leading to both A′ and A′′. For both samples the best match between computed and experimental spectra is obtained for the A → A′ transition (Figure 4). We thus conclude that for both DASAs, photoexcitation leads to the formation of the A′ photoproduct, which, in turn, excludes the hula-twist mechanism.
Figure 4.
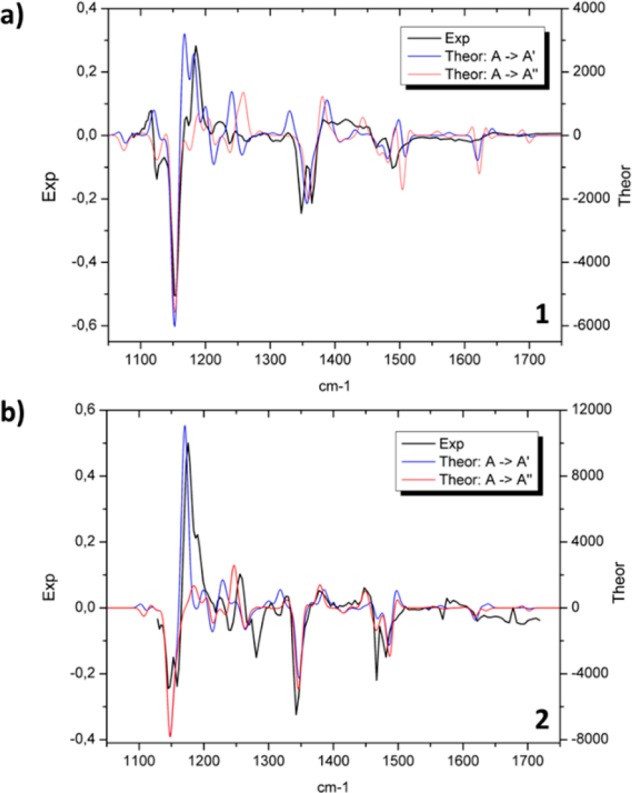
Comparison of the experimental and DFT-computed long-lasting IR spectral component of 1 (a) and 2 (b) in chloroform. The conversion of A → A′ (blue) and A → A′′ (hula-twist, red) are depicted.
The TRIR spectra confirm the time scale of photoisomerization: the intermediate bands in the 1100–1200 cm–1 fingerprint region appear on a 2 ps time scale for 1 and on a 24 ps time scale for 2. In case of 1 additional positive bands appear in the second EADS, as for instance at 1139 and 1331 cm–1. These signals decay in the following 10 ps evolution, suggesting that they are not attributable to A′ but possibly to the hot ground state of the A species. Target analysis31 of both the visible and IR transient data of 1 assuming a branched decay of A* in both A′ and the hot ground state of A successfully disentangles the individual spectra of these two species (SI section 4.4). In contrast, the initial (vibrational) relaxation of 2 occurs on the A* excited-state surface. Notably, in the first spectral component of both samples, assigned to the A* – A difference spectrum, only a few low intensity positive bands are noticed, indicating much weaker IR absorption in A*. The most prominent band shifts in the initial EADS are in the 1600 cm–1 region, attributable to carbonyl stretch vibrations according to the DFT calculations.
Besides the analysis of the IR spectra, our calculations of relative energies, electron density differences (EDD) and geometrical changes upon excitation provide additional information supporting the proposed mechanism. The EDD plots (Figure 1c and SI Figure S6.9) show that for both samples the electron density changes upon excitation are localized on the π-conjugated linker. In the excited-state electron density flows toward the carbon atom functionalized by an OH group, resulting in decreased electron density on “double” bonds. Upon excitation, the structure remains planar (SI Figure S6.10), but the bond lengths increase by ca. 0.01–0.02 Å along the chain, except for the central C3–C4 bond, in line with the EDD analysis. The experimental and theoretical red shift of 2 vs 1 in the excitation energy for A → A* (Figure 2 and SI Table S6.2) is related to an extension of the π-conjugation to the indoline moiety. The energy level diagram also reveals that the A′ → A′′ barrier is ca. 2 kcal/mol smaller for 2, suggesting faster kinetics for the C3–C4 bond rotation step, which is related to a less distorted TS structure (difference of ca. 5°) for this derivative.
Finally, we photo-accumulated intermediate A′ of compound 2 at 233 K under continuous illumination at 520 nm in deuterated dichloromethane (SI section 5). Irradiation at 660 nm resulted in back-switching to the elongated A species as assessed by TRIR spectra (SI Figure S5.2), suggesting that reversible isomerization can be induced by selectively pumping A or A′, while ring-closure to form B is only possible via thermal pathway.
The electrocyclization time scale is several orders of magnitude longer than the actinic step, thus being the rate-determining step as tentatively concluded before.27 Our study identifies key factors for improving switching characteristics, for instance, increasing the photochemical quantum yield by optimizing A′ properties and disfavor the reverse isomerization process. It also suggests that the reason why first-generation DASAs do not cyclize in chloroform or dichloromethane, as opposed to second-generation DASAs, is solely a question of energy levels and barriers involved in the thermally induced 4π-electrocyclization, and is not due to the actinic step. The presented results elucidate the time scale of the actinic step and bode well for implementation of photoswitch improvement.
Acknowledgments
The authors gratefully acknowledge financial support from Laserlab-Europe (LENS002289), the Ministry of Education, Culture and Science (Gravitation program 024.001.035), The Netherlands Organization for Scientific Research (NWO-CW, Top grant to B.L.F., VIDI grant no. 723.014.001 for W.S.), the European Research Council (Advanced Investigator Grant, no. 227897 to B.L.F.), and the Royal Netherlands Academy of Arts and Sciences Science (KNAW). A.D.L., D.J., and M.M. thank the Campus France and the Slovak Research and Development Agency for supporting their long-standing collaboration in the framework of Stefanik PHC program (BridgET project, No. 35646SE and SK-FR-2015-0003, respectively), the Czech Science Foundation (project no. 16-01618S), and the Ministry of Education, Youth and Sports of the Czech Republic (grant LO1305). This research used computational resources of the GENCI-CINES/IDRIS, Centre de Calcul Intensif des Pays de Loire (CCIPL), a local Troy cluster, and the HPCC of the Matej Bel University in Banska Bystrica by using the infrastructure acquired in projects ITMS 26230120002 and 26210120002 supported by the Research and Development Operational Programme funded by the ERDF. The Swiss Study Foundation is acknowledged for a fellowship to M.M.L.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09081.
Experimental procedures and characterization of compounds; additional UV/vis absorption spectra and photoswitching studies; ultrafast visible and mid-IR spectroscopy; evolution associated difference spectra obtained by global analysis; species associated spectra obtained by target analysis and DFT computations (PDF)
Author Contributions
○ M.D.D. and M.M.L. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Molecular Switches, 2nd ed.; Feringa B. L., Browne W. R., Eds.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Erbas-Cakmak S.; Leigh D. A.; McTernan C. T.; Nussbaumer A. L. Chem. Rev. 2015, 115, 10081. 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natali M.; Giordani S. Chem. Soc. Rev. 2012, 41, 4010. 10.1039/c2cs35015g. [DOI] [PubMed] [Google Scholar]
- Russew M. M.; Hecht S. Adv. Mater. 2010, 22, 3348. 10.1002/adma.200904102. [DOI] [PubMed] [Google Scholar]
- Tian H.; Zhang J.. Photochromic Materials: Preparation, Properties and Applications; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Angew. Chem., Int. Ed. 2016, 55, 10978. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- Broichhagen J.; Frank J. A.; Trauner D. Acc. Chem. Res. 2015, 48, 1947. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Acc. Chem. Res. 2015, 48, 2662. 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Hecht S. Angew. Chem., Int. Ed. 2015, 54, 11338. 10.1002/anie.201500628. [DOI] [PubMed] [Google Scholar]
- Barachevsky V. A. Rev. J. Chem. 2017, 7, 334. 10.1134/S2079978017030013. [DOI] [Google Scholar]
- Helmy S.; Leibfarth F. A.; Oh S.; Poelma J. E.; Hawker C. J.; De Alaniz J. R. J. Am. Chem. Soc. 2014, 136, 8169. 10.1021/ja503016b. [DOI] [PubMed] [Google Scholar]
- Helmy S.; Oh S.; Leibfarth F. A.; Hawker C. J.; Read De Alaniz J. J. Org. Chem. 2014, 79, 11316. 10.1021/jo502206g. [DOI] [PubMed] [Google Scholar]
- Laurent A. D.; Medved’ M.; Jacquemin D. ChemPhysChem 2016, 17, 1846. 10.1002/cphc.201600041. [DOI] [PubMed] [Google Scholar]
- Belhboub A.; Boucher F.; Jacquemin D. J. Mater. Chem. C 2017, 5, 1624. 10.1039/C6TC05297E. [DOI] [Google Scholar]
- Hemmer J. R.; Poelma S. O.; Treat N.; Page Z. A.; Dolinski N. D.; Diaz Y. J.; Tomlinson W.; Clark K. D.; Hooper J. P.; Hawker C.; Read De Alaniz J. J. Am. Chem. Soc. 2016, 138, 13960. 10.1021/jacs.6b07434. [DOI] [PubMed] [Google Scholar]
- Mallo N.; Brown P. T.; Iranmanesh H.; MacDonald T. S. C.; Teusner M. J.; Harper J. B.; Ball G. E.; Beves J. E. Chem. Commun. 2016, 52, 13576. 10.1039/C6CC08079K. [DOI] [PubMed] [Google Scholar]
- Singh S.; Friedel K.; Himmerlich M.; Lei Y.; Schlingloff G.; Schober A. ACS Macro Lett. 2015, 4, 1273. 10.1021/acsmacrolett.5b00653. [DOI] [PubMed] [Google Scholar]
- Sinawang G.; Wu B.; Wang J.; Li S.; He Y. Macromol. Chem. Phys. 2016, 217, 2409. 10.1002/macp.201600351. [DOI] [Google Scholar]
- Mason B. P.; Whittaker M.; Hemmer J.; Arora S.; Harper A.; Alnemrat S.; McEachen A.; Helmy S.; Read De Alaniz J.; Hooper J. P. Appl. Phys. Lett. 2016, 108, 041906. 10.1063/1.4940750. [DOI] [Google Scholar]
- Jia S.; Du J. D.; Hawley A.; Fong W. K.; Graham B.; Boyd B. J. Langmuir 2017, 33, 2215. 10.1021/acs.langmuir.6b03726. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Hansen M. J.; Velema W. A.; Szymanski W.; Feringa B. L. Nat. Commun. 2016, 7, 12054. 10.1038/ncomms12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F.-Y.; Hou J.-N.; Liang K.-X.; Liu Y.; Deng L.; Liu Y.-N. New J. Chem. 2017, 41, 6071. 10.1039/C7NJ00815E. [DOI] [Google Scholar]
- Balamurugan A.; Lee H. Il Macromolecules 2016, 49, 2568. 10.1021/acs.macromol.6b00309. [DOI] [Google Scholar]
- Diaz Y. J.; Page Z. A.; Knight A. S.; Treat N. J.; Hemmer J. R.; Hawker C. J.; Read de Alaniz J. Chem. - Eur. J. 2017, 23, 3562. 10.1002/chem.201700368. [DOI] [PubMed] [Google Scholar]
- Helmy S.; Read de Alaniz J. Adv. Heterocycl. Chem. 2015, 117, 131. 10.1016/bs.aihch.2015.05.003. [DOI] [Google Scholar]
- Poelma S. O.; Oh S. S.; Helmy S.; Knight A. S.; Burnett G. L.; Soh H. T.; Hawker C. J.; Read de Alaniz J. Chem. Commun. 2016, 52, 10525. 10.1039/C6CC04127B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch M. M.; Wezenberg S. J.; Szymanski W.; Feringa B. L. J. Am. Chem. Soc. 2016, 138, 6344. 10.1021/jacs.6b01722. [DOI] [PubMed] [Google Scholar]
- Liu R. S. H. Acc. Chem. Res. 2001, 34, 555. 10.1021/ar000165c. [DOI] [PubMed] [Google Scholar]
- Nibbering E. T. J.; Fidder H.; Pines E. Annu. Rev. Phys. Chem. 2005, 56, 337. 10.1146/annurev.physchem.56.092503.141314. [DOI] [PubMed] [Google Scholar]
- Berera R.; van Grondelle R.; Kennis J. T. M. Photosynth. Res. 2009, 101, 105. 10.1007/s11120-009-9454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stokkum I. H. M.; Larsen D. S.; Van Grondelle R. Biochim. Biophys. Acta, Bioenerg. 2004, 1657, 82. 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Herbst J. Science 2002, 297, 822. 10.1126/science.1072144. [DOI] [PubMed] [Google Scholar]
- Kukura P. Science 2005, 310, 1006. 10.1126/science.1118379. [DOI] [PubMed] [Google Scholar]
- van Wilderen L. J. G. W.; van der Horst M. A.; van Stokkum I. H. M.; Hellingwerf K. J.; van Grondelle R.; Groot M. L. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15050. 10.1073/pnas.0603476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. 10.1063/1.464913. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623. 10.1021/j100096a001. [DOI] [Google Scholar]
- Ditchfield R.; Hehre W. J.; Pople J. A. J. Chem. Phys. 1971, 54, 724. 10.1063/1.1674902. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.