

Abstract
Background and Purpose
Valvular heart disease (VHD) is highly prevalent in industrialized countries. Chronic use of anorexigens, amphetamine or ergot derivatives targeting the 5‐HT system is associated with VHD. Here, we investigated the contribution of 5‐HT receptors in a model of valve degeneration induced by nordexfenfluramine, the main metabolite of the anorexigens, dexfenfluramine and benfluorex.
Experimental Approach
Nordexfenfluramine was infused chronically (28 days) in mice ((WT and transgenic Htr 2B ‐/‐, Htr 2A ‐/‐ , and Htr 2B/2A ‐/‐) to induce mitral valve lesions. Bone marrow transplantation was also carried out. Haemodynamics were measured with echocardiography; tissues and cells were analysed by histology, immunocytochemistry, flow cytometry and RT –qPCR. Samples of human prolapsed mitral valves were also analysed.
Key Results
Chronic treatment of mice with nordexfenfluramine activated 5‐HT2B receptors and increased valve thickness and cell density in a thick extracellular matrix, mimicking early steps of mitral valve remodelling. Lesions were prevented by 5‐HT2A or 5‐HT2B receptor antagonists and in transgenic Htr 2B −/− or Htr 2A/2B −/− mice. Surprisingly, valve lesions were mainly formed by numerous non‐proliferative CD34+ endothelial progenitors. These progenitors originated from bone marrow (BM) as revealed by BM transplantation. The initial steps of mitral valve remodelling involved mobilization of BM‐derived CD34+CD31+ cells by 5‐HT2B receptor stimulation. Analysis of human prolapsed mitral valves showing spontaneous degenerative lesions, demonstrated the presence of non‐proliferating CD34+/CD309+/NOS3+ endothelial progenitors expressing 5‐HT2B receptors.
Conclusions and Implications
BM‐derived endothelial progenitor cells make a crucial contribution to the remodelling of mitral valve tissue. Our data describe a new and important mechanism underlying human VHD.
Abbreviations
- 5‐HIAA
5‐hydroxyindoleacetic acid
- BM
bone marrow
- NOS3
endothelial NO synthase
- pCPA
para‐chlorophenylalanine
- VHD
valvular heart disease
- WT
wild type
Introduction
Several drugs, such as ergot derivatives (pergolide, cabergoline and ergotamine) (Van Camp et al., 2004; Zanettini et al., 2007) and anorectic compounds (fenfluramine and dexfenfluramine) (Connolly et al., 1997), have been associated with remodelling of the left cardiac (mitral and aortic) valves. These drugs interact with the serotonergic system by, more precisely, targeting the 5‐HT2 receptor subtypes (5‐HT2B and 5‐HT2A) (Fitzgerald et al., 2000; Rothman et al., 2000), which belong to the superfamily of Gq‐protein‐coupled receptors that activate phospholipase C. There is much evidence for the involvement of 5‐HT2 receptors in drug‐induced valvular heart disease (VHD). Stimulation of 5‐HT2 receptors leads to up‐regulation of target genes involved in proliferation and stimulation of valvular interstitial cells (VICs) through activation of protein kinase C, Src‐protein, phosphorylation of ERK1/2 and TGF‐β receptor activation (Jian et al., 2002; Xu et al., 2002; Hutcheson et al., 2012). Phosphorylated ERK is thought to induce TGF‐β signalling and transcription of effector genes mediating myxomatous pathology (Disatian and Orton, 2009). Overexpression of TGF‐β1 has also been implicated in cardiac diseases where fibrosis is a prominent feature. The activation of 5‐HT2B receptors in human VICs is mitogenic, resulting in ERK1/2 phosphorylation and incorporation of [3H]‐deoxythymidine (Setola et al., 2003) and has also been implicated in 5‐HT‐induced valvulopathy in experimental animals (Elangbam et al., 2008). The 5‐HT2B receptor appears enriched in heart valves from various species including dog (Oyama and Chittur, 2006; Lu et al., 2015; Cremer et al., 2015a), rat (Elangbam et al., 2005), pig (Fitzgerald et al., 2000; (Cremer et al., 2015b) and human (Fitzgerald et al., 2000), mainly in healthy tissues. Nevertheless, 5‐HT receptors other than the 5‐HT2B subtype have been reported in valve tissue (Lu et al., 2015; Cremer et al., 2015b), and the involvement of the 5‐HT transporter (SERT) in VHD has been suggested by the development of valvular fibrosis in SERT knockout (KO) mice (Mekontso‐Dessap et al., 2006). Moreover, the well‐known ‘carcinoid heart’, a valvulopathy associated with excess of plasma (free) 5‐HT secreted by neuroendocrine tumours, highlights the important role of this biogenic amine in VHD (Lundin et al., 1988). Although earlier work has linked 5‐HT2 receptors to VHD, these links are not as strong as they may appear. For instance, many experiments were performed with healthy tissues (Fitzgerald et al., 2000; Setola et al., 2003), making difficult the extrapolation to pathological mechanisms. Stimulation with 5‐HT was performed with high concentrations (≥106 M) over a long period (Jian et al., 2002; Xu et al., 2002; Elangbam et al., 2008) and 5‐HT2 receptor agonists applied in vitro to valve tissues induced relatively small effects (Barzilla et al., 2010). Taking all these limitations into consideration, we hypothesized that the 5‐HT2 receptor stimulation involved in VHD could take place in sites other than the valve itself and that receptors other than the 5‐HT2B subtype could also be involved.
Cardiac valves are made of valvular endothelial (VECs) and interstitial cells (VICs). Both cell types are required for the homeostatic maintenance of valve tissue, which is exposed to high haemodynamic constraints. These two cell populations are heterogeneous. VICs can be found quiescent with a fibroblast phenotype or activated with various markers typical of myofibroblasts, stem cells and osteoblasts (Liu et al., 2007). Progenitor VICs were also observed in response to injury, but their origin and the mechanism underlying the repair are not yet known (Liu et al., 2007). Some VECs are able to synthesize extracellular cell matrix (ECM) and to populate and assemble all three valve layers. Valve endothelium is absolutely required to renew the population of VICs from endothelial progenitors (Paruchuri et al., 2006). Nevertheless, the origin of these last cells and the mechanism(s) by which they reach the valve is also not known. Here, we have tested the possibility that 5‐HT stimulation could be involved in VHD through the mobilization of endothelial progenitors in cardiac valve tissue. In fact, some authors described rapid migration of bone marrow (BM)‐derived cells in mitral valve tissue after engraftment of fluorescent cells in wild‐type (WT) mice (Visconti et al., 2006) showing, in basal homeostatic conditions, the recruitment of precursor cells to the valvular tissue. These cells could constitute part of a so‐called ‘VECs reservoir’ of endothelial progenitors that would then transdifferentiate via the general process of endothelial to mesenchymal transition (Paranya et al., 2001; Paruchuri et al., 2006). In the first approach of this study, using pharmacological and transgenic mice experiments, we investigated the contribution of 5‐HT2B receptors in drug‐induced valvulopathy, in terms of endothelial progenitor mobilization. In the second part, the association between mitral valve prolapse and activation of platelets (Walsh et al., 1981; Martini et al., 1996), as the main cellular store of 5‐HT in the periphery, led us to investigate if similar mechanisms could be involved in human VHD.
Methods
Animals
All animal care and experimental procedures were performed in accordance with the guidelines for animal experimentation of the European Communities Council Directive EU/63/2010 and appropriate approval with the authorization number AL/81/88/02/13. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Studies were performed in 12‐week‐old male mice. WT 129S2/SvPasCrl mice were purchased from Charles River Laboratories (L'Arbresle, France). Htr 2A −/− mice (González‐Maeso et al., 2003) were obtained from Dr René Hen's laboratory, and Htr 2B −/− (Nebigil et al., 2000) and Htr 2A/2B −/− mice were obtained from Dr Luc Maroteaux's laboratory and left on a pure 129S2/SvPasCrl background.
Experimental procedures
To induce valvulopathy, mice were treated with a 28‐day‐long infusion of nordexfenfluramine (NdF; 1 mg·kg−1·day−1) delivered by micro‐osmotic pumps (model 1004, Alzet, Cupertino, CA, USA), implanted s.c. under isoflurane anaesthesia (Aerrane, Baxter, Meyzieu, France). Seven groups of WT mice were prepared: (i) vehicle; (ii) NdF; (iii) NdF + the 5‐HT2 receptor antagonist, ritanserin (2 mg·kg−1·day−1); (iv) NdF + the selective 5‐HT2A receptor antagonist, sarpogrelate (2 mg·kg−1·day−1); (v) NdF + the selective 5‐HT2B receptor antagonist, SB204741 (1 mg·kg−1·day−1); (vi) NdF + the 5‐HT2B/2C receptor antagonist, SB206553 (1 mg·kg−1·day−1); and (vii) NdF + the non‐selective tryptophan hydroxylase inhibitor, para‐chlorophenylalanine (pCPA) (100 mg·kg−1·day−1). Ritanserin dose and administration route were validated by a platelet aggregation test (Supporting Information Figure S1). Ritanserin and pCPA were added to food pellets. Sarpogrelate, SB204741 and SB206553 were infused by the mean of a second osmotic pump. Food and water intake were controlled together with body weight to adjust p.o. delivered drugs (Supporting Information Figure S2).
Six groups of transgenic mice were studied: (i) Htr 2A −/− vehicle, (ii) Htr 2A −/− NdF, (iii) Htr 2B −/− vehicle, (iv) Htr 2B −/− NdF, (v) Htr 2A/2B −/− vehicle and (vi) Htr 2A/2B −/− NdF. All groups were monitored weekly for systolic BP and heart rate (HR) by tail‐cuff photoplethysmography (BP‐2000, Visitech, Apex, NC, USA) (Supporting Information Figure S4). Transthoracic echocardiography was performed 1 day before the beginning of the experiments (day −1) and after 28 days of treatment (in WT and NdF mice groups, Table 1). At day 27, urinary 5‐hydroxyindoleacetic acid (5‐HIAA) was measured from 0.5 to 1.0 mL urine collected in a tube protected against light and containing a drop of 5 N HCl in mice housed individually in metabolic cages overnight (18:00 to 08:00 h). At day 28, at the end of the last echocardiography and during isoflurane anaesthesia, 0.5 mL blood was collected by intracardiac puncture, before giving a lethal i.p. injection of pentobarbital (150 mg·kg−1, CEVA Santé Animale, Libourne, France). Hearts harvested from mice were immediately rinsed, weighed (Supporting Information Figure S3) and fixed with 10% formalin solution before being processed.
