

ABSTRACT
Localization of the interface between the candidate antibody and its antigen target, commonly known as epitope mapping, is a critical component of the development of therapeutic monoclonal antibodies. With the recent availability of commercial automated systems, hydrogen / deuterium eXchange (HDX) is rapidly becoming the tool for mapping epitopes preferred by researchers in both industry and academia. However, this approach has a significant drawback in that it can be confounded by ‘allosteric’ structural and dynamic changes that result from the interaction, but occur far from the point(s) of contact. Here, we introduce a ‘kinetic’ millisecond HDX workflow that suppresses allosteric effects in epitope mapping experiments. The approach employs a previously introduced microfluidic apparatus that enables millisecond HDX labeling times with on-chip pepsin digestion and electrospray ionization. The ‘kinetic’ workflow also differs from conventional HDX-based epitope mapping in that the antibody is introduced to the antigen at the onset of HDX labeling. Using myoglobin / anti-myoglobin as a model system, we demonstrate that at short ‘kinetic’ workflow labeling times (i.e., 200 ms), the HDX signal is already fully developed at the ‘true’ epitope, but is still largely below the significance threshold at allosteric sites. Identification of the ‘true’ epitope is supported by computational docking predictions and allostery modeling using the rigidity transmission allostery algorithm.
KEYWORDS: Allosteric Effects, Binding Dynamics, Biopharmaceuticals, Effective Mixing, Epitope Mapping, Hydrogen Deuterium Exchange, Laminar Flow, Microfluidic Chip, Monoclonal Antibody, Time-Resolved Electrospray Ionization Mass Spectrometry
Introduction
Monoclonal antibodies (mAbs) are the largest and fastest-growing class of protein therapeutics in the pharmaceutical industry.1-5 With this rapidly rising interest has come a need to enhance bioanalytical methods for characterizing mAb structure and activity on the molecular level.6,7 Techniques that can rapidly map binding epitopes are of particular interest because this information can guide early-stage protein therapeutic development, provide critical information to support advancement decisions and, ultimately, be incorporated into regulatory agency fillings as mechanism of action (MoA) data. For biosimilar products, epitope mapping is critical for assessment of bioequivalency. Knowledge of a potential mAb drug's epitope can thus both accelerate development and mitigate risk.
A number of methods have been established for determining epitopes, including X-ray crystallography,8 nuclear magnetic resonance (NMR) spectroscopy,9 immunochemical analysis,10 limited proteolysis,11 site-directed mutagenesis,12 computational docking.13 X-ray crystallography and NMR are uniquely powerful, as they can directly provide epitope maps at atomic resolution, but both also have significant drawbacks. The main issue with X-crystallography is the challenge of producing high quality co-crystals,14 while NMR suffers from an inherent analyte size limitation that is generally below that of an intact antibody (resulting in the use of antigen-binding fragments to acquire NMR epitope maps).15 The remaining experimental approaches are lower resolution, susceptible to false positives (and negatives), and tend to be labor intensive.16
One emerging technique for epitope mapping is hydrogen / deuterium exchange (HDX) with detection by electrospray ionization mass spectrometry (ESI-MS).17-22 Briefly, this approach measures the rate at which peptide backbone amide hydrogens are exchanged with deuterium from D2O solvent.23-25 The exchange process is structure-sensitive because hydrogen bonding (such as occurs in secondary structure) and solvent access (which depends largely on tertiary structure) attenuate the rate of exchange. Regions with low exchange rates are thus structured or solvent inaccessible, while regions with high exchange rates are less structured (dynamic) and solvent exposed. In the case of a binding interaction, both new hydrogen bonding contacts and physical blocking of solvent access at the binding site will typically lower deuterium uptake in the ‘bound’ state compared to the ‘unbound state’. In principle, this makes HDX a facile and potentially very powerful approach to map epitopes.26
Allosteric effects, however, can be a substantial challenge for HDX-based epitope mapping.27 These effects occur when binding at one site causes a substantial change in conformation or dynamics at a distant site. Since allosteric conformational changes also influence hydrogen bonding and solvent access for backbone amides, they are often indistinguishable from the true binding site in conventional HDX measurements. This is a particular problem for antibody /antigen interactions, where allosteric effects may appear as a discontinuous epitope.
Here, we introduce a technique based on millisecond time-resolved electrospray ionization mass spectrometry with HDX (TRESI-HDX) that selectively attenuates or eliminates signals from allosteric effects in epitope mapping experiments. TRESI-HDX is analogous to conventional ‘bottom-up’ HDX experiments (in which the labeled protein is digested using an acid protease prior to analysis), but uses millisecond time-scale deuterium labeling, which allows investigations on rapid conformational changes,28 weak binding interactions29 and intrinsically disordered proteins.30,31 The experiment also differs from conventional epitope mapping in that the antibody is not pre-incubated with the antigen, but is instead introduced to the antigen concomitantly with D2O solvent so that binding and labeling occur simultaneously. The result is a method that combines aspects of continuous and pulsed HDX labeling, allowing the development of allostery that occurs shortly after binding to influence the evolution of the deuterium uptake profile. Because conformational equilibria are not fully established at early (millisecond) labeling timepoints, we call this approach ‘kinetic’ HDX labeling. Using myoglobin (Mb) / anti-myoglobin antibody (anti-Mb) as a model system, we find that with ‘kinetic’ HDX, allosteric effects are specifically suppressed for the first roughly 200 ms of HDX labeling, which allows higher-confidence determination of the ‘true’ epitope.
Results
Microfluidics-enabled TRESI-HDX
A schematic of TRESI-HDX apparatus and workflow is shown in Fig. 1. Millisecond hydrogen-deuterium exchange occurs in the concentric capillary TRESI mixer that is incorporated into a poly(methyl methacrylate) (PMMA) microfluidic chip as described previously.31,32 In this implementation of the TRESI-HDX chip, microstructures that disturb laminar flow were included after the acid quenching channels to improve the mixing between the deuterated protein and the quench solution. The efficient mixing with acetic acid (pH 2.5) and rapid transfer from the labeling step through pepsin digestion to ionization allows this setup to largely avoid back exchange, which is often a significant problem in conventional bottom-up HDX systems, particularly when liquid chromatography (LC) separation is incorporated.23, 33
Figure 1.
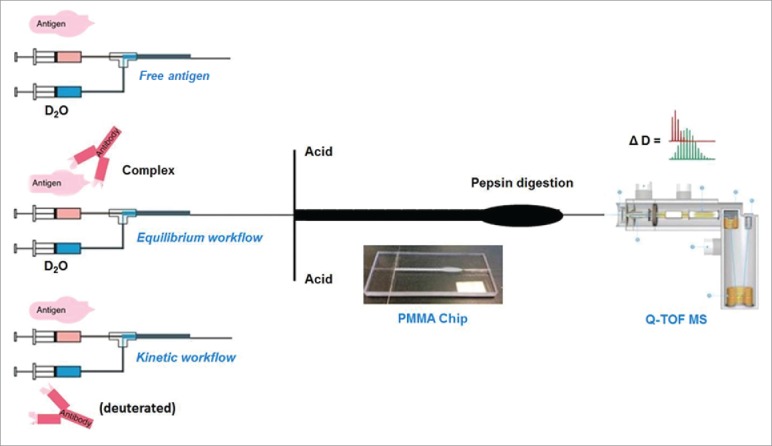
Schematic of the three TRESI-HDX experiments carried out in this work. All three workflows incorporate millisecond HDX labeling followed by acid quenching, digestion, on-chip electrospray ionization, MS detection and data analysis. (Top) TRESI-HDX of free antibody. Peptide-specific uptake data from this experiment are subtracted from‘equilibrium'or ‘kinetic’ experiment data to provide HDX difference profiles associated with complexation. (Middle) ‘Equilibrium’ workflow: Antibody and antigen are pre-equilibrated prior to analysis. (Bottom) ‘Kinetic’ workflow: Antibody is introduced to the antigen through the TRESI mixer, so that binding and labeling are initiated simultaneously.
Characterization of the Mb:anti-Mb complex by native-PAGE and SEC-MS
To confirm the formation of the Mb:anti-Mb complex, native polyacrylamide gel electrophoresis (PAGE) and size-exclusion chromatography (SEC)–MS were used. In the native-PAGE gels (Fig. 2A), a slight retardation was observed for the bound complex (myoglobin-mAb, 167 KDa, lanes 4 and 5) compared to the free antibody (150 kDa, lane 3), which is consistent based on the small size increase. Under the same conditions, Mb (17 KDa, lane 2) had migrated off of the gel. SEC-MS provided a more definitive picture of complexation. The SEC-MS total ion current (TIC) chromatograms are shown in Fig. 2B. Ubiquitin (8.5 kDa), the internal standard, eluted at the same time (56.6 ± 0.1 min) for all runs. Without anti-Mb, Mb (17 kDa) eluted at 54.2 ± 0.1 min. Upon addition of anti-Mb, the Mb signal was detected exclusively at 44.3 ± 0.2 min, indicating complete complexation, which is expected for the concentrations used (KD = 1.78 nM).34
Figure 2.
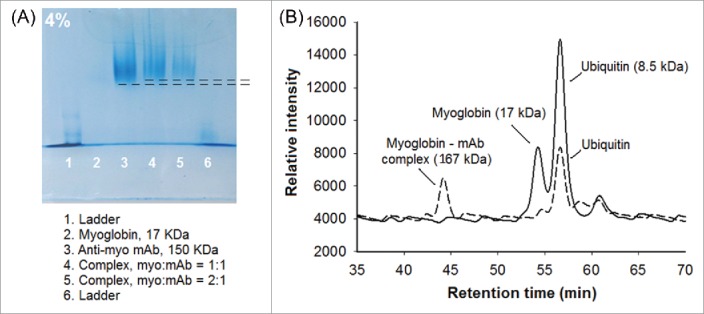
Detection of the Mb:anti-Mb complex. (A) Mb:anti-Mb complexation is detectable in native-PAGE as a slight retardation of migration (lanes 4 and 5) compared to the free antibody (lane 3). (B) SEC-MS chromatograms showing the elution of myoglobin in the absence and presence of anti-Mb (solid line and dashed line, respectively). Ubiquitin was used as an internal standard.
Equilibrium epitope mapping
For equilibrium epitope mapping experiments, anti-Mb was pre-incubated with Mb in a 1:1 ratio (10 μM) and allowed to equilibrate for 30 mins at room temperature. Once the complex was fully equilibrated, the solution was transferred into the microfluidic device for millisecond timescale HDX labeling, quench, digestion and online MS analysis of the resulting peptides. Peptides from Mb were identified by MS/MS and their deuterium uptake levels were calculated using an in-house built program as described previously.31 When anti-Mb was present, peptides from the mAb were observed, but coverage was insufficient to directly identify the paratope via HDX. This is likely because of inefficient digestion by pepsin due to the presence of disulfides. A putative paratope was ultimately determined by docking hyper-variable region peptides to the epitope ultimately determined (see subsequent sections).
In HDX experiments, peptic digestion of Mb resulted in 98% coverage of the Mb sequence with an average segment length of 6 residues. The results of epitope mapping on the equilibrium binding complex are shown in Fig. 3 (left column), which plots the deuterium uptake difference between the Mb:anti-Mb complex and free Mb (i.e., HDXMb:anti-Mb – HDXMb). For the vast majority of peptides, HDX decreased upon complexation, resulting in negative difference values. Using the average change in uptake (15%) as a significance threshold, regions corresponding to 49–70, 108–116, and 140–147 showed an above-average effect, and might tentatively be assigned as corresponding to an epitope. As expected, these results were essentially independent of labeling time, although most regions showed a subtle difference increase over time, which can be attributed to widespread, subtle changes in dynamics upon binding (the subtler the change in HDX accessibility, the longer it takes to ‘develop’ in the HDX difference plot).
Figure 3.

Differences in deuterium uptake (%) between myoglobin-antibody complex and native myoglobin at different reaction times detected by TRHDX-MS. X-axis shows the sequence of the localized peptides. (A-C) Equilibrium workflow: Mb pre-incubated with anti-Mb for 30 minutes prior to HDX-MS analysis. (D-F) Kinetic workflow: Mb was introduced to anti-Mb antibody concomitantly with the onset of labeling in the TRESI mixer.
The identified regions agreed quite well with the closest matching epitopes from literature, which reported regions of 15–21, 56–62, 94–100, 113–119, 146–151 on Mb.35 However, these results were obtained for sperm-whale myoglobin using a polyclonal system and a pepscan approach. To the best of our knowledge, our study is the first to provide epitope information for human myoglobin interacting with a mouse mAb.
Kinetic epitope mapping
The results of the kinetic experiment (described schematically in Fig. 1) are shown in Fig. 3 (right column). At the earliest mixing time of 0.20 s, a 15% difference threshold identifies regions 49–51, 57–60, 112–116, and 140–147, with substantially weaker attenuated uptake in the 49–70 region compared to the equilibrium measurement. Decreases in the 112–116 and 140–147 regions, however, are similar in magnitude to those observed in the equilibrium data. In contrast to the equilibrium experiment, the kinetic experiment profile changes over time, with uptake differences ‘building in’ to ultimately produce a profile that is similar to the equilibrium one after only a few hundred ms of labeling. At 3.92 s, the equilibrium and kinetic profiles are similar enough that they generate essentially identical epitope maps when the ‘greater than average’ 15% cutoff is used, identifying 41–70, 108–116, and 137–147 as potential epitopes.
One interesting feature of the ‘kinetic’ data is the occurrence of relatively faster uptake in some regions shortly after binding. These effects, while not above the ‘hit’ threshold in magnitude, are nonetheless statistically significant and sometimes persistent over the timescale of the measurements, particularly for the short segments 133 – 136 and 148 – 154. Similar increases have been detected previously in the context of ligand binding in HDX pulse labeling experiments,36 and may reflect slow rearrangement of the target protein conformational ensemble to the ‘bound configuration’ after binding.
Peptide mapping of antibody
LC-MS was applied to identify peptides for possible complementarity-determining regions (CDRs) on the antibody. The anti-Mb antibody was digested with trypsin and Glu-C proteases. The resultant peptides were separated by LC-MS and matched to a Mus musculus database. Based on matched peptides, 76% of the secreted form of IgG-1 chain C region sequence was covered. After filtering for common contaminants, 4 peptides were identified that were correctly sequenced, but did not match the Mus musculus database. Specifically, these peptides were: Peptide 1 – mouse IgG1 hypervariable region (Ighv1-62-2) [Uniprot: A0A075B680]; peptide 2 – mouse Ig heavy chain V region [Uniprot: P01741]; peptide 3 – mouse Ighv1-77 [Uniprot: A0A0B4J1M0]; peptide 4 – mouse Igkv14-100 [Uniprot: A0A075B5K7]. These peptides were submitted for epitope-constrained docking simulations to determine if they corresponded to the anti-Mb paratope.
Docking simulations
Docking simulations were carried out using the PatchDock, ZDOCK and Molecular Operating Environment (MOE) software packages. Ten molecular dynamics-minimized docking poses were evaluated for PatchDock and ZDOCK, while five poses were selected from MOE. Simulations were carried out either with no constraints or with ‘site specified’ bias or volume constraints based on the HDX data. Optimal poses were determined by measuring the number of energetically favorable contacts between the peptide and Mb. A residue would be regarded as a ‘hit’ when it was one of the 13 HDX-predicted epitope residues on myoglobin and located within 4.5 Å of a residue from the antibody peptide.
The results showed that for all three software packages, computational docking with HDX-MS constraints produced more hit residues than docking without HDX-MS constraints. Similarly, more ‘hit’ residues were detected for docking at the HDX-specified site compared to randomly selected sites. In three-dimensional (3D)-modeling of the optimal poses for peptides 1 – 3, however, it quickly became evident that these peptides could only recognize the putative epitope region I112-L116 on myoglobin, i.e., they were simply not large enough to create a discontinuous epitope for any pair of sites identified by HDX and they lacked specificity for the other two sites. On the other hand, peptide 4 showed recognition to two identified regions corresponding to I112-L116 and R140-Y147 on myoglobin. Therefore, peptide 4 was chosen to generate a predicted binding complex model (Fig. 4). The other site identified in equilibrium and longer-labeling kinetic experiments (49 – 70) is located on the opposite face of Mb and showed substantially lower scores for all peptides.
Figure 4.

Computational docking on myoglobin with the peptides from the anti-myoglobin mAb. The software of MOE, PatchDock, and ZDOCK were used to generate the binding models. (A) The peptide 4 (Uniprot: A0A075B5K7) showed a significant difference in docking with and without HDX constraints by using ZDOCK and (B) the binding model of the peptide 4 with myoglobin.
Computational predictions of allostery
To probe the presence of allosteric effects, we applied the rigidity-transmission allostery (RTA) algorithm, which can assess the extent to which binding events at one site on the protein allosterically influence remote regions. RTA analysis, which utilizes rigidity theory37 and the FIRST computational method,38 predicts the extent to which local mechanical perturbation of rigidity (mimicking ligand binding) at one region can transmit across a protein structure and cause a change in rigidity and conformational degrees of freedom at a second distant region. RTA has been shown to effectively capture allosteric effects in various protein structures, including enzymes and membrane receptors.39
We first used the FIRST program to decompose the myoglobin structure into flexible and rigid regions (Figure 5A, B) and then applied the RTA algorithm to evaluate if perturbation of rigidity in regions 112–116 and 140–147 caused changes in rigidity and conformational degrees of freedom in 41–70 (Figure 5C). The RTA algorithm calculates the available conformational degrees of freedom at residues 41–70 before and after perturbation of rigidity in the 112–116 and 140–147 regions. In this way, any subsequent change in conformational degrees of freedom due to the initial perturbation is obtained. Mechanically, the presence of rigidity-based allostery (positive degree of freedom transmission) means that a change in shape or rigidity at one site will lead to a change in shape or rigidity at the second site.
Figure 5.
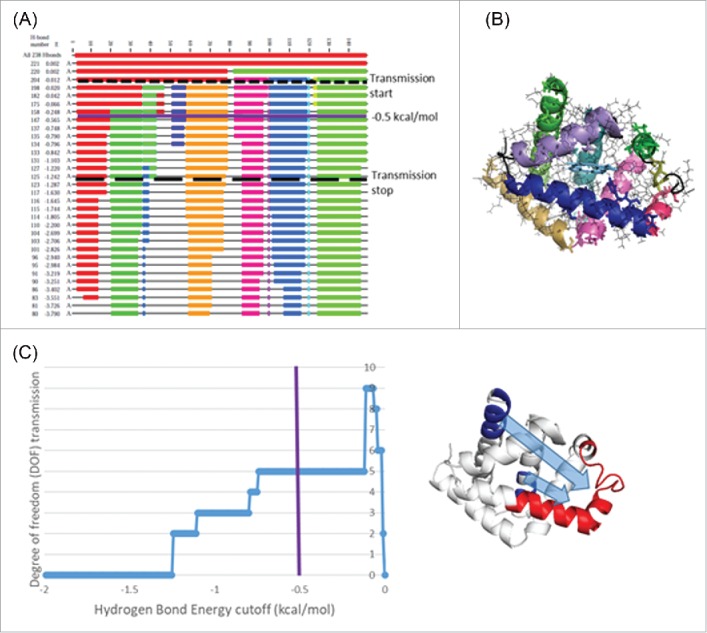
Computational allostery predictions using RTA analysis. (A) Dilution plot of myoglobin (3rgk) using FIRST. The horizontal axis represents the residue numbers and vertical axis the current hydrogen bond energy cutoff in kcal/mol. Flexible regions of the polypeptide chain appear as black thin lines and rigid clusters as colored blocks. Dash line represent the energy cutoff associated with start of allosteric transmission and long dash line represents the end of allosteric transmission (as shown in C). As hydrogen bonds cutoff is decreased, rigid clusters break up and other parts of the chain become flexible. (B) Rigid clusters mapped onto 3D protein structure at -0.5 kcal/mol. (C) The transmission of conformational degrees of freedom (DOF) from region 112–116 and 140–147 (shown in blue) to region 41–70 (shown in red) (i.e., a change in rigidity at site one propagating to modify rigidity at site two) as a function of energy cutoff. Positive DOF transmission demonstrates the presence of allosteric transmission between the two regions.
Binding events at residues 112–116 and 140–147 introduce new local constraints, and RTA analysis shows that this initial perturbation of rigidity resulted in a change in the available degrees of freedom specifically in the 41–70 region (Figure 5C), hence computationally demonstrating that allosteric transmission propagates between the two sites. Perturbation of rigidity of both 112–116 and 140–147 regions is required to observe the observed allosteric effect in 41–70 region; however, the RTA analysis predicts allosteric transmission is slightly more dependent on the binding events in 112–116 region (data not shown). Furthermore, allosteric transmission persists for a wide range of energy cutoffs, which indicates ‘robust’ allosteric communication and means that small changes in the hydrogen bonding network (i.e., hydrogen bond flickering) will likely not significantly affect the allosteric transmission.
Discussion
In this work, two epitope mapping workflows of TRESI-HDX, ‘equilibrium’ and ‘kinetic’, were compared using human myoglobin with monoclonal mouse anti-Mb as a model. Based on a ‘greater than average decrease’ criterion, peptide regions with over 15% decrease in deuterium uptake were selected as potential epitopes. The epitopes identified by each workflow at different D2O labeling times (0.20, 0.94 and 3.92 s) are highlighted as blue regions on the myoglobin 3D structures in Fig. 6. It is clear from this representation of the data that the kinetic workflow ultimately produced similar epitope predictions (namely 49–70, 108–116, and 137–147) to the equilibrium workflow. The essential difference is that, while the equilibrium workflow generated an unchanging profile as a function of labeling time (Fig. 6A-C), short interaction time profiles from the kinetic experiment exhibit a substantial bias towards the 108–116 and 137–147 regions (Fig. 6D). The detection of transient conformational changes in mAb:target binding could be critical for the understanding of the mechanism and physiological consequences of a wide array of antibody:antigen interactions. The same workflow can be applied to the investigation of other antigens and antibodies, as well as other types of interactions, such as enzyme-substrate and drug-target reactions.
Figure 6.
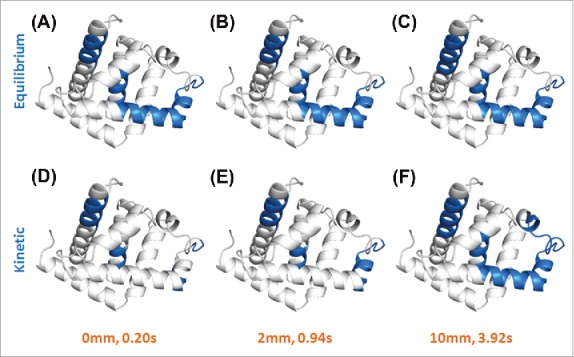
Epitopes on human myoglobin were indicated as blue regions on its 3D structures. The binding complexes were obtained by both equilibrium (A-C) and kinetic (D-F) workflows with TRHDX-MS detection at different D2O labeling time (0.20, 0.94 and 3.92 s). Figures were drawn using PyMOL software. Compared to the equilibrium workflow, the kinetic workflow can clearly show the dynamics of conformational changes on myoglobin during the mAb binding process on a sub-second time scale.
Allosteric effects, which involve conformational or dynamic changes remote from the active (or binding) site40 are critical to a wide array of protein functions.27, 41 However, in an epitope mapping experiment, allosteric effects are essentially false positives.16 In conventional HDX-MS epitope mapping (with 10 s to multiple hours labeling times), and even for the TRESI-MS ‘equilibrium’ experiments conducted here, pre-incubation for extended periods ensures not only that complexation is equilibrated, but also that allosteric effects have had orders of magnitude more time than needed to fully develop prior to analysis. In the ‘kinetic’ workflow demonstrated here, complexation and HDX labeling are initiated simultaneously, which allows the HDX signal to develop as allostery is propagated after a binding event. The result is a novel molecular probe of allosteric propagation with a critical capability, at least in the system studied here, of helping to distinguish between binding and allostery in epitope mapping experiments.
While the results of the ‘kinetic’ workflow experiment are unambiguous and reproducible, it is difficult to connect what we observe – the development of an ‘allosteric’ HDX signature over a period of milliseconds – to what is commonly understood about the biophysics of allostery from other measurements. Computational predictions in particular suggest that the transmission of conformational changes associated with allostery should occur on the ns – low μs timescale.42, 43 Nonetheless, some studies have reported substantially slower allosteric effects in small proteins,44 including pulse-labeling experiments from our group,36 and the occurrence of allosteric conformational changes on the seconds timescale has long been established in large (usually transmembrane) systems.45, 46
Exploration of the physical basis of the slow (ms) allostery we report here, as well as the application of the ‘kinetic’ workflow to trace allosteric pathways in other systems like enzymes and non-antibody protein complexes, are clear avenues for further work. We have here shown that millisecond HDX using the ‘kinetic’ workflow may provide a path to HDX-based epitope mapping experiments in which allosteric effects are selectively suppressed. This would represent a substantial improvement for a technique that is rapidly being adopted both in academic and industrial research.
Method and materials
Materials
Human myoglobin (ab96036, ∼ 17 KDa) and anti-myoglobin mAb (ab19607, ∼ 150 KDa) were purchased from Abcam (Cambridge, MA, USA). Pepsin (P6887), ubiquitin (U6253), deuterium oxide (D2O, 99.9%, 151882) and high purity acetic acid (>99.7%, 695092) were purchased from Sigma-Aldrich (St. Louis, MO). Endoproteinase GluC (P8100S) was purchased from New England Biolabs (Hitchin, UK). Agarose resin (NHS-activated, 26196) was purchased from Thermo Scientific (Rockford, IL). HPLC-grade ammonium acetate, acetonitrile, methanol, water and methyl yellow (151400250) were purchased from Fisher Chemical (Fair Lawn, NJ).
Native gel separation of the myoglobin-mAb complex
The binding complex of myoglobin and anti-Mb mAb was separated by native polyacrylamide gel electrophoresis (PAGE) following the protocol from the Institute of Molecular Development LLC (http://www.molecularinfo.com/). Ten μL of proteins (1.7 μg) and complex (15 μg) samples were loaded in each lane and a voltage of 120 volts was applied for 100 min. The results were indicated by Coomassie brilliant blue R250 staining and photos were taken by AlphaImager HP imaging system (Alpha, San Jose, CA).
SEC-MS of the myoglobin-mAb complex
Twenty μL of 10 μM myoglobin, anti-Mb mAb, and myoglobin-antibody complex with ubiquitin as the internal standard were characterized using an Agilent 1200 HPLC (Santa Clara, CA) with BioSep-SEC-S4000 column (5 μm, 300 × 7.8 mm, Phenomenex, Torrance, CA) coupled with the Synapt G1 HDMS (Waters, Milford, MA). The mobile phase was 200 mM ammonium acetate (pH 7.4) with a flow rate of 0.2 mL/min. Data was collected and analyzed using Waters MassLynx 4.1 software.
Microfluidic chip fabrication for HDX-MS
The chip device was fabricated on a rectangular piece of PMMA with dimensions of 8.9 cm × 3.7 cm × 0.5 cm. The micro-channels, microstructures, and digestion well within the chip were designed using CorelDRAW X3 software (Ottawa, ON) and were engraved on PMMA using the VersaLaser (Universal Laser Systems, Scottsdale, AZ). The solutions were driven by syringes (Hamilton, Reno, NV) on infusion pumps (Harvard, Holliston, MA).
For HDX labeling, the TRESI apparatus was assembled as described previously.31, 32 Briefly, a polyimide-coated fused silica capillary (I.D. 75 μm, O.D. 150 μm, Polymicro, Phoenix, AZ) was inserted into a stainless metal capillary (I.D. 178 μm, O.D. 356 μm, McMaster-Carr, Aurora, OH) via a three-way PEEK union (IDEX, Lake Forest, IL). Mixing of the two solutions occurred within the intercapillary space when the solution was released from a notch cut 2 mm from the sealed inner capillary end. HDX profiles were acquired by steadily pulling back the inner capillary to increase the reaction volume that resulted in the increase of the labeling times. Under these conditions, back exchange has been shown to be negligible (< 5%) due to the rapid (< 4 s) transfer from labeling to ionization under quench conditions.31
TRHDX-MS for equilibrium and kinetic epitopes
Proteins were desalted before MS detection by buffer exchange with 200 mM ammonium acetate (pH 7.0) using Zeba Spin Desalting columns (89890, 7K, Thermo) on Allegra 25R centrifuge (Beckman, Mississauga, ON) by centrifugation at 1000 g/min at 4°C.
In the equilibrium workflow, 10 μM myoglobin was incubated with 10 μM anti-Mb mAb in 200 mM ammonium acetate (pH 7.4) for 30 min at room temperature. The myoglobin-antibody complex was then labeled with D2O in the TRESI device from 0.20 s to 3.92 s. The HDX reaction was quenched in the chip by rapidly mixing with 5% acetic acid of pH 2.5. The binding complex was then digested by pepsin-linked agarose resin in the digestion well on the chip. The peptides were electrosprayed into a QStar Elite Qq-TOF mass spectrometer (AB Sciex, Concord, ON). The MS data was analyzed using AB Sciex Analyst 2.0 software. By comparison between the equilibrium complex and native myoglobin, the regions of antibody-bound myoglobin showing significantly less D2O uptake were identified as equilibrium epitopes.
In the kinetic workflow, the mAb was initially incubated with excess D2O for 30 min at room temperature. The deuterated antibody was then mixed with myoglobin at the same labeling times from 0.20 s to 3.92 s. The myoglobin-mAb complex was kinetically formed while the D2O labeling was initiated. Similarly, HDX was stopped by acid quenching, followed by protease digestion and MS detection. Kinetic epitopes were recognized by comparison between the kinetically formed Mb-mAb complex and native myoglobin. The regions showing significantly less exchange were identified as kinetic epitopes.
HDX data analysis
The peptides from the digested myoglobin were screened using FindPept on the ExPASy Server (Swiss Institute of Bioinformatics, Basel, Switzerland). Deuterium uptake of each peptide was calculated using an in-house written FORTRAN software for isotopic distribution analysis and was normalized to the maximum D2O uptake of 50%. HDX on myoglobin structures were rendered using PyMOL 1.8 software.
Digestion of the anti-Mb mAb
Dual-protease digestion was carried out to map peptides of the anti-Mb mAb. Twenty μL of 15 μM anti-Mb mAb was reduced with 20 mM dithiothreitol in 0.5 M Tris-HCl (pH 8.0) for 60 min at 37°C. After that, the antibody was alkylated with 10 mg/mL iodoacetamide (1/1, v/v) in 10 mM ammonium bicarbonate at room temperature for 10 min in the dark. The sample was then treated with 0.1 mg/mL trypsin with a 20:1 protein:protease w/w ratio at 37°C overnight. For further digestion, the sample was added to 0.1 mg/mL Glu-C with a 40:1 protein:protease w/w ratio at 37°C for an additional 2 hours. The enzymatic activity was quenched by adding 1 μL 10% acetic acid. The digested anti-Mb mAb peptides were desalted using C18 ZipTip pipette tips (EMD Millipore, Etobicoke, ON) prior to LC-MS analysis. Briefly, ZipTips were cleaned with acetonitrile and equilibrated with water. The sample was bound to the ZipTips and washed with 0.1% formic acid in water. The peptides were eluted using 20 µL of 80% acetonitrile, dried using a speed vacuum centrifuge, and reconstituted in 0.1% formic acid.
LC-MS/MS for antibody peptide mapping
The peptides were separated by reversed-phase chromatography using an Agilent 1200 HPLC with Aeris Peptide XB-C18 column (Phenomenex) (3.6 μm, 150 × 4.6 mm) coupled with Waters Synapt G1 HDMS or Thermo Orbitrap Elite MS (Thermo, Waltham, MA). Samples were loaded onto the column using a loading buffer composition of 5% acetonitrile and 0.1% formic acid at a flow rate of 200 µL/min for 15 min. The acetonitrile composition was linearly increased to 100% in 80 min followed by a column re-equilibration step for 30 min. The LC-MS/MS data were processed using Waters PLGS 3.0 software or using Thermo Proteome Discoverer 1.4 software. The data were matched to a Mus musculus database (Uniprot, April 20th, 2016) with the addition of porcine trypsin and common human contaminants using a 10 ppm mass accuracy threshold, the oxidation of methionine residues as a variable modification and the carbamidomethylation of cysteine residues as a fixed modification. The protein matches were processed with a 1% false-discovery rate and a 99% peptide confidence cutoff.
Computational docking
The structure of myoglobin was extracted and modified from the crystal structure of human myoglobin (PDB code: 3RGK). Based on the segments of antibody structure, the structures of four peptides from anti-Mb mAb were generated with MOE 2013.08.47 Three approaches, ZDOCK,48 PatchDock49 and MOE, were utilized to dock myoglobin with the four peptides. In the docking, the modes with or without HDX-MS constraints were both applied. In docking without HDX-MS constraints, there was no constraint for all three software programs, where the docking results were only dependent upon the docking algorithms. While in docking with HDX-MS constraints, the epitope residues of I112-L116 and R140-Y147 of myoglobin were set as the docking sites in PatchDock and MOE, and as volume constraint pharmacophores in MOE. In ZDOCK, non-epitope residues of myoglobin were blocked as a scoring penalty.
Computational allostery prediction
Starting with a crystal structure of myoglobin (PDB code: 3rgk), missing hydrogen atoms were added using the WHAT IF web server (http://swift.cmbi.ru.nl/ servers/html/htopo.html). FIRST was run as previously described.38 FIRST generates a constraint network consisting of nodes (atoms) and edges (i.e., constraints representing covalent bonds, hydrogen bonds, hydrophobic and electrostatic interactions). Each potential hydrogen bond is assigned an energy strength dependent on donor-hydrogen acceptor geometry (reference), and a hydrogen bond energy cutoff value is selected where all bonds weaker than this cutoff are ignored in the network. The resulting protein network is then decomposed into rigid clusters and flexible regions. The patterns of rigidity loss during progressive removal of hydrogen bonds is visualized on ‘dilution plot’ (Fig. 5A).38 For dilution, FIRST performs rigidity analysis with all potential hydrogen bonds of energies 0 kcal/mol or lower. Hydrogen bonds are then removed in order of increasing strength gradually reducing the rigidity of the structure. Rigidity calculation over the main-chain and side-chain and rigid cluster decomposition is performed every time a hydrogen bond is removed from the list of constraints.
Once the output of FIRST rigid cluster decomposition is generated, the rigidity-transmission allostery (RTA) algorithm is applied to probe the allosteric transmission between regions 112–16, 140–147 and region 41–70.39 The number of conformational degrees of freedom at 41–70 region was calculated before and after perturbation of rigidity of 112–116 and 140–147, and subsequent degree of freedom transmission is obtained as a function of hydrogen bond energy cutoff as described by Kim et al.39 Positive degree of freedom transmission indicates presence of rigidity-based allostery.
Disclosure of potential conflicts of interest
Authors declare that no potential conflicts of interest.
Acknowledgments
The Wilson lab is supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada Discovery (504027), Engage (492095-15) and Collaborative Research and Development (CRD) (485321-15) Programs. Additional support is provided by the Krembil Foundation, the Canadian Alzheimer's Society, Mathematics of Information Technology and Complex Systems (MITACS) and the Ontario Ministry of Research and Innovation (MRI).
References
- 1.Buss NAPS, Henderson SJ, McFarlane M, Shenton JM, de Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 2012;12:615-22. doi: 10.1016/j.coph.2012.08.001. PMID:22920732 [DOI] [PubMed] [Google Scholar]
- 2.Rodgers KR, Chou RC. Therapeutic monoclonal antibodies and derivatives: Historical perspectives and future directions. Biotechnol Adv. 2016;34:1149-58. doi: 10.1016/j.biotechadv.2016.07.004. PMID:27460206 [DOI] [PubMed] [Google Scholar]
- 3.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301-16. doi: 10.1038/nri2761. PMID:20414204 [DOI] [PubMed] [Google Scholar]
- 4.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317-27. doi: 10.1038/nri2744. PMID:20414205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol. 2014;32:992-1000. doi: 10.1038/nbt.3040. PMID:25299917 [DOI] [PubMed] [Google Scholar]
- 6.Barlow DJ, Edwards MS, Thornton JM. Continuous and discontinuous protein antigenic determinants. Nature. 1986;322:747-8. doi: 10.1038/322747a0. PMID:2427953 [DOI] [PubMed] [Google Scholar]
- 7.Gershoni JM, Roitburd-Berman A, Siman-Tov DD, Tarnovitski Freund N, Weiss Y. Epitope mapping: the first step in developing epitope-based vaccines. BioDrugs Clin Immunother Biopharm Gene Ther. 2007;21:145-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen E, Salinas ND, Huang Y, Ntumngia F, Plasencia MD, Gross ML, Adams JH, Tolia NH. Broadly neutralizing epitopes in the Plasmodium vivax vaccine candidate Duffy Binding Protein. Proc Natl Acad Sci U S A. 2016;113:6277-82. doi: 10.1073/pnas.1600488113. PMID:27194724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bardelli M, Livoti E, Simonelli L, Pedotti M, Moraes A, Valente AP, Varani L. Epitope mapping by solution NMR spectroscopy. J Mol Recognit JMR. 2015;28:393-400. doi: 10.1002/jmr.2454. PMID:25726811 [DOI] [PubMed] [Google Scholar]
- 10.Atassi MZ. Antigenic structure of myoglobin: the complete immunochemical anatomy of a protein and conclusions relating to antigenic structures of proteins. Immunochemistry. 1975;12:423-38. doi: 10.1016/0019-2791(75)90010-5. PMID:53189 [DOI] [PubMed] [Google Scholar]
- 11.Suckau D, Köhl J, Karwath G, Schneider K, Casaretto M, Bitter-Suermann D, Przybylski M. Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A. 1990;87:9848-52. doi: 10.1073/pnas.87.24.9848. PMID:1702219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin null, Perdue null . Site-Directed Mutagenesis in Epitope Mapping. Methods San Diego Calif. 1996;9:508-15. doi: 10.1006/meth.1996.0058 [DOI] [PubMed] [Google Scholar]
- 13.Grellier B, Le Pogam F Vitorino M, Starck J-P, Geist M, Duong V, Haegel H, Menguy T, Bonnefoy J-Y, Marchand J-B, et al.. 3D modeling and characterization of the human CD115 monoclonal antibody H27K15 epitope and design of a chimeric CD115 target. mAbs. 2014;6:533-46. doi: 10.4161/mabs.27736. PMID:24492308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197-208. doi: 10.1038/nrm1589. PMID:15738986 [DOI] [PubMed] [Google Scholar]
- 15.Frueh DP, Goodrich AC, Mishra SH, Nichols SR. NMR methods for structural studies of large monomeric and multimeric proteins. Curr Opin Struct Biol. 2013;23:734-9. doi: 10.1016/j.sbi.2013.06.016. PMID:23850141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandit D, Tuske SJ, Coales SJ, E SY, Liu A, Lee JE, Morrow JA, Nemeth JF, Hamuro Y. Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J Mol Recognit JMR. 2012;25:114-24. doi: 10.1002/jmr.1169. PMID:22407975 [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Cui W, Gross ML. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Lett. 2014;588:308-17. doi: 10.1016/j.febslet.2013.11.027. PMID:24291257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leurs U, Mistarz UH, Rand KD. Getting to the core of protein pharmaceuticals–Comprehensive structure analysis by mass spectrometry. Eur J Pharm Biopharm Off J Arbeitsgemeinschaft Für Pharm Verfahrenstechnik EV. 2015;93:95-109 [DOI] [PubMed] [Google Scholar]
- 19.Jardine I. Electrospray ionization mass spectrometry of biomolecules. Nature. 1990;345:747-8. doi: 10.1038/345747a0 [DOI] [Google Scholar]
- 20.Loo JA. Electrospray ