

ABSTRACT
Ras proteins are considered as the founding members of a large superfamily of small GTPases that control fundamental cellular functions. Mutationally activated RAS genes were discovered in human cancer cells more than 3 decades ago, but intensive efforts on Ras structure, biochemistry, function and signaling continue even now. Because mutant Ras proteins are inherently difficult to inhibit and have yet been therapeutically conquered, it was designated as “the Everest of oncogenes” in the cancer genome landscape, further promoting a “renaissance” in RAS research. Different paths to directly or indirectly targeting mutant Ras signaling are currently under investigation in the hope of finding an efficacious regimen. Inhibitors directly binding to KRASG12C to block its downstream signaling have been revealed, supporting the notion of Ras' druggability. An alternative indirect approach by targeting synthetic lethal interactors of mutant RAS is underway. We recently employed a synthetic lethal drug screen plus a combinatorial strategy using a panel of clinical agents and discovered that KRAS-mutant cancers were fragile to the combined inhibition of polo-like kinase 1 (Plk1) and RhoA/Rho kinase (ROCK). The combined regimen of BI-2536 (a Plk1 inhibitor) and fasudil (a ROCK inhibitor) promoted a significant inhibition of patient-derived lung cancer xenografts and prolonged the survival of LSL-KRASG12D mice. In this commentary, we will summarize the state-of-the art for the direction of synthetic lethality, and also speculate on the future development of this approach.
KEYWORDS: combination therapy, KRAS mutations, Ras, synthetic lethality, synthetic lethal screens
Anti-RAS: The battle continues
According to the Ras history, RAS genes were first identified as viral genes, and Ras proteins are founding members of a large superfamily of small GTPases, including Ras, Rho, Rab, Arf and Ran families.1 With some variation and exceptions, Ras superfamily proteins function as GDP/GTP-regulated binary on-off switches. Ras proteins (KRAS4A, KRAS4B, NRAS and HRAS) control cytoplasmic signaling networks and regulate diverse normal cellular functions. Mutationally activated RAS genes were discovered in human cancer cells in 1982, but drugging the Ras proteins was considered as “the Everest” to climb.2 Gain-of-function missense mutations in RAS genes occur in approximately a third of all human solid tumors. As the principal of the 3 isoforms of RAS, KRAS (KRAS4A and KRAS4B) mutations are particularly prevalent in malignancies with the highest mortality rates, such as pancreatic (90%), colorectal (30–40%) and lung (15–20%) tumors.3 Due to the high frequency of RAS mutations in a wide spectrum of human cancers, intensive efforts on Ras structure, biochemistry and biology have been made during the last 3 decades for “anti-RAS” therapy.
However, a clinically effective Ras inhibitor has eluded drug-discovery efforts for many years. Ras proteins lack pockets to which small molecules bind with high affinity, and also, a large family of related protein members share similar GDP/GTP-binding domain, making Ras therapeutic attack extremely challenging. Six strategies for targeting Ras signaling have been proposed4: targeting Ras proteins directly, upsetting Ras membrane association, exploiting synthetic lethal partners of mutant RAS, targeting Ras downstream pathways, disrupting the greedy metabolic habit of RAS-mutant cells, and harnessing the immune response. These anti-RAS strategies could be generally divided into “direct” and “indirect” approaches. KRASG12C selective inhibitors that target the thiol to inhibit GTP binding were recently identified.5-7 Binding of these inhibitors to KRASG12C subverts the native nucleotide preference to favor GDP over GTP, therefore preventing subsequent downstream activation. Most recently, a novel cell-active and mutant-specific inhibitor targeting the allosteric switch II pocket of KRASG12C showed improved potency and selectivity, further revealing that KRASG12C rapidly cycles its nucleotide substrates.8,9 These breakthroughs provide novel insights into the function of KRASG12C. Although further work is still needed to determine whether these inhibitors suppress the in vivo growth of KRASG12C-driven tumors, the development of KRASG12C selective inhibitors opens a landmark discovery that changes the perception of Ras proteins from “undruggability” to “druggability.” Ras mimetics has also been recently revealed that the clinical small-molecule inhibitor rigosertib disrupts the association of Ras with Raf and other effector proteins.10 The discovery of new mechanism of rigosertib shed light on the translation of a clinical drug to directly treat KRAS-mutant cancers. Other drug development of targeting mutant RAS by inhibiting proteins that facilitate Ras trafficking to the plasma membrane has progressed as well. Two classes of inhibitors that direct bound to the prenyl-binding pocket of PDEδ were synthesized by the same group.11,12 Additionally, based on the rationality that the Ras protein degradation occurs together with the β-catenin degradation, novel chemical molecules that bound directly to the regulators of G-protein signaling domain of axin and promoted Ras degradation were discovered.13 Collectively, the “best” path to inhibit Ras has yet to be determined, as these molecules through directly targeting Ras proteins or Ras signaling have a long way before going into the clinic. Attempting to find “synthetic lethal” interactions between activated mutant RAS and other genes to which the cancer cells heavily addict shows enormous potential in recent years.
Synthetic lethal strategy: Challenging but promising
Synthetic lethal studies, the indirect strategy to defeat RAS-mutant tumors, have made another big splash, changing the landscape by uncovering vulnerabilities in tumors with RAS mutations. The existence of oncogene-specific synthetic lethal interaction is supported by the notion that oncogenic transformation substantially alters the cell phenotype.14 Synthetic lethal screens uncover multiple “oncogene addiction” and “non-oncogene addiction” pathways that were required for the survival of RAS-mutant cells.15 This direction has been raised on the concept of synthetic lethality that was described in invertebrate genetics one century years ago. At its simplest, the 2 or more separate genes (or pathways) are synthetic lethal if the mutations in any one of them will not change the viability of a cell but simultaneous mutations in both of them will result in a lethal phenotype. Therefore, targeting a gene that is synthetic lethal to a cancer-relevant mutation should kill only cancer cells and spare normal ones. Based on this principle, mutations in candidate genes might be either loss-of-function or gain-of-function defects. This approach has been inspired most strongly by the successful use of poly (adenosine diphosphate [ADP]-ribose) polymerase inhibitors to treat BRCA defective cancers in the clinic.16,17 During the last decade, synthetic lethality has been intensively exploited for identification of novel anticancer targets, development of new genotype-selective anticancer agents, and characterization of genes associated with drug responses.18,19
In the Ras research community, several studies from different groups have identified synthetic lethal interactors with mutant KRAS by using large-scale RNA interference (RNAi) screens.14,20,21 Since normal cells lack mutant KRAS, genes or inhibitors identified in this manner should in principle be selectively lethal for tumors but not for untransformed cells. Although the first generation of screens led to more novel acknowledgment of the dependency of mutant KRAS but not many tractable drug targets, potential interactors of mutant KRAS identified by synthetic lethal RNAi screens, such as the anti-apoptotic protein BCL-XL, cyclin-dependent kinase CDK4 and serine/threonine-protein kinase TBK1, have already been translated to clinical settings to treat KRAS-mutant cancers.3
RNAi-based synthetic lethal screens
Synthetic lethal screens have now progressed from drosophila model system to genome-wide short hairpin RNA- and small interfering RNA-mediated drug-sensitization screens and novel small-molecule inhibitor screens. This approach is particularly attractive for those oncogenic drivers or tumor suppressors, such as KRAS, MYC and TP53, which were usually thought “undruggable.”
During the last 10 years, intensive studies have taken advantage of synthetic lethal approach to identify genes that maintain the tumor phenotype with “addiction” to mutant KRAS. Genome-wide RNAi screens and other technologies have identified a list of candidate genes, including TANK-binding kinase 1,20 serine/threonine-protein kinase 33,22 heat-shock protein 90,23 polo-like kinase 1 and cell mitotic regulators,14 the cyclin-dependent kinase 1 and 4,24,25 transcription factor GATA2,21 evolutionarily conserved gene enhancer of rudimentary homolog,26 transforming growth factor β-activated kinase 1,27 anti-apoptotic BH3 family member BCL-XL,28 proteasome and topoisomerase components,14,29 genes that are involved in glucose metabolism30 and SUMO E2 ligase Ubc9.31 These hit genes span diverse different cellular regulations, including cell mitosis, cell apoptosis, cell growth, cell metabolism, and gene replication, transcription and modification (Fig. 1). These important works broaden the view of the biology and function of mutant KRAS. In our recent paper, a combinatorial clinical drug screen based on synthetic lethality revealed a preclinical feasibility for combining a polo-like kinase 1 inhibitor and a Rho signaling inhibitor to conquer KRAS-mutant lung cancer.32 Such combined regimen activates the cyclin-dependent kinase inhibitor p21WAF1/CIP1, a critical regulator of cell cycle and cell mitosis. Because mutations in KRAS have been suggested to contribute to chromosome instability and mitotic stress,33 further load of mitotic stress by activating p21WAF1/CIP1 would cause susceptibility to apoptosis in KRAS-mutant cells.
Figure 1.
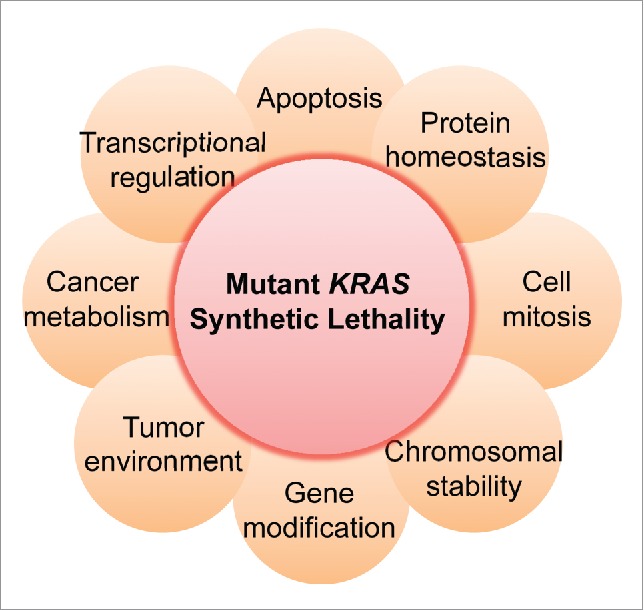
The dependency of mutant KRAS on diverse cellular regulations. Synthetic lethal interactors of mutant KRAS identified by several synthetic lethal screens span different cell phenotypes, such as cell apoptosis (e.g. BCL-XL), cell mitosis (e.g., PLK1, CDK4 and CDKN1A), transcriptional regulation (e.g. GATA2) and protein homeostasis (e.g., HSP90). Clinical inhibitors targeting some of these synthetic lethal partners of mutant KRAS have been tested in clinical settings for KRAS-mutant cancers through a combinatorial strategy with a MEK inhibitor (e.g. NCT02079740 NCT02258607 and NCT02022982).
Synthetic lethal studies based on KRAS mutations illustrate the great potential to improve our understanding of Ras signaling and open new possibilities for blocking the Ras' functions. Although only a modest overlap is found among these identified synthetic lethal targets, and some targets are intractable and still lack effective inhibitors, the strategy based on synthetic lethality dose open up a new avenue to understand the dependency features of KRAS-mutant cancers.
Chemical-based synthetic lethal screens
To advance the discovery of drugs against KRAS-driven tumors, synthetic lethal small-molecule inhibitor screens have concomitantly emerged and represent a complementary approach to directly identify drugs that target the essential signaling for the growth of KRAS-mutant tumors. Compared with RNAi-based screens that only downregulate gene expression, chemical-based screens show great advantages in regulating genes' functions. For example, drug-induced inhibition of enzymatic activity functions distinctly from loss of expression of a protein. At this point, chemical drugs are likely to be better at regulating multiple related isoforms of a protein. Therefore, chemical screens are expected to give significantly different insights to functional genomic screens.34 Additionally, the use of well-known, clinical approved agents in a synthetic lethal screen in KRAS-mutant and wildtype cells has the advantage of sorting out both gain-of-function as well as loss-of-function candidate factors for synthetic lethal interaction with mutant KRAS. Such approach provides a shortcut to utilize drugs that are already in clinical use. Here we will briefly list some novel-structure compounds and clinical drug combinations discovered based on synthetic lethality to treat KRAS-mutant cancers.
Chemical libraries were usually utilized by the synthetic lethality technology to identify novel-structure anticancer drugs for killing RAS-mutant cells.35 Novel compounds triphenyl tetrazolium and a sulfinyl cytidine derivative were identified through a drug screen based on isogenic cells lines of KRAS-mutant colon cancer. This class of compounds displayed selective activity in vitro against tumor cells and inhibited tumor xenografts containing mutant Ras.36 Compound erastin exhibited lethal selectivity in human tumor cells harboring mutations in the HRAS, KRAS or BRAF oncogenes by modulating mitochondrial voltage-dependent anion channels.37 Other compounds, such as oncrasin-1,38 lanperisone,39 and oncrasin analogs40 all induced cytotoxic effects in KRAS-mutant tumor cells by triggering oxidative stress and regulating oxidative stress-related pathways. However, the precise target and target specificity of these compounds are still unclear.
Tumor initiation, progression and high heterogeneity are primarily driven by multiple genetic mutations rather than by a single defect.41 Resistance and partial responses to targeted monotherapy are major obstacles in cancer treatment. To achieve more potent efficacy and less toxicity, a “cocktail” of drugs is now often given.42 Based on this rationale, many significant works have been conducted to search for effective drug combinations to defeat KRAS-mutant cancers through synthetic lethality. Inhibition of KRAS-mutant tumors by using surrogate drugs already approved for clinical use is the fast way for translational research. By RNAi library screening, transcriptional factor GATA2 was identified as a synthetic lethal gene of mutant KRAS. Pharmacological inhibition of GATA2-mediated pathways with bortezomib (a proteasome inhibitor) and fasudil (a ROCK inhibitor) results in dramatic tumor inhibition.21 By far, MEK inhibitors have showed potential effectiveness in patients with KRAS-mutant cancer and have been approved by the US Food and Drug Administration. A large-scale screen of short hairpin RNA with MEK inhibitor as a backbone identified that BCL-XL inhibitor ABT-263 in combination with a MEK inhibitor led to dramatic apoptosis in many KRAS-mutant cell lines from different tissue types.28 This study promotes the initial clinical trial for KRAS-mutant cancer (NCT02079740) by using a combined regimen of navitoclax (an inhibitor of BCL-2 and BCL-XL) and trametinib (an MEK inhibitor).3 In a similar screen system, the tyrosine kinase TBK120 and interphase cyclin-dependent kinase CDK424 were identified as synthetically lethal partners of mutant KRAS. These works lead to ongoing clinical trials combing momelotinib (a dual JAK2 and TBK1 inhibitor; NCT02258607) or palbociclib (a CDK4/6 inhibitor; NCT02022982) with a MEK inhibitor for treating KRAS-mutant cancers. Another study showed that agents enhancing proteotoxic stress, such as the Hsp90 inhibitor IPI-504, induce tumor regression in aggressive mouse models when combined with rapamycin (a mTOR inhibitor).33 Our recent work show that dual inhibition of polo-like kinase 1 and RhoA/Rho kinase leads to the synergistic effects in KRAS-mutant lung cancer. Mechanism study revealed a new synthetic lethal interaction between KRAS and CDKN1A (encoding p21),32 as genetic or pharmacological increase of p21WAF1/CIP1 level preferentially impairs the growth of KRAS-mutant cells. Most recently, a combinatorial strategy by combining a FGFR inhibitor and a MEK inhibitor for treating KRAS-mutant lung cancer has been reported.43 These synthetic lethal chemical screens based on available clinical drugs and drug combinations unveil new armamentaria to fight KRAS-mutant cancer.
Synthetic lethality: System development
As mentioned above, synthetic lethal partners of mutant KRAS identified by the first generation of gene screens lacks of reproducibility. Changes in context of cell model and analytic approaches could easily affect the outcome of the results, making the efforts difficult to bear fruits. Considering the complexities of the Ras proteins and several problems in the current experimental system of synthetic lethal screens, several optimization strategies have been addressed.
KRAS specific mutations and cancer sub-classification
There is now emerging recognition that the human Ras proteins are not functionally identical, and there are mutation-specific consequences on Ras structure, biochemistry and biology.44 Although KRAS mutation is prevalently present in pancreatic, colon and lung cancers, the hot mutations and mutation-specific signaling pathways of KRAS in these cancer types are dramatically different. Not all mutant K-Ras proteins affect patient survival or downstream signaling in a similar way. Most mutations of KRAS occur at codons 12 and 13, and the KRASG12C mutation is the most common mutation in lung cancer, which is quite different from other cancer types. Difference in mutation frequency may reflect different biological characteristics of a mutant protein. For example, KRASG12C and KRASG12V mutations in lung adenocarcinoma preferentially activate the RalGDS pathway, whereas KRASG12D prefers the MAPK and PI3K pathways.45,46 The heterogeneous behavior of mutant K-Ras proteins suggests that therapeutic interventions may need to take into account the specific mutant KRAS expressed by the tumor. Therefore, mutation-selective cell models are needed for setting up a synthetic lethal screen. Recent identification of KRASG12C inhibitors also inspires us at this point.
Multiple cancers have altered metabolic processes, and oncogenic KRAS has been shown to be a key player in promoting metabolic rewiring. It has been demonstrated that mutant KRASG12D is responsible for orchestrating metabolic phenotype of pancreatic cancer cells in part through its role in reprogramming anabolic glucose metabolism.47 Mutation in KRAS has also been reported to facilitate pancreatic cancer cells to addict to glutamine for maintaining their redox homeostasis.48 However, the specific actions of oncogenic KRAS on metabolic regulation may differ depending on tumor types and genetic context (including difference in mutant KRAS copy number).49 For example, the in vivo evidences of metabolic rewiring during lung cancer malignant progression showed that mutant KRASG12D homozygous cells exhibited an increase in glucose metabolism toward the tricarboxylic acid cycle and glutathione synthesis, leading to enhanced glutathione-mediated detoxification.49 Therefore, effective sub-classification of KRAS-mutant cancers will be required to improve anti-RAS therapy through personalized medicine in the near future.
Cell models
Cell model is another limitation in the first generation of mutant KRAS synthetic lethal screens. Most of previous screens relied on isogenic cell lines or a panel of laboratory cancer cell lines, which could hardly mimic tumor heterogeneity. Isogenic cell lines were usually generated by ablation or overexpression of mutant KRAS. It is quite possible that such gene editing causes adaptive alterations in other oncogene drivers or other signaling pathways. As our experiences, it is better to use early passages of isogenic cells generated by the CRISPR/Cas9 system-mediate-knockout or by adeno-associated virus-mediated overexpression of a KRAS mutation. Additionally, it is also strongly suggested to rigorously detect the activation of Ras proteins (Ras-GTP state) in isogenic cell lines during synthetic lethal screens or in cell functional assays. RNAi screening technology with low potency of knockdown usually causes a high level of false negatives and off-target effects. The recently developed gene editing technology, the CRISPR/Cas9 system, could be applied to KRAS synthetic lethal screens by using a genome-scale lentiviral single guide RNA library.34 Additionally, all previous screens have utilized in vitro anchorage-dependent culture conditions. Future synthetic lethal screens will benefit from ex vivo organoid cultures or in vivo xenograft tumor assays, which more accurately model tumor heterogeneity and tumor microenvironment.4 Nowadays, patient-derived xenograft models are largely acceptable for cancer research, and it offers a powerful tool for developing anticancer therapies and personalized medicine for cancer patients.50 It is strongly recommended to utilize patient-derived xenograft models to validate the efficacy of anticancer drugs or drug combinations identified by synthetic lethal chemical screens for targeting mutant KRAS.
Data validation and mechanism elucidation
The hits identified from the first generation of synthetic lethal screens for mutant KRAS span many different cellular processes, including protein homeostasis, mitotic modulation, chromosomal stability, transcriptional regulation, gene modification, apoptosis and cancer metabolism (Fig. 1), revealing targeting “non-oncogene addition” for efficacious cancer therapies.15 However, most of these hits are not been validated by rescue experiments and other rigorous experimental assays. It seems that analytic approaches could easily affect the outcome of the results, therefore making it hard to achieve reliable candidate hits. Additionally, the mechanism for synthetic lethal interaction of mutant KRAS and these identified hits are largely unknown. Although some synthetic lethal hits are roughly mapped to the Ras signaling pathway network,19 the detailed interaction between these hits and mutant KRAS are still elusive. Therefore, the candidate genes from the next generation of screens should be rigorously validated, and how these genes to map to mutant KRAS signaling also should be clarified.
Conclusions
Despite more than 3 decades of intense efforts, an effective anti-RAS therapy has yet to reach the clinic. A better understanding of Ras structure, biochemistry, processing and signaling will open the novel possibility to defeat RAS-driven tumors. Several strategies to target mutant Ras proteins are progressing, but each of them has pitfalls and the best way has not been determined. It has been gradually recognized that all the human Ras proteins are not functionally identical, and therefore, mutation-selective therapeutic strategies are accordingly appreciated. Recent studies of small-molecule approaches to directly inhibit oncogenic KRASG12C have invigorated the Ras community, raising the possibility of drugging Ras that has been long considered “undruggable.” However, these direct inhibitors are more considered as chemical probes to understand Ras' biology, rather agents reaching clinical application. First-generation synthetic lethal screens have identified several synthetic lethal interactors of mutant KRAS, and these efforts are extremely important to dissect the signaling addiction of KRAS-mutant cells. Some inhibitors of KRAS synthetic lethal partners, such as CDK4, TBK1 and BCL-XL, are currently tested in clinical trials in combination with a MEK inhibitor to treat KRAS-driven cancers. Given the complexity of Ras proteins in different tumor tissues and the less reproducibility of those identified KRAS synthetic lethal interactors, the experimental system of synthetic lethal screens should be properly improved by taking into account KRAS specific mutations, cancer sub-classification, cell models, data validation and mechanism elucidation. The major challenge with current cancer treatments is drug resistance, making combination therapies by simultaneously targeting multiple cancer-associated pathways a necessary for efficacious anti-RAS treatments.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The work was supported by the Science and Technology Commission of Shanghai Municipality (Grant 16ZR1410400) and the State Scholarship Council (Fellowship 201506145040).
References
- [1].Cox AD, Der CJ. Ras history: The saga continues. Small GTPases 2010; 1:2-27; PMID:21686117; https://doi.org/ 10.4161/sgtp.1.1.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Russo M, Di Nicolantonio F, Bardelli A. Climbing RAS, the everest of oncogenes. Cancer Discov 2014; 4:19-21; PMID:24402942; https://doi.org/ 10.1158/2159-8290.CD-13-0906 [DOI] [PubMed] [Google Scholar]
- [3].Singh H, Longo DL, Chabner BA. Improving Prospects for Targeting RAS. J Clin Oncol 2015; 33:3650-9; PMID:26371146; https://doi.org/ 10.1200/JCO.2015.62.1052 [DOI] [PubMed] [Google Scholar]
- [4].Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers (Basel) 2016; 8:45-66; PMID:27096871; https://doi.org/ 10.3390/cancers8040045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503:548-51; PMID:24256730; https://doi.org/ 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, et al.. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl 2014; 53:199-204; PMID:24259466; https://doi.org/ 10.1002/anie.201307387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, et al.. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A 2014; 111:8895-900; PMID:24889603; https://doi.org/ 10.1073/pnas.1404639111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016; 351:604-8; PMID:26841430; https://doi.org/ 10.1126/science.aad6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al.. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov 2016; 6:316-29; PMID:26739882; https://doi.org/ 10.1158/2159-8290.CD-15-1105 [DOI] [PubMed] [Google Scholar]
- [10].Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MV, Ueno L, Hart JR, et al.. A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell 2016; 165:643-55; PMID:27104980; https://doi.org/ 10.1016/j.cell.2016.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, et al.. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013; 497:638-42; PMID:23698361; https://doi.org/ 10.1038/nature12205 [DOI] [PubMed] [Google Scholar]
- [12].Papke B, Murarka S, Vogel HA, Martin-Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmermann G, Heinelt K, et al.. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat Commun 2016; 7:11360; PMID:27094677; https://doi.org/ 10.1038/ncomms11360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cha PH, Cho YH, Lee SK, Lee J, Jeong WJ, Moon BS, Yun JH, Yang JS, Choi S, Yoon J, et al.. Small-molecule binding of the axin RGS domain promotes beta-catenin and Ras degradation. Nat Chem Biol 2016; 12:593-600; https://doi.org/ 10.1038/nchembio.2103 [DOI] [PubMed] [Google Scholar]
- [14].Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137:835-48; PMID:19490893; https://doi.org/ 10.1016/j.cell.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009; 136:823-37; PMID:19269363; https://doi.org/ 10.1016/j.cell.2009.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913-7; PMID:15829966; https://doi.org/ 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- [17].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al.. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917-21; PMID:15829967; https://doi.org/ 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- [18].McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med 2014; 371:1725-35; PMID:25354106; https://doi.org/ 10.1056/NEJMra1407390 [DOI] [PubMed] [Google Scholar]
- [19].Fang B. Development of synthetic lethality anticancer therapeutics. J Med Chem 2014; 57:7859-73; PMID:24893124; https://doi.org/ 10.1021/jm500415t [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al.. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009; 462:108-12; PMID:19847166; https://doi.org/ 10.1038/nature08460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E, Lassailly F, Matthews N, Nye E, et al.. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 2012; 149:642-55; PMID:22541434; https://doi.org/ 10.1016/j.cell.2012.02.059 [DOI] [PubMed] [Google Scholar]
- [22].Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, et al.. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009; 137:821-34; PMID:19490892; https://doi.org/ 10.1016/j.cell.2009.03.017 [DOI] [PubMed] [Google Scholar]
- [23].Azoitei N, Hoffmann CM, Ellegast JM, Ball CR, Obermayer K, Gossele U, Koch B, Faber K, Genze F, Schrader M, et al.. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J Exp Med 2012; 209:697-711; PMID:22451720; https://doi.org/ 10.1084/jem.20111910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaría D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010; 18:63-73; PMID:20609353; https://doi.org/ 10.1016/j.ccr.2010.05.025 [DOI] [PubMed] [Google Scholar]
- [25].Costa-Cabral S, Brough R, Konde A, Aarts M, Campbell J, Marinari E, Riffell J, Bardelli A, Torrance C, Lord CJ, et al.. CDK1 Is a Synthetic Lethal Target for KRAS Mutant Tumours. PLoS One 2016; 11:e0149099; PMID:26881434; https://doi.org/ 10.1371/journal.pone.0149099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Weng MT, Lee JH, Wei SC, Li Q, Shahamatdar S, Hsu D, Schetter AJ, Swatkoski S, Mannan P, Garfield S, et al.. Evolutionarily conserved protein ERH controls CENP-E mRNA splicing and is required for the survival of KRAS mutant cancer cells. Proc Natl Acad Sci U S A 2012; 109:E3659-67; PMID:23236152; https://doi.org/ 10.1073/pnas.1207673110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012; 148:639-50; PMID:22341439; https://doi.org/ 10.1016/j.cell.2011.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et al.. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013; 23:121-8; PMID:23245996; https://doi.org/ 10.1016/j.ccr.2012.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J, et al.. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res 2012; 22:1227-45; PMID:22613949; https://doi.org/ 10.1038/cr.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al.. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009; 325:1555-9; PMID:19661383; https://doi.org/ 10.1126/science.1174229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yu B, Swatkoski S, Holly A, Lee LC, Giroux V, Lee CS, Hsu D, Smith JL, Yuen G, Yue J, et al.. Oncogenesis driven by the Ras/Raf pathway requires the SUMO E2 ligase Ubc9. Proc Natl Acad Sci U S A 2015; 112:E1724-33; PMID:25805818; https://doi.org/ 10.1073/pnas.1415569112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, Lu W, Liu J, Pang X, Liu M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun 2016; 7:11363; PMID:27193833; https://doi.org/ 10.1038/ncomms11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V, et al.. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011; 20:400-13; PMID:21907929; https://doi.org/ 10.1016/j.ccr.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Downward J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin Cancer Res 2015; 21:1802-9; PMID:25878361; https://doi.org/ 10.1158/1078-0432.CCR-14-2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689-98; PMID:16110319; https://doi.org/ 10.1038/nrc1691 [DOI] [PubMed] [Google Scholar]
- [36].Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol 2001; 19:940-5; PMID:11581659; https://doi.org/ 10.1038/nbt1001-940 [DOI] [PubMed] [Google Scholar]
- [37].Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al.. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447:864-8; PMID:17568748; https://doi.org/ 10.1038/nature05859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res 2008; 68:7403-8; PMID:18794128; https://doi.org/ 10.1158/0008-5472.CAN-08-1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N, Jacks T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A 2011; 108:8773-8; PMID:21555567; https://doi.org/ 10.1073/pnas.1105941108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guo W, Wei X, Wu S, Wang L, Peng H, Wang J, Fang B. Antagonistic effect of flavonoids on NSC-741909-mediated antitumor activity via scavenging of reactive oxygen species. Eur J Pharmacol 2010; 649:51-8; PMID:20854805; https://doi.org/ 10.1016/j.ejphar.2010.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A 2003; 100:776-81; PMID:12552134; https://doi.org/ 10.1073/pnas.0334858100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13:714-26; PMID:24060863; https://doi.org/ 10.1038/nrc3599 [DOI] [PubMed] [Google Scholar]
- [43].Manchado E, Weissmueller S, Morris JP, Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al.. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016; 534:647-51; PMID:27338794; https://doi.org/ 10.1038/nature18600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016; 129:1287-92; PMID:26985062; https://doi.org/ 10.1242/jcs.182873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell 2014; 25:272-81; PMID:24651010; https://doi.org/ 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- [46].Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, et al.. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 2012; 104:228-39; PMID:22247021; https://doi.org/ 10.1093/jnci/djr523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al.. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435; https://doi.org/ 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al.. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496:101-5; PMID:23535601; https://doi.org/ 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016; 531:110-3; PMID:26909577; https://doi.org/ 10.1038/nature16967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res 2013; 73:5315-9; PMID:23733750; https://doi.org/ 10.1158/0008-5472.CAN-13-1069 [DOI] [PMC free article] [PubMed] [Google Scholar]