Table 1.
Effects of NdF on haemodynamic and morphometric parameters in WT mice
| Parameters | Day 1 | Day 28 | ||||
|---|---|---|---|---|---|---|
| WT‐C (n = 10) | WT‐NdF (n = 10) | P value | WT‐C (n = 10) | WT‐NdF (n = 10) | P value | |
| BW (mg) | 30 ± 0.6 | 28 ± 0.6 | ns | 30 ± 0.9 | 29 ± 0.9 | ns |
| HW/BW (mg·g−1) | ‐ | ‐ | ‐ | 5.38 ± 0.07 | 5.42 ± 0.09 | ns |
| SAP (mmHg) | 96 ± 2.5 | 101 ± 2 | ns | 123 ± 1.5 | 126 ± 2 | ns |
| HR (bpm) | 480 ± 20 | 441 ± 10 | ns | 575 ± 17 | 570 ± 13 | ns |
| PWT (mm) | 0.76 ± 0.02 | 0.77 ± 0.02 | ns | 0.73 ± 0.02 | 0.65 ± 0.04 | ns |
| SWT (mm) | 0.79 ± 0.02 | 0.78 ± 0.02 | ns | 0.78 ± 0.02 | 0.67 ± 0.03 | ns |
| LV mass (mg) | 103 ± 8 | 97 ± 4 | ns | 103 ± 8 | 103 ± 9 | ns |
| LV mass index (mg·g−1) | 3.43 ± 0.20 | 3.56 ± 0.23 | ns | 3.33 ± 0.29 | 3.76 ± 0.13 | ns |
| EDLVD (mm) | 3.77 ± 0.19 | 3.68 ± 0.14 | ns | 4.10 ± 0.09 | 4.28 ± 0.08 | ns |
| ESLVD (mm) | 2.22 ± 0.16 | 2.55 ± 0.11 | ns | 2.23 ± 0.11 | 2.5 ± 0.11 | ns |
| SF (%) | 38 ± 2.5 | 40 ± 2 | ns | 38 ± 1.5 | 42 ± 1.5 | ns |
| CO (mL·min−1) | 50 ± 5.5 | 46 ± 5.5 | ns | 55 ± 3.5 | 53 ± 2.5 | ns |
Values are means ± SEM, '‐ ' represents undefined parameters. n, number per group; ns, not significant, p >0.05, unpaired Student's t‐test; BW, body weight; HW, heart weight; SAP, systolic arterial pressure; HR, heart rate; PWT, posterior wall thickness; SWT, septum wall thickness; LV, left ventricle; EDLVD, end diastolic left‐ventricular diameter; ESLVD, end systolic left‐ventricular diameter; SF, shortening fraction; CO, cardiac output.
Biochemical measurement
Whole blood 5‐HT and urinary 5‐HIAA concentrations were obtained by HPLC (Plateau Technique de Biologie, Nouvel Hôpital Civil, Strasbourg, France).
Echocardiography
Animals were analysed for cardiac anatomy and function on a Sonos 5500 (Hewlett Packard, Palo Alto, CA, USA) with a 15 MHz linear transducer (15L6). All the examinations were performed in mice anaesthetized with 1–1.5% isoflurane. The heart was first imaged in the two‐dimensional (2D) mode in the parasternal long‐axis view to obtain the aortic root dimensions. The aortic flow velocity and the HR were measured with pulsed‐wave Doppler on the same section. The cardiac output (CO) was calculated from the following equation: CO = 0.785 × D2 × VTI × HR, where D is the diameter of the aortic root and VTI is the velocity‐time integral of the Doppler aortic spectrum. Left ventricular cross‐sectional internal diameters in end‐diastole (EDLVD) and end‐systole (ESLVD) were obtained by an M‐mode analysis of a 2D short‐axis view at the papillary muscle level. The shortening fraction was calculated as SF = (EDLVD − ESLVD∕EDLVD) × 100. From this view, the diastolic septum (S) and posterior wall (PW) thicknesses were measured. The left ventricular mass (LVM) was calculated with the following formula: LVM = 1.055 × [(S + PW + EDLVD)3 − (EDLVD)3]. All the measurements were performed on, at least three beats, according to the guidelines of the American Society of Echocardiography.
Histology and quantification of mitral valve lesions
Fixed mouse hearts were paraffin embedded, sectioned at 4 μm (using HM 355S Automatic Microtome) and stained with haematoxylin and eosin. Sections were performed so as to obtain four‐chamber views of the heart with long‐axis section of the tricuspid, mitral and aortic leaflets. A single operator (R.L.), blinded to the experimental groups, performed valve morphometric analysis with a microscope (Carl Zeiss, Jena, Germany) equipped with a 40× calibrated objective giving; at this magnification, a large square of 250 × 250 μm divided in 100 equal parts of 25 × 25 μm each. NdF induced non‐reproducible lesions of the aortic valve and no alteration of the tricuspid one in our conditions. Therefore, we decided to focus on the mitral valve. For quantification, the whole mitral valve leaflet section was divided in three equal segments: proximal (near the insertion), medial and distal (including the tip). The thickness of each segment is the mean value obtained in three distinct sites. The surface cell density was determined on the whole segment (mean value of three distinct sites), and the result expressed in number of cells per 0.01 mm2.
Immunohistochemistry was performed on paraffin‐embedded samples with anti‐Ki‐67 (Mib1, 1/100, USBiological, Salem, MA, USA) and anti‐CD34 (EP373Y, 1/100, Genetex, Irvine, CA, USA) antibodies.
Bone marrow (BM) transplantation
As previously described, thirteen 8‐week‐old WT male mice were subjected to 9.5 Gray lethal total body irradiation (Potteaux et al., 2006). The following day, mouse BM was reconstituted by direct i.v. injection with 2.5 × 106 cells of freshly isolated BM from femurs and tibias of age and sex‐matched WT or Htr 2B −/− mice. All lethally irradiated and transplanted mice survived, revealing the efficiency of BM reconstitution by either WT (n = 6) or Htr 2B −/− (n = 7) BM. After 4 weeks of recovery, transplanted mice were then exposed to NdF and followed the same protocol applied to other groups.
Blood mobilization of progenitors and bone marrow analysis
To analyse the effects of NdF on the mobilization of progenitor cells in the blood and BM, WT control (WT‐C) and Htr 2B −/− mice received a single 3 mg·kg−1 NdF s.c. injection. They were compared with vehicle‐treated animals. Blood was collected, by intracardiac puncture in animals anaesthetized with sodium pentobarbital (40 mg·kg−1 i.p.), for flow cytometric analysis (LSRII, H48700015; software BD FACSDiva 6.1.2) with the following antibodies: CD31 (130‐102‐571, Miltenyi, Bergisch Gladbach, Germany) and CD34 (553930, BD Pharmingen, Le Pont de Claix, France). Blood was treated with a solution of 0.83% NH4Cl to induce erythrocytes lysis and then washed before labelling. BM cells were isolated from tibias and femurs, filtered, treated with a solution of 0.83% NH4Cl to induce erythrocytes lysis and labelled with the following antibodies: CD31 (130‐102‐571, Miltenyi, Bergisch Gladbach, Germany), CD34 (553930, BD Pharmingen, Le Pont de Claix, France) and phalloidin (P1951, Sigma‐Aldrich, Lyon, France), for flow cytometric analysis (LSRII, H48700015; software BD FACSDiva 6.1.2). Phalloidin analysis was evaluated by FACS analysis on a CANTOII BD. Analysis was performed on the FlowJo® software.
Human mitral valve tissues
Eighteen human degenerative heart valves were obtained from patients referred to the Department of Cardiovascular Surgery, University Hospitals of Strasbourg, France, for a cardiac valve replacement (mitral valve prolapsed diagnosis). All patients authorized the subsequent use of their valve tissues for research purpose and the protocol was approved by the Ethics Committee of the Faculty of Medicine of Strasbourg (6 December 2011).
Valve histology and immunohistochemistry analysis
Human mitral valve tissues (n = 4) were fixed with formalin solution for 2 days and embedded in paraffin; 4 μm slices were stained with Alcian blue and haematoxylin and eosin to identify valve lesions and evaluate cellular density. Immunostaining was performed to localize 5‐HT2B receptors (A72‐1, BD Pharmingen, BD Biosciences, Le Pont de Claix, France) and CD34 progenitors (QBEnd 10, Dako®, Glostrup, Denmark). Finally, Ki‐67 was used as a marker of cell proliferation (MIB‐1, Dako, Santa Clara, CA, USA). Immunostaining was performed using a biotin‐labelled peroxidase‐conjugated secondary antibody incubated with diaminobenzidinetetrahydrochloride as a final chromogen (Ultraview DAB Detection Kit, Roche®, Tucson, Arizona, USA). Labelling control was performed by incubating only the secondary antibody. Cell count was performed with a microscope (Carl Zeiss, Jena, Germany) equipped with a 40× calibrated objective giving, at this magnification, a large square of 250 × 250 μm divided in 100 equal parts of 25 × 25 μm each. The surface cell density was determined at the endothelium, sub‐endothelium and spongiosa levels, and the results expressed in number of cells per 0.01 mm2. To quantify immunostained cells, results were expressed as a percentage of positive cells compared with total cells per 0.01 mm2 (5‐HT2B receptor, CD34 and Ki‐67). The number of total or labelled cells in valve lesions is the mean value obtained in three distinct sites per valve.
Mitral valvular cells isolation and flow cytometry and magnetic cell sorting
To remove cells from mitral valve tissues, specimens were immersed in a collagenase solution (NB8 Broad range 1 mg·mL−1; Serva, Coger, Paris, France) for 2 h at 37°C. Tissue pieces were scrubbed through a 70 μm strainer, and cell suspension was filtered to remove debris. In a first set of experiments, using eight mitral valves, single‐cell suspensions were labelled with CD34‐Alexa Fluor 647‐conjugated antibody (ICO115, Santa Cruz; Dallas, Texas, USA) and CD31‐FITC‐conjugated antibody (MEM‐05, Exbio, Vestec, Czech Republic), before cell sorting on a BD FACSAria® II flow cytometer. In a second set of experiments, single‐cell suspension was stained with a CD34 antibody conjugated to magnetic beads [magnetic cell sorting (MACS)] and passed on a midi column (CD34 MicroBead Kit UltraPure human, Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's protocol (three mitral valves were used). This technology was used to select and purify CD34+ cells only, representing more than 90% of the whole cell population after MACS procedure. These cells were characterized by FACS using a double labelling with CD34‐FITC‐conjugated (CD34, Miltenyi Biotec, Bergisch Gladbach, Germany) and CD309‐PE‐conjugated (VEGFR2, BD Pharmingen, Le Pont de Claix, France) antibodies. Acquisition was carried out on a BD LSR flow cytometer (BD BiosciencesLe Pont de Claix, France). Percentages shown in all figures are per cent of single cells fell within gates determined using a negative control (isotype specific IgG) stained cell population. Analysis was performed on the FlowJo software.
RNA extraction and gene expression by RT‐qPCR on selected valve cell populations
Total mRNA was extracted using Tri Reagent (Life Technologies, Grand Island, NY, USA) according to the manufacturer's recommended protocol. All RNA samples were initially quantified via spectrophotometer. RNA samples were converted to cDNAs using Kit iScript cDNA Synthesis BioRad®, Hercules, CA, USA. Semi‐quantitative RT‐PCR was produced on an amplification system (Light Cycler® 2.0 Instrument, Penzberg, Germany) with the kit Light Cycler Fast Start DNA Master Plus SYBR Green I® (Roche Diagnostics, Mannheim, Germany). Forward and reverse primer sequences for all analysed genes were obtained by Qiagen® (Hs_HTR2A_1_SG/Hs_HTR2B_1_SG/Hs_NOS3_1_SG, Hs_RRN18S_1_SG, Qiagen, Courtaboeuf, France).
Immunocytochemical staining
To identify the expression of 5‐HT2B receptors in endothelial cells, the whole cell suspension obtained after human mitral valve collagenase treatment was stained with antibodies against the 5‐HT2B receptor and some other surface markers (n = 3 mitral valves). Briefly, cells were plated and cultured overnight at 37°C, 5% CO2 with DMEM/F12 (1:1) medium containing antibiotics (100 U·mL−1 penicillin and 0.1 mg·mL−1 streptomycin). Cells were then fixed with 4% paraformaldehyde and incubated with a rabbit anti‐human CD34 antibody (PA1334, Boster Biological Technology, Fremont, CA, USA) and a mouse anti‐human 5‐HT2B receptor antibody (BD Pharmingen, BD Biosciences, Le Pont de Chaix, France) in 1% BSA solution. Cells were then incubated with fluorescent‐tagged secondary antibodies, goat anti‐rabbit antibody – Alexa Fluor® 488 (AP132JA4, Millipore, Darmstadt, Germany) and goat anti‐mouse antibody‐Cy3 (AP124C, Millipore, Darmstadt, Germany) and finally covered with a Dako Fluorescence mounting medium (S3023, Dako, Carpinteria, CA, USA). Nuclei were stained with the Hoechst 33342 dye (H1399, Invitrogen, Paisley, United Kingdom). Double positive cells (CD34–5‐HT2B receptor) (average of five fields per valve) were counted by bright field and UV illumination.
Data analysis and statistics
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). For data from mice, values are expressed as mean ± SEM. Statistical comparisons between two or more groups were performed when appropriate using Student's unpaired t‐tests or one‐way ANOVA followed by post hoc analysis with Dunnett's or Fisher's least significant difference test (GraphPad Software version 6.0). P < 0.05 was considered statistically significant.
For data from the human samples, values are expressed as mean ± SEM. Statistical comparisons between two or more groups were performed when appropriate using Student's unpaired t‐tests or non‐parametric tests (Kruskal–Wallis) (GraphPad Software version 6.0). P < 0.05 was considered statistically significant.
Materials
All drugs and chemicals used in these experiments were purchased from Sigma‐Aldrich (Saint‐Quentin‐Fallavier, France), unless otherwise indicated.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/2016 (Alexander et al., 2015a,b,c).
Results
Chronic administration of nordexfenfluramine induces mitral valve remodelling
Infusion of NdF for 28 days did not modify body weight, systolic arterial BP, HR and echocardiographic parameters (Table 1 and Supporting Information Figures S2–S4). In particular, no cardiac remodelling and/or systolic dysfunction was observed. Whole blood 5‐HT and urinary 5‐HIAA assays revealed an increase in 5‐HT turnover with a twofold augmentation of 5‐HIAA, while blood 5‐HT concentration remained unchanged (Table 2). WT‐C mice presented a homogenous and thin valve architecture bordered by regular endothelial CD31+ lining (data not shown) and only few CD34+ cells (Figure 1F). Conversely, we observed segmental thickened (84%) and retracted leaflets in WT‐NdF animals (Figure 1A, B) with increased endothelial cell number bordering the valves (Figure 1A, C). Concerning interstitial cell density, no significant change was observed between WT‐NdF and WT‐C (Figure 1A, D). Finally, clusters of rounded shape cells positive for the CD34 antigen were observed (Figure 1F). However, similarly to the WT‐C group, in WT‐NdF mitral valves, only few nuclei were Ki‐67 positive indicating a low rate of proliferation (Figure 1E).
Table 2.
Blood 5‐HT and urinary 5‐HIAA in WT mice
| WT‐C (n = 9) | WT‐NdF (n = 10) | WT‐S (n = 9) | WT‐SB204 (n = 10) | WT‐SB206 (n = 10) | WT‐R (n = 10) | WT‐P (n = 9) | |
|---|---|---|---|---|---|---|---|
| Blood 5‐HT (μg·L−1) | 1930 ± 162 | 1801 ± 81 | 1646 ± 163 | 2160 ± 93 | 2213 ± 89 | 1877 ± 76 | 596 ± 48 * , # |
| Urinary 5‐HIAA (μmol·L−1) | 36 ± 7 | 64 ± 5 * | 39 ± 4 # | 46 ± 6 | 75 ± 10 * | 44 ± 5 | 16 ± 3 # |
Values are means ± SEM. Seven groups of WT mice: WT mice infused with vehicle (WT‐C), or treated for 28 days with NdF (WT‐NdF), or NdF with sarpogrelate (WT‐S), or NdF with SB204741 (WT‐SB204), or NdF with SB206553 (WT‐SB206), or NdF with ritanserin (WT‐R), and NdF with pCPA (WT‐P).
P < 0.05, significantly different from WT‐C; ANOVA and post hoc Dunnett's multiple comparison test.
P < 0.05, significantly different from WT‐NdF; ANOVA and post hoc Dunnett's multiple comparison test.
Figure 1.
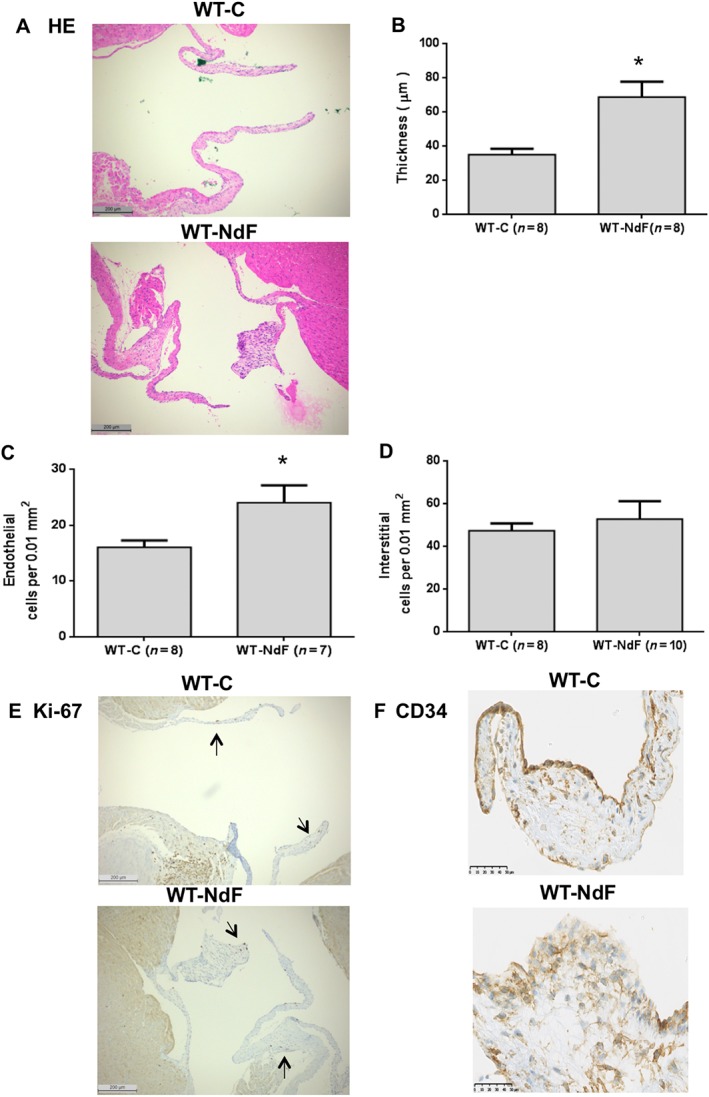
Chronic administration of NdF induces mitral valve remodelling in mice. Mitral valve architecture was assessed in WT‐C mice or treated for 28 days with NdF (1 mg·kg−1·day−1). (A) Haematoxylin and eosin (HE)‐stained slides were used to quantify (B) thickness, (C) endothelial cellularity and (D) interstitial cellularity in WT‐C and WT‐NdF (using the same grid‐line reticule) (scale bar = 200 μm). Immunohistochemical staining (scale bar = 50 μm) for (E) Ki‐67 and (F) CD34; arrows indicate positive nuclei for Ki‐67. Values are means ± SEM (n = number per group). * P < 0.05, significantly different from control; unpaired Student's t‐test.
Involvement of 5‐HT2A and 5‐HT2B receptors but not 5‐HT in NdF‐induced valvulopathy
Two sets of experiments using pharmacological antagonists and KO mice were used to investigate the role of 5‐HT2A and 5‐HT2B receptors. Haemodynamic and physical parameters of these experimental groups are presented in Supporting Information Figures S2–S4. To test the contribution of the 5‐HT2A receptor, we evaluated NdF effects in WT mice co‐treated with sarpogrelate, a selective 5‐HT2A receptor antagonist, or in Htr 2A −/− mice. Sarpogrelate treatment prevented the NdF‐induced increase in urinary 5‐HIAA and blood 5‐HT levels (Table 2). No difference was observed regarding blood 5‐HT and urine 5‐HIAA in NdF‐treated Htr 2A −/− mice (Table 3). Sarpogrelate treatment also prevented valve injuries induced by NdF (Figure 2). However, we detected a significant increase thickness with Htr 2A −/− mice under NdF treatment (31%) (Figure 3B, G). To test the contribution of 5‐HT2B receptors, we evaluated NdF effects in WT mice co‐treated with SB204741 or SB206553, two 5‐HT2B receptor antagonists or in Htr 2B −/− mice. In WT mice, NdF‐induced urine 5‐HIAA increase was prevented by SB204741 co‐treatment, but not blocked by SB206553. Blood 5‐HT concentration was not statistically different from WT‐NdF group (Table 2). Htr 2B −/− mice had higher basal urinary 5‐HIAA, which was not affected by NdF treatment (Table 3). SB204741 and SB206553 treatments prevented valve injuries induced by NdF (Figure 2). Furthermore, no lesion was detectable in Htr 2B −/− mice under NdF treatment (Figure 3C, D, H, K, N). To test the contribution to lesions of both 5‐HT2B and 5‐HT2A receptor blockade, we evaluated NdF effects in WT mice co‐treated with ritanserin, a non‐selective murine 5‐HT2 receptor antagonist or in Htr 2A/2B −/− double mutants. NdF‐induced urine 5‐HIAA increase was blocked by ritanserin co‐treatment. Peripheral blood 5‐HT concentration was not statistically different (Table 2). Htr 2A/2B −/− double mutant mice had a twofold increase of urine 5‐HIAA under NdF treatment and significantly lower blood 5‐HT compared with their control group (Table 3). Histological lesions were fully prevented by ritanserin (Figure 2). Histological and morphometric analysis of Htr 2A/2B −/− mice mitral valve did not reveal any lesion (Figure 3E, F, I, L, O). These results confirm the role of both 5‐HT2A and 5‐HT2B receptors in valve remodelling with a major contribution of the 5‐HT2B receptor subtype, based on the increased thickness of valve tissue observed in Htr 2A −/− mice.
Table 3.
Blood 5‐HT and urinary 5‐HIAA in transgenic mice
| Htr 2A −/− | Htr 2B −/− | Htr 2A/2B −/− | ||||
|---|---|---|---|---|---|---|
| C (n = 9) | NdF (n = 11) | C (n = 7) | NdF (n = 9) | C (n = 10) | NdF (n = 9) | |
| Blood 5‐HT (μg·L−1) | 1750 ± 69 | 1774 ± 102 | 1656 ± 109 | 1834 ± 79 | 2144 ± 53 | 1795 ± 75 * |
| Urinary 5‐HIAA (μmol·L−1) | 52 ± 4 | 51 ± 7 | 74 ± 7 | 57 ± 6 | 43 ± 4 | 85 ± 5 * |
Values are means ± SEM. Six groups of transgenic mice (Htr 2A −/−, Htr 2B −/− and Htr 2A/2B −/−): transgenic mice infused with vehicle (‐C), or treated for 28 days with NdF (‐NdF).
P < 0.05, significantly different from corresponding vehicle value; unpaired Student's t‐test.
Figure 2.
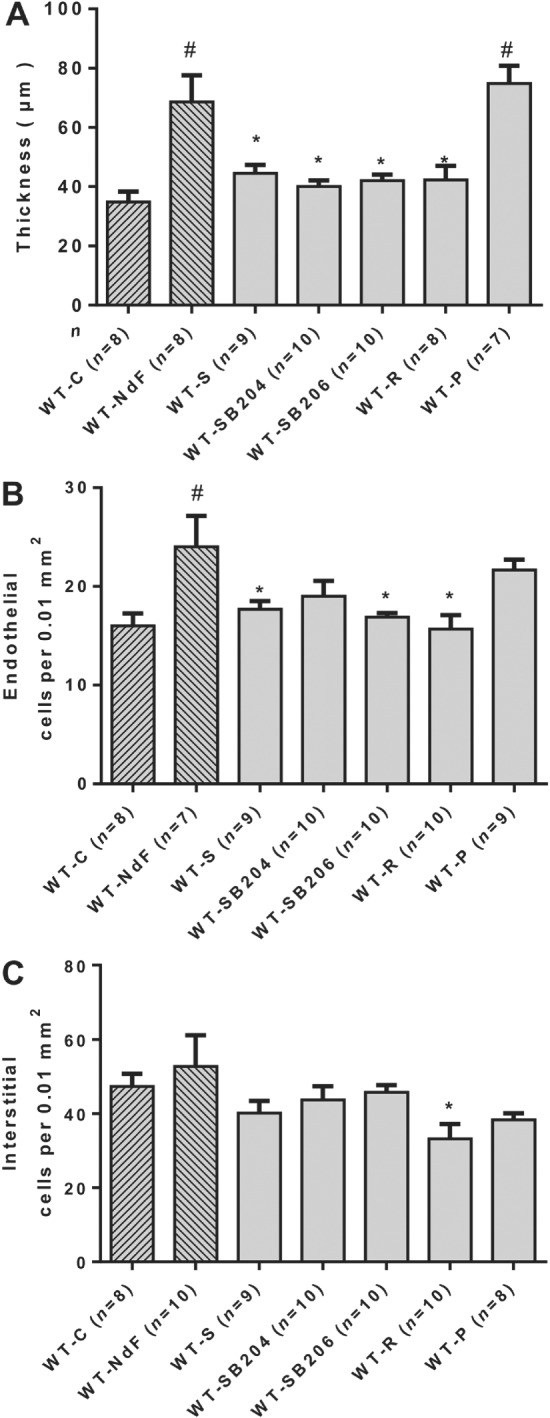
Effect of 5‐HT receptor blockade in WT mitral valves treated with NdF in vivo. Mitral valve architecture was determined in NdF‐treated mice in the presence of various 5‐HT receptor blockers. (A) Thickness, (B) endothelial cells and (C) interstitial cells numbers in WT mice treated for 28 days with NdF (1 mg·kg−1·day−1) (WT‐NdF), or NdF with sarpogrelate (2 mg·kg−1·day−1) (WT‐S), or NdF with SB204741 (1 mg·kg−1·day−1) (WT‐SB204), or NdF with SB206553 (1 mg·kg−1·day−1) (WT‐SB206), or NdF with ritanserin (2 mg·kg−1·day−1) (WT‐R), and NdF with pCPA (100 mg·kg−1·day−1) (WT‐P). Values are means ± SEM (n = number of animals per group). # P < 0.05, significantly different from WT‐C; * P < 0.05, significantly different from WT‐NdF; ANOVA and Dunnett's post hoc tests.
Figure 3.
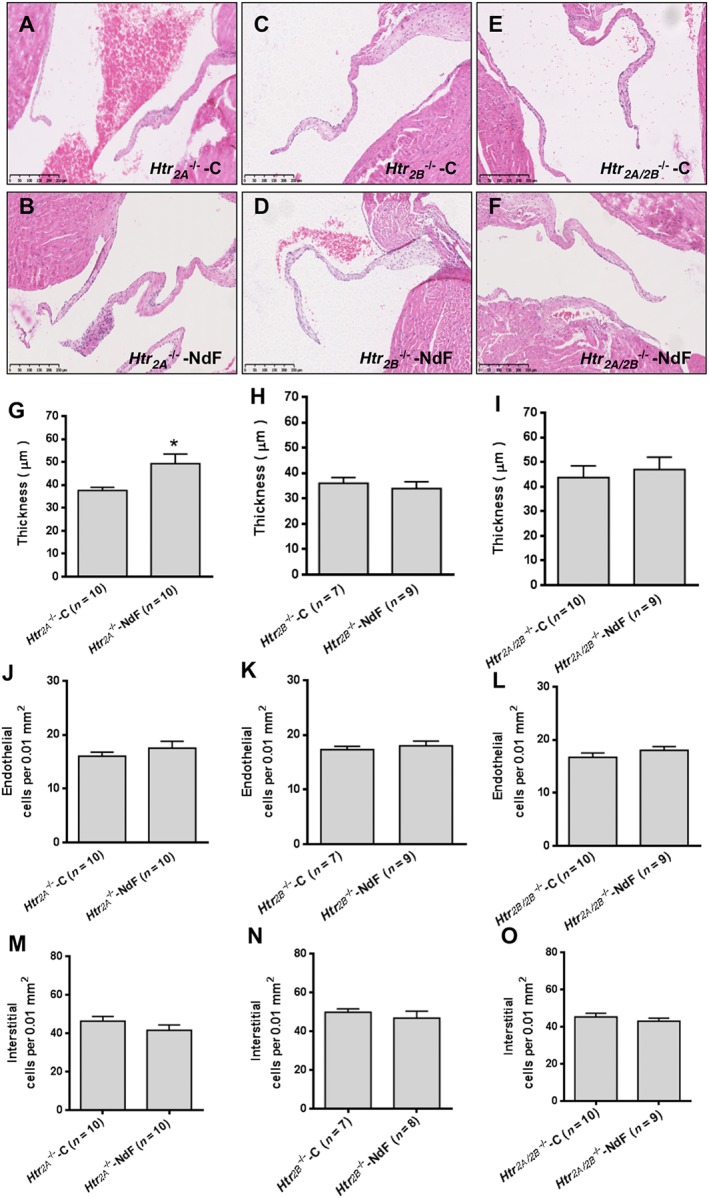
Effects of NdF in Htr 2A −/−, Htr 2B −/− and Htr 2A/2B −/− mice. (A) to (F) correspond to typical histological aspects of the different Htr 2 KO mice lines (Htr 2A −/−, Htr 2B −/− and Htr 2A/2B −/−) (controls and treated for 28 days with NdF (1 mg·kg−1·day−1) stained with haematoxylin and eosin (scale bar = 250 μm); (G), (H) and (I) represent measured thickness; (J), (K) and (L) represent endothelial cells count; and (M), (N) and (O) represent counted interstitial cells count. Values are means ± SEM (n = number of animals per group). Statistical significant difference by unpaired t‐test versus KO control is indicated by * P < 0.05, significantly different from the corresponding KO control; unpaired Student's t‐test.
Taking into account that NdF induces 5‐HT release by platelets through its interaction with the SERT, we investigated whether the levels of free 5‐HT contributes to NdF‐induced valve remodelling. We induced a peripheral 5‐HT depletion by treating mice with 100 mg·kg−1·day−1 of pCPA, a tryptophan hydroxylase inhibitor. Chronic pCPA markedly decreased blood 5‐HT (3×) and urinary 5‐HIAA (Table 2). Nevertheless, this massive depletion failed to prevent the remodelling of valve leaflets induced by NdF (Figure 2). This result indicates that increased peripheral 5‐HT is not necessary for NdF‐induced mitral valve injuries and that the direct 5‐HT2B receptor stimulation by NdF is sufficient to trigger valve remodelling.
Taking into account that mitral valve lesions show a low rate of proliferation but are highly cellularized (CD34+ cells), we hypothesized that these cells could be recruited at the valve surface from the blood stream after being mobilized from the BM.
5‐HT2B receptors are involved in the mobilization of endothelial progenitors from the bone marrow
To test this hypothesis, we compared the effect of a chronic NdF treatment in lethally irradiated WT mice engrafted with a Htr 2B −/− BM with the effects of a similar treatment in WT mice engrafted with a WT BM. Mice transplanted with WT BM exhibited histological lesions with a significant increase of the thickness (28.4%) and endothelial cell density in the mitral valves (Figure 4A, C). By contrast, histological lesions were totally absent from mice that received BM from Htr 2B −/− mice (Figure 4B, C). Therefore, the ablation of 5‐HT2B receptors, restricted to BM, prevents the increase in thickness and endothelial cellularity of mitral valves, supporting the contribution of 5‐HT2B receptors to the mobilization of BM‐derived endothelial progenitors.
Figure 4.
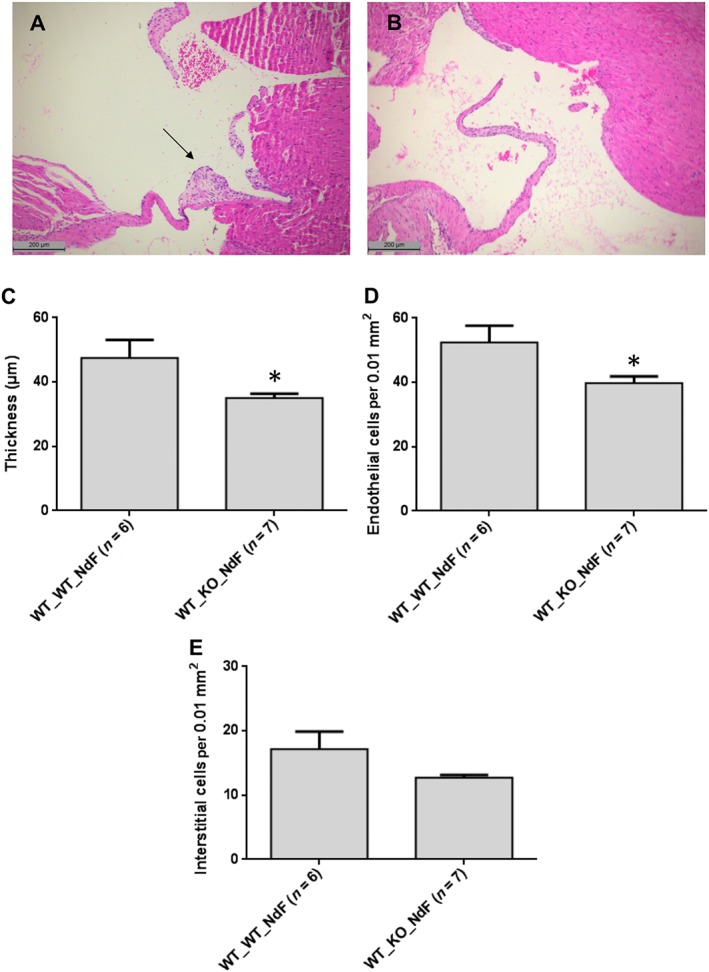
Mice with ablation of 5‐HT2B receptors, restricted to the BM, are resistant to NdF‐induced valvulopathy. Reduced mitral valve thickness and endothelial cellularity after chronic NdF administration (1 mg·kg−1·day−1, during 28 days, s.c.) in lethally irradiated mice transplanted with BM cells from Htr 2B −/− (B: WT_KO_NdF) compared with mice transplanted with WT BM (A: WT_WT_NdF) (scale bar = 200 μm); arrow indicates characteristic valvular lesions. Mitral valve thickness (C), endothelial (D) and interstitial cellularity (E) of transplanted mice exposed to NdF. Values are means ± SEM (n = number of animals per group). *P < 0.05, significantly different from mice transplanted with WT BM; unpaired Student's t‐test.
Whole‐body irradiation probably affects the repopulation of the BM in non‐haematopoietic cells leading to a reduction of the BM mobilization capabilities of endothelial progenitors in WT_WT mice.
Stimulation of BM 5‐HT2B receptors by NdF mobilizes CD34+/CD31+ cells into the blood
To investigate whether 5‐HT2B receptor stimulation affects the mobilization of BM‐derived endothelial progenitors, we measured CD34+/CD31+ cells by FACS, 15 min after a single NdF (3 mg·kg−1) injection. In the BM of WT mice, the single NdF injection induced, a significant increase in the number of free (i.e. released from the ECM) CD34+/CD31+ cells (Figure 5A), which was associated with an increase in actin polymerization in these cells, as shown by phalloidin labelling (Figure 5B). Simultaneously, a significant increase in circulating CD34+/CD31+ cells was observed in the blood of these animals (Figure 5C). Conversely, NdF did not produce these events in Htr 2B −/− mice. This reduction of migration capabilities is probably at the origin of the higher number of these cells found in the BM in basal conditions. These data support the idea that NdF induces endothelial progenitors mobilization by a 5‐HT2B receptor‐dependent mechanism and that NdF triggers the acute mobilization of CD34+/CD31+ cells from BM into the blood. These cells then home into target tissues that require homeostatic repair.
Figure 5.

Stimulation of 5‐HT2B receptors triggers the mobilization of CD34+/CD31+ cells from BM into the blood. After a single s.c. NdF injection (3 mg·kg−1), the number of free CD34+/CD31+ cells (corresponding to endothelial progenitors, flushed from the femur and tibia and not sequestered in osteoblastic niches), increased significantly after 15 min in the BM of WT mice, but not in the BM of Htr 2B −/− mice, as shown by the quantification in per cent of total BM cells (A). In the BM of WT mice, following the NdF injection, an increase in actin polymerization was observed in CD34+/CD31+ cells, whereas no change was noticed in cells of Htr 2B −/− mice (B). Simultaneously, in the WT mice, the single NdF injection induced a significant increase in endothelial progenitors, whereas the amount of these progenitors in the blood of Htr 2B −/− mice did not change (C). These data suggest that endothelial progenitors mobilization by NdF is a 5‐HT2B receptor‐dependent mechanism and originates from the BM. Values are means ± SEM (n = number per group). non‐parametric Mann–Whitney U‐test is shown by * P < 0.05 and # P < 0.05, significantly different from WT‐C. NS, not significant.
In the second part of this work, we assessed the possibility of a similar mechanism, triggered by endogenous 5‐HT, being involved in spontaneous human mitral valve degeneration.
Identification of CD34+ cells sharing the 5‐HT2B receptor in human mitral valve lesions
We analysed mitral valve tissue obtained from four patients with a mitral valve prolapse, a common acquired disorder (Figure 6). Mitral valve leaflets show typical fibromyxoid lesions with high cellular density at the valve surface (37.5 ± 6.8 cells per 0.01 mm2), sub‐endothelium area (25.4 ± 3.4 cells per 0.01 mm2), and into the spongiosa layer (26.2 ± 3 cells per 0.01 mm2) (Figure 6). Surprisingly, despite the high cellular density, only few nuclei were Ki‐67 positive (2.6 ± 0.4% in MV) indicating a low rate of cell proliferation in valve lesions (Figure 6D, G). Interestingly, the cellular areas of the sub‐endothelium surface and the spongiosa layer were densely populated with CD34+ cells (48.0 ± 1%) (Figure 6E, G). 5‐HT2B receptors were found not only in endothelial cells (at the valve surface) but also inside the valve lesions (40.2 ± 4.2% of 5‐HT2B receptor‐positive cells) (Figure 6F, G).
Figure 6.

Human mitral valve lesions contain numerous 5‐HT2B‐positive cells and show a low proliferation rate. (A) Alcian blue staining shows severe deposition of glycosaminoglycans (blue) in the spongiosa and disrupted, and disorganized collagen (pink) (scale bar = 500 μm). (B) Haematoxylin and eosin (HE) staining shows cell‐rich areas (scale bar = 500 μm). In valve lesions, immunostaining for Ki‐67 reveals a low rate of proliferation (D); arrows indicate positive nuclei for Ki‐67 (scale bar = 250 μm). Immunostaining for CD34 (E) and 5‐HT2B receptors (F) revealed cells located in the surface and under the surface of valve leaflets (F) (scale bar = 250 μm). (G) Immunostaining for Ki‐67, CD34 and 5‐HT2B receptors were quantified (using the same grid‐line reticle and focusing valvular lesions) and shown as the percentage of cells located in valvular lesions; Values are means ± SEM, obtained in three distinct sites from four human mitral valves. In ( C) the labelling control (CTL) represents same mitral valve tissue processed with the same methods but without primary antibodies.
We hypothesized that the large number of CD34+ cells identified in myxomatous lesions could be precursors of the endothelial lineage. To confirm this hypothesis, we used flow cytometry to profile the distribution of the double surface markers: PECAM1 (CD31) and CD34. After collagenase treatment, 59.8% mitral valve cells were CD34+. Among the CD34+ cell population, only 12% were CD31+ (Figure 7A, B). The CD34/CD31 labelling identified three major cell populations, that is, CD34+/CD31−, CD34+/CD31+ and CD34−/CD31−. In these three populations, we investigated by RT‐qPCR, the expression of mRNA from HTR 2B and HTR 2A and endothelial NO synthase (NOS3). Interestingly, NOS3 mRNA was expressed in all CD34+ but not CD34− cells, supporting the endothelial lineage commitment of CD34+ cells. Moreover, these CD34+ cells expressed both HTR 2A and HTR 2B mRNA subtypes (Figure 7C). In isolated valve cells, using double immunocytochemical staining with anti‐5‐HT2B receptors and anti‐CD34 antibodies, we confirmed at the protein level that all CD34+ cells express 5‐HT2B receptors (Figure 7D). To confirm the endothelial commitment of the CD34+ cell population, we used MACS with a CD34 antibody conjugated to magnetic beads. We obtained a homogenous cell population with more than 95% CD34+ cells. Among this population, the co‐labelling using another anti‐CD34 and an anti‐CD309 (VEGFR2) antibodies revealed about 80% double positive cells in mitral valves (Figure 7E, F). These results confirm the endothelial lineage of the CD34+ valvular cells and support the proposition that endothelial progenitor cells, expressing both 5‐HT2A and 5‐HT2B receptors, contribute to cardiac valve remodelling in human mitral valve prolapse.
Figure 7.

5‐HT2B‐positive cells extracted from human mitral valve lesions are endothelial progenitors. In mitral valve prolapse (n = 8), after collagenase treatment, 60% of single cells sorted by FACS are CD34+(B). Values are means ± SEM, and representative data from one experiment are displayed (A). (C) Expression of HTR 2A, HTR 2B, NOS3 and 18S RNAs isolated from three major valvular cell populations was analysed by RT‐qPCR, showing the expression of HTR 2A and HTR 2B mRNA in CD34+ cells. (D) Double staining of mitral valvular cells (n = 3) with antibodies directed against CD34 (green) and 5‐HT2B receptors (5‐HT2BR – red). The merged pictures show co‐localization of CD34 and 5‐HT2B receptor proteins, and quantification shows about 60% of co‐staining. Size bar = 10 μm. Flow cytometric analysis of CD34 and CD309 expression after magnetic sorting of CD34+ cells isolated from mitral valve prolapse (n = 3), after collagenase treatment. The CD34 and CD309 double positive cells were counted, after MACS isolation (F). Values are means ± SEM, and representative data from one experiment are displayed (E).
Discussion
The present work demonstrates that (i) cardiac mitral valve remodelling occurs after chronic administration of NdF and is characterized by segmental increased valve thickness and endothelial cell density, with no change in interstitial cell number; (ii) lesions display low proliferation (Ki‐67) but numerous CD34+ cells; (iii) both 5‐HT2A and 5‐HT2B receptor blockade by antagonists or in transgenic Htr 2B −/− or Htr 2A/2B −/− mice successfully prevent NdF‐induced valve lesions; (iv) ablation of 5‐HT2B receptors, restricted to the BM, prevents NdF‐induced valve lesions; and (v) upon acute NdF exposure, BM mobilization of CD34+/CD31+ precursors is dependent on 5‐HT2B receptors. Moreover, the analysis of samples of prolapsed human mitral valves, showing spontaneous degenerative lesions, demonstrated the presence of non‐proliferating CD34+/CD309+/NOS3+ endothelial progenitors expressing 5‐HT2B receptors.
5‐HT2B receptors have been implicated in the fibrosis of various organs (Maroteaux et al., 2017). In the liver, genetic suppression or antagonists of 5‐HT2B receptors limited CCL4‐induced fibrogenesis by blocking TGF‐β secretion by stellate cells (Ebrahimkhani et al., 2011). Similarly, in the skin, pharmacological inactivation of 5‐HT2B receptors reduces dermal fibrosis in models where a contribution from platelet 5‐HT is highly likely (Dees et al., 2011). Nevertheless, fibrotic side effects induced by stimulation of 5‐HT2B receptors were more frequently reported in lung and cardiac valve tissues. The hepatic de‐ethylated dexfenfluramine metabolite, NdF, shows a high affinity for the 5‐HT2B receptor (K i = 11.2 ± 4.3 nM), 5‐HT2C receptor (K i = 324 ± 7.1 nM) and a lower affinity towards the 5‐HT2A subtype (K i = 1516 ± 88 nM) (Rothman et al., 2000). Given chronically to mice, this compound induced VHD, comparable to the valve lesions induced by the ergot derivatives. NdF induced non‐reproducible lesions of the aortic valve in this model. Species differences, haemodynamics and/or embryological origin of valve cells could explain some differences related to the response of these two valves to the treatments. Here, we show that these lesions appear as a result of the stimulation, chiefly of 5‐HT2B receptors, with some contribution of 5‐HT2A receptors as demonstrated by antagonist treatments (SB204741, SB206553, sarpogrelate and ritanserin) and transgenic mice experiments (Htr 2B −/− and Htr 2A/2B −/−). These data confirm previous results showing that cyclic stretch in porcine valve cusps increases proliferation and extracellular matrix remodelling, through up‐regulation of the mRNA for 5‐HT2A and 5‐HT2B receptors (Balachandran et al., 2011) and emphasize that both receptors are required for VHD. A low level of increased mitral valve thickness persisted in Htr 2A −/− transgenic mice. Taking into account that the same endothelial cellularity was measured in Htr 2A −/− transgenic mice, with or without treatment with NdF, it is possible that a local 5‐HT2B compensatory overexpression could contribute to collagen synthesis and extracellular matrix remodelling in these particular animals (Balachandran et al., 2012). Some authors have suggested that fenfluramine effects could be, at least in part, mediated by peripheral and central 5‐HT release following direct action on SERT (Rothman et al., 2010). This endogenous, locally released 5‐HT could explain non‐5‐HT2B receptor mediated effects in valve tissue. Nevertheless, in our study, marked depletion of peripheral 5‐HT induced by pCPA failed to prevent remodelling of valve leaflets induced by NdF, indicating that peripheral 5‐HT was not necessary for NdF to induce mitral valve damage and that direct stimulation of 5‐HT2A/2B receptor by NdF is sufficient to trigger valve remodelling.
In this study, we showed, in mice, that the valve lesions contain a large number of CD34+ cells, confirming previous studies showing that haematopoietic stem cells contribute to the fibroblast population of adult valves (Visconti et al., 2006). 5‐HT can promote the differentiation of progenitor cells (Hirota et al., 2014). These data support the idea that 5‐HT through 5‐HT2B receptor stimulation could induce maturation of progenitor cells in the valve tissue and/or their mobilization from the BM followed by their recruitment into the valves. We investigated the last hypothesis in the NdF mouse model.
To confirm the BM origin of progenitor cells and the role of 5‐HT2B receptors, we performed Htr 2B +/+ and Htr 2B −/− BM transplantation in WT mice. The absence of 5‐HT2B receptors in BM prevented NdF‐induced mitral lesions. This result showed the crucial contribution of BM‐derived endothelial progenitors to remodelling of mitral valve tissue and clearly demonstrated the role of 5‐HT2B receptors in this process triggered by 5‐HT receptor agonists. In addition, this work makes a parallel with pulmonary hypertension in which a similar mechanism involving BM progenitors was previously demonstrated (Launay et al., 2012). The contribution of circulating cells originating from the BM to fibrosis has been suggested in tumours (Quante et al., 2011) but also in a non‐tumour tissue such as the liver (Abe et al., 2001; Castilho‐Fernandes et al., 2011). In this last tissue, myofibroblasts originate at least in part from cells coming from the BM (Brenner et al., 2012).
The migration of BM‐derived endothelial progenitors is supported by results from earlier studies. Vaturi et al. (2011) observed a reduction of blood endothelial progenitors in patients with aortic stenosis that was interpreted as a deffect on the repair process. Nevertheless, we could postulate that this decrease is in fact due to the recruitment of these cells in the injured tissue. Similarly, in severe aortic stenosis, blood endothelial progenitors decrease to below the control level (Matsumoto et al., 2009). Stimulation by 5‐HT could favour mobilization and/or recruitment of endothelial progenitors, as reported in accelerated mitral valve bioprosthesis degeneration which was induced by benfluorex, an NdF precursor (Ayme‐Dietrich et al., 2012). The non‐cellularized collagen matrix was rapidly colonized and developed typical myxomatous lesions. In our work, a single NdF injection rapidly triggered the increase of CD34+/CD31+ cells in BM and blood. These cells were released from the BM matrix and migrated after cytoskeleton activation. The NdF‐induced blood mobilization from BM was prevented in Htr 2B −/− mice. Both of these effects involved 5‐HT2B receptors as they were totally absent from Htr 2B −/− mice. Interestingly, BMPR2, a TGF‐β1 high‐affinity receptor, mutations are associated with heritable pulmonary arterial hypertension (Deng et al., 2000; International PPH Consortium et al., 2000). Mice bearing the R899X mutation in the BMPR2 gene (loss of function) develop spontaneous pulmonary hypertension that is prevented by the 5‐HT2B receptor antagonist, SB204741 (West et al., 2016), or by transplantation of WT BM (Yan et al., 2016). Similarly, animals lacking BPMR2 in endothelial progenitors develop a valvulopathy that closely mimics the one we obtained following NdF infusion (Beppu et al., 2009). This common mechanism of 5‐HT2B receptor‐mediated mobilization of endothelial progenitors from BM could explain why valvulopathies are frequently associated with pulmonary hypertension. Therefore, we propose that 5‐HT2B receptors by regulating BMPR2/Src signalling regulates cytoskeletal genes and function in endothelial progenitor cells, leading to their mobilization from the BM and their subsequent recruitment to pulmonary vessels and cardiac valves.
In samples of degenerated valves from humans, 40% of all cells expressed the glycoprotein CD34, a progenitor cell marker. Stem cells (CD34+, CD133+ and CD45+) residing in the BM niche give rise to a population of cells that differentiate into a progenitor subtype (CD34+, CD133+ and VEGFR2+) considered as endothelial (Balaji et al., 2013) or haematopoietic, when they lack vessel‐forming activity (Case et al., 2007). The CD34 transmembrane molecule is expressed by precursors of both endothelial cells and fibroblasts (Lanza et al., 2001). Few reports had described CD34+ labelling in the spongiosa and fibrosa layers of the valves (Barth et al., 2005) and assumed that it corresponds to mesenchymal cells proliferating in the tissue. In the same paper, these authors also analysed 10 so‐called normal mitral valves obtained from 41‐ to 85‐year‐old patients who had died from non‐cardiovascular causes. Immunohistochemistry identified CD34+ cells in the spongiosa. Their photomicrographs also showed a positive staining at the endothelial surface, arguing in favour of a recruitment followed by migration in the spongiosa. So, it is likely that CD34+ cells are present in the ‘normal’ mitral tissue and their number is increased in the pathological state. The low rate of cell proliferation supported that the high cell content of the pathological samples was due to migration instead of local proliferation. Based on CD34 expression, we identified two main cell populations, from collagenase‐treated human mitral valve samples, that is, CD34+/CD31− and CD34−/CD31−. The CD34+/CD31+ cells probably correspond to differentiating endothelial cells (Murohara, 2001). The most prevalent CD34+/CD31− population comprised 60–85% of cells that co‐expressed CD309/VEGFR2 and NOS3, strongly suggesting that they are endothelial cell progenitors (Schatteman et al., 2007). Nevertheless, the definition of an endothelial progenitor is still matter of debate and is used to describe cell types included in the ‘proangiogenic haematopoietic progenitor cells’ family (Asahara et al., 1997). Some of these cells are involved in neoangiogenesis but others lack pro‐angiogenic capabilities (Wara et al., 2011) and would be involved in tissue repair. Our immunohistochemistry experiments revealed α‐SMA+ cells around cushions made of CD34+ cells at sites of myxomatous lesions. In these CD34+ cells, we found expression of 5‐HT2B receptors both at mRNA and protein levels, leading to the conclusion that the degenerated mitral valves from humans, contain a large number of endothelial progenitors which also expressed 5‐HT2B receptors. These cells could transdifferentiate to α‐SMA+ myofibroblasts (Wylie‐Sears et al., 2011).
In conclusion, our present results demonstrate that 5‐HT and 5‐HT2B receptors are involved in early processes of valve remodelling. The cellular mechanism involves mobilization from the BM of endothelial progenitors expressing the 5‐HT2B receptor. These cells are then recruited to the valve where they later undergo differentiation to myofibroblast or other cell types to propagate valve lesions, in an irreversible step. This work opens new fields with potential clinical consequences. First, it strongly suggests that the screening of drugs and their metabolites acting on BM‐derived endothelial progenitors via 5‐HT2B receptors or other targets could be useful to predict valve and pulmonary side effects. Such screening would be done in preclinical models (Whitebread et al., 2016). Second, it opens the way to search for antagonists of the 5‐HT2A and/or 5‐HT2B receptors to protect, reverse or slow down early steps of cardiac valve degeneration, in patients at risk.
Author contributions
E.A.D. performed and analysed the molecular and biochemical experiments of the human part of the study, analysed histological human samples, performed and analysed study of blood mobilization progenitors in mice; R.L. performed mice experimental procedures and treatment, analysed histological mice samples, performed and analysed echocardiography experiments; C.D.T. contributed to mice experimental procedures and treatment; S.D.S analysed histological graft mice samples; C.E. gave technical support for flow cytometry sorting and analysis; B.H. and C.G. provided advice for ritanserin dose using platelets aggregation tests; H.R. and J.S. prepared all humans and mice samples for histological analysis; E.Q. prepared transfected cells for Western blot controls; S.B. prepared two groups of transplanted mice; F.D. and N.F. provided advices for progenitors mobilization; B.G. trained E.A.D. and R.L. for histological analysis and provided advice; J.P.M. provided human resected valve samples; F.C. and O.H. designed, performed and analysed the study of BM progenitors in mice; E.A.D., R.L., L.Ma and L.Mo designed the study and wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Inhibition of platelet aggregation by ritanserin.
Figure S2 Physical parameters.
Figure S3 Heart to body weight ratio from all experimental mice groups.
Figure S4 Haemodynamic parameters in all experimental mice groups.
Acknowledgements
Hugues Jacob and the Imaging Department (Mouse Clinical Institute) are thanked for their technical assistance.
This study was supported by the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, the Paris Université Pierre et Marie Curie, the University of Strasbourg, the Centre Hospitalier et Universitaire de Strasbourg and by grants from the Fondation de France, the French Ministry of Research, Agence Nationale pour la Recherche [ANR‐12‐BSV1‐0015‐01]. L. Maroteaux's team is supported by the Fondation pour la Recherche Médicale ‘Equipe FRM DEQ2014039529’ and is part of the École des Neurosciences de Paris Ile‐de‐France network and of the Bio‐PsyLabex, Investissements d'Avenir programme ANR‐11‐IDEX‐0004‐02). We also acknowledge in this study the private sponsorship of CEVA Santé Animale.
Ayme‐Dietrich, E. , Lawson, R. , Côté, F. , de Tapia, C. , Da Silva, S. , Ebel, C. , Hechler, B. , Gachet, C. , Guyonnet, J. , Rouillard, H. , Stoltz, J. , Quentin, E. , Banas, S. , Daubeuf, F. , Frossard, N. , Gasser, B. , Mazzucotelli, J.‐P. , Hermine, O. , Maroteaux, L. , and Monassier, L. (2017) The role of 5‐HT2B receptors in mitral valvulopathy: bone marrow mobilization of endothelial progenitors. British Journal of Pharmacology, 174: 4123–4139. doi: 10.1111/bph.13981.
References
- Abe R, Donnelly S, Peng T, Bucala R, Metz CN (2001). Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 166: 7556–7562. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G‐protein coupled receptors. Br J Pharmacol 172: 5761–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, Zee R, van der Li T et al (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science 275: 964–966. https://doi.org/10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Ayme‐Dietrich E, Lawson R, Gasser B, Dallemand R, Bischoff N, Monassier L (2012). Mitral bioprosthesis hypertrophic scarring and native aortic valve fibrosis during benfluorex therapy. Fundam Clin Pharmacol 26: 215–218. https://doi.org/10.1111/j.1472‐8206.2012.01027. [DOI] [PubMed] [Google Scholar]
- Balachandran K, Bakay MA, Connolly JM, Zhang X, Yoganathan AP, Levy RJ (2011). Aortic valve cyclic stretch causes increased remodeling activity and enhanced serotonin receptor responsiveness. Ann Thorac Surg 92: 147–153. https://doi.org/10.1016/j.athoracsur.2011.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran K, Hussain S, Yap C‐H, Padala M, Chester AH, Yoganathan AP (2012). Elevated cyclic stretch and serotonin result in altered aortic valve remodeling via a mechanosensitive 5‐HT(2A) receptor‐dependent pathway. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 21: 206–213. https://doi.org/10.1016/j.carpath.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Balaji S, King A, Crombleholme TM, Keswani SG (2013). The role of endothelial progenitor cells in postnatal vasculogenesis: implications for therapeutic neovascularization and wound healing. Adv Wound Care 2: 283–295. https://doi.org/10.1089/wound.2012.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PJ, Köster H, Moosdorf R (2005). CD34+ fibrocytes in normal mitral valves and myxomatous mitral valve degeneration. Pathol Res Pract 201: 301–304. https://doi.org/10.1016/j.prp.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barzilla JE, Acevedo FE, Grande‐Allen KJ (2010). Organ culture as a tool to identify early mechanisms of serotonergic valve disease. J Heart Valve Dis 19: 626–635. [PubMed] [Google Scholar]
- Beppu H, Malhotra R, Beppu Y, Lepore JJ, Parmacek MS, Bloch KD (2009). BMP type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Dev Biol 331: 167 https://doi.org/10.1016/j.ydbio.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DA, Kisseleva T, Scholten D, Paik YH, Iwaisako K, Inokuchi S (2012). Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue repair 5: S17 https://doi.org/10.1186/1755‐1536‐5‐S1‐S17. eCollection 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J, Mead LE, Bessler WK, Prater D, White HA, Saadatzadeh MR et al (2007). Human CD34+AC133+VEGFR‐2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol 35: 1109–1118. https://doi.org/10.1016/j.exphem.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Castilho‐Fernandes A, de Almeida DC, Fontes AM, Melo FU, Picanço‐Castro V, Freitas MC et al (2011). Human hepatic stellate cell line (LX‐2) exhibits characteristics of bone marrow‐derived mesenchymal stem cells. Exp Mol Pathol 91: 664–672. https://doi.org/10.1016/j.yexmp.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD et al (1997). Valvular heart disease associated with fenfluramine‐phentermine. N Engl J Med 337: 581–588. https://doi.org/10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- Cremer SE, Moesgaard SG, Rasmussen CE, Zois NE, Falk T, Reimann MJ et al (2015a). Alpha‐smooth muscle actin and serotonin receptors 2A and 2B in dogs with myxomatous mitral valve disease. Res Vet Sci 100: 197–206. https://doi.org/10.1016/j.rvsc.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Cremer SE, Zois NE, Moesgaard SG, Ravn N, Cirera S, Honge JL et al (2015b). Serotonin markers show altered transcription levels in an experimental pig model of mitral regurgitation. Vet J 203: 192–198. https://doi.org/10.1016/j.tvjl.2014.12.016. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A et al (2011). Platelet‐derived serotonin links vascular disease and tissue fibrosis. J Exp Med 208: 961–972. https://doi.org/10.1084/jem.20101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G et al (2000). Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor‐II gene. Am J Hum Genet 67: 737–744. https://doi.org/10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disatian S, Orton EC (2009). Autocrine serotonin and transforming growth factor beta 1 signaling mediates spontaneous myxomatous mitral valve disease. J Heart Valve Dis 18: 44–51. [PubMed] [Google Scholar]
- Ebrahimkhani MR, Oakley F, Murphy LB, Mann J, Moles A, Perugorria MJ et al (2011). Stimulating healthy tissue regeneration by targeting the 5‐HT2B receptor in chronic liver disease. Nat Med 17: 1668–1673. https://doi.org/10.1038/nm.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elangbam CS, Job LE, Zadrozny LM, Barton JC, Yoon LW, Gates LD et al (2008). 5‐Hydroxytryptamine (5HT)‐induced valvulopathy: compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague‐Dawley rats. Exp Toxicol Pathol 60: 253–262. https://doi.org/10.1016/j.etp.2008.03. 005. [DOI] [PubMed] [Google Scholar]
- Elangbam CS, Lightfoot RM, Yoon LW, Creech DR, Geske RS, Crumbley CW et al (2005). 5‐Hydroxytryptamine (5HT) receptors in the heart valves of cynomolgus monkeys and Sprague‐Dawley rats. J Histochem Cytochem 53: 671–677. https://doi.org/10.1369/jhc.4A6500.2005. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA et al (2000). Possible role of valvular serotonin 5‐HT2B receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol 57: 75–81. [PubMed] [Google Scholar]
- González‐Maeso J, Yuen T, Ebersole BJ, Wurmbach E, Lira A, Zhou M et al (2003). Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5‐hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J Neurosci 23: 8836–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota N, McCuaig S, O'Sullivan MJ, Martin JG (2014). Serotonin augments smooth muscle differentiation of bone marrow stromal cells. Stem Cell Res 12: 599–609. https://doi.org/10.1016/j.scr.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Hutcheson JD, Ryzhova LM, Setola V, Merryman WD (2012). 5‐HT2B antagonism arrests non‐canonical TGF‐β1‐induced valvular myofibroblast differentiation. J Mol Cell Cardiol 53: 707–714. https://doi.org/10.1016/j.yjmcc.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International PPH Consortium , Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA et al (2000). Heterozygous germline mutations in BMPR2, encoding a TGF‐beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81–84. https://doi.org/10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Jian B, Xu J, Connolly J, Savani RC, Narula N, Liang B et al (2002). Serotonin mechanisms in heart valve disease I: serotonin‐induced up‐regulation of transforming growth factor‐beta1 via G‐protein signal transduction in aortic valve interstitial cells. Am J Pathol 161: 2111–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza F, Healy L, Sutherland DR (2001). Structural and functional features of the CD34 antigen: an update. J Biol Regul Homeost Agents 15: 1–13. [PubMed] [Google Scholar]
- Launay J‐M, Hervé P, Callebert J, Mallat Z, Collet C, Doly S et al (2012). Serotonin 5‐HT2B receptors are required for bone‐marrow contribution to pulmonary arterial hypertension. Blood 119: 1772–1780. https://doi.org/10.1182/blood‐2011‐06‐358374. [DOI] [PubMed] [Google Scholar]
- Liu AC, Joag VR, Gotlieb AI (2007). The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol 171: 1407–1418. https://doi.org/10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C‐C, Liu M‐M, Culshaw G, Clinton M, Argyle DJ, Corcoran BM (2015). Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: a microarray study. Vet J 204: 23–31. https://doi.org/10.1016/j.tvjl.2015.02.021. [DOI] [PubMed] [Google Scholar]
- Lundin L, Norheim I, Landelius J, Oberg K, Theodorsson‐Norheim E (1988). Carcinoid heart disease: relationship of circulating vasoactive substances to ultrasound‐detectable cardiac abnormalities. Circulation 77: 264–269. https://doi.org/10.1161/01.CIR.77.2.264. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Ayme‐Dietrich E, Aubertin‐Kirch G, Banas SM, Quentin E, Lawson R et al (2017). New therapeutic opportunities for 5‐HT2 receptor ligands. Pharmacol Ther 170: 14–36. https://doi.org/10.1016/j.pharmthera.2016.10.008. [DOI] [PubMed] [Google Scholar]
- Martini F, Zuppiroli A, Gori A, Chiarantini E, Fedi S, Prisco D et al (1996). Platelet and blood clotting activation in patients with mitral valve prolapse. Thromb Res 83: 299–306. https://doi.org/10.1016/0049‐3848(96)00138‐7. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Adams V, Walther C, Kleinecke C, Brugger P, Linke A et al (2009). Reduced number and function of endothelial progenitor cells in patients with aortic valve stenosis: a novel concept for valvular endothelial cell repair. Eur Heart J 30: 346–355. https://doi.org/10.1093/eurheartj/ehn501. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekontso‐Dessap A, Brouri F, Pascal O, Lechat P, Hanoun N, Lanfumey L et al (2006). Deficiency of the 5‐hydroxytryptamine transporter gene leads to cardiac fibrosis and valvulopathy in mice. Circulation 113: 81–89. https://doi.org/10.1161/CIRCULATIONAHA.105.554667. [DOI] [PubMed] [Google Scholar]
- Murohara T (2001). Therapeutic vasculogenesis using human cord blood‐derived endothelial progenitors. Trends Cardiovasc Med 11: 303–307. https://doi.org/10.1016/S1050‐1738(01)00128‐1. [DOI] [PubMed] [Google Scholar]
- Nebigil CG, Choi D‐S, Dierich A, Hickel P, Le Meur M, Messaddeq N et al (2000). Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci U S A 97: 9508–9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama MA, Chittur SV (2006). Genomic expression patterns of mitral valve tissues from dogs with degenerative mitral valve disease. Am J Vet Res 67: 1307–1318. https://doi.org/10.2460/ajvr.67.8.1307. [DOI] [PubMed] [Google Scholar]
- Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E et al (2001). Aortic valve endothelial cells undergo transforming growth factor‐β‐mediated and non‐transforming growth factor‐β‐mediated transdifferentiation in vitro. Am J Pathol 159: 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paruchuri S, Yang J‐H, Aikawa E, Melero‐Martin JM, Khan ZA, Loukogeorgakis S et al (2006). Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to VEGF‐A and TGFβ2. Circ Res 99: 861–869. https://doi.org/10.1161/01.RES.0000245188.41002.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potteaux S, Combadière C, Esposito B, Lecureuil C, Ait‐Oufella H, Merval R et al (2006). Role of bone marrow‐derived CC‐chemokine receptor 5 in the development of atherosclerosis of low‐density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol 26: 1858–1863. https://doi.org/10.1161/01.ATV.0000231527.22762.71. [DOI] [PubMed] [Google Scholar]
- Quante M, Tu SP, Tomita H, Gonda T, Whang SS, Takashi S et al (2011). Bone marrow‐derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19: 257–272. https://doi.org/10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Blough BE, Jacobson AE, Rice KC, Partilla JS (2010). Evidence for non‐competitive modulation of substrate‐induced serotonin release. Synapse (N Y N) 64: 862–869. https://doi.org/10.1002/syn.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ et al (2000). Evidence for possible involvement of 5‐HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102: 2836–2841. https://doi.org/10.1161/01.CIR.102.23.2836. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Dunnwald M, Jiao C (2007). Biology of bone marrow‐derived endothelial cell precursors. Am J Physiol Heart Circ Physiol 292: H1–H18. https://doi.org/10.1152/ajpheart.00662.2006. [DOI] [PubMed] [Google Scholar]
- Setola V, Hufeisen SJ, Grande‐Allen KJ, Vesely I, Glennon RA, Blough B et al (2003). 3,4‐Methylenedioxymethamphetamine (MDMA, “ecstasy”) induces fenfluramine‐like proliferative actions on human cardiac valvular interstitial cells in vitro. Mol Pharmacol 63: 1223–1229. https://doi.org/10.1124/mol.63.6.1223. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 130 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Camp G, Flamez A, Cosyns B, Weytjens C, Muyldermans L, Van Zandijcke M et al (2004). Treatment of Parkinson's disease with pergolide and relation to restrictive valvular heart disease. Lancet 363: 1179–1183. https://doi.org/10.1016/S0140‐6736(04)15945‐X. [DOI] [PubMed] [Google Scholar]
- Vaturi M, Perl L, Leshem‐Lev D, Dadush O, Bental T, Shapira Y et al (2011). Circulating endothelial progenitor cells in patients with dysfunctional versus normally functioning congenitally bicuspid aortic valves. Am J Cardiol 108: 272–276. https://doi.org/10.1016/j.amjcard.2011.03.039. [DOI] [PubMed] [Google Scholar]
- Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M et al (2006). An in vivo analysis of hematopoietic stem cell potential hematopoietic origin of cardiac valve interstitial cells. Circ Res 98: 690–696. https://doi.org/10.1161/01. RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- Walsh PN, Kansu TA, Corbett JJ, Savion PJ, Goldburgh WP, Schatz NJ (1981). Platelets, thromboembolism and mitral valve prolapse. Circulation 63: 552–559. https://doi.org/10.1161/01.CIR.63.3.552. [DOI] [PubMed] [Google Scholar]
- Wara AK, Croce K, Foo S, Sun X, Icli B, Tesmenitsky Y et al (2011). Bone marrow‐derived CMPs and GMPs represent highly functional proangiogenic cells: implications for ischemic cardiovascular disease. Blood 118: 6461–6464. https://doi.org/10.1182/blood‐2011‐06‐363457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JD, Carrier EJ, Bloodworth NC, Schroer AK, Chen P, Ryzhova LM et al (2016). Serotonin 2B receptor antagonism prevents heritable pulmonary arterial hypertension. PLoS One 11: e0148657 https://doi.org/10.1371/journal.pone.0148657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitebread S, Dumotier B, Armstrong D, Fekete A, Chen S, Hartmann A et al (2016). Secondary pharmacology: screening and interpretation of off‐target activities‐focus on translation. Drug Discov Today 21: 1232–1242. https://doi.org/10.1016/j.drudis. 2016.04.021. [DOI] [PubMed] [Google Scholar]
- Wylie‐Sears J, Aikawa E, Levine RA, Yang J‐H, Bischoff J (2011). Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol 31: 598–607. https://doi.org/10.1161/ATVBAHA.110.216184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Jian B, Chu R, Lu Z, Li Q, Dunlop J et al (2002). Serotonin mechanisms in heart valve disease II. Am J Pathol 161: 2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Chen X, Talati M, Nunley BW, Gladson S, Blackwell T et al (2016). Bone marrow‐derived cells contribute to the pathogenesis of pulmonary arterial hypertension. Am J Respir Crit Care Med 193: 898–909. https://doi.org/10.1164/rccm.201502‐0407OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G (2007). Valvular heart disease and the use of dopamine agonists for Parkinson's disease. N Engl J Med 356: 39–46. https://doi.org/10.1056/NEJMoa054830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Inhibition of platelet aggregation by ritanserin.
Figure S2 Physical parameters.
Figure S3 Heart to body weight ratio from all experimental mice groups.
Figure S4 Haemodynamic parameters in all experimental mice groups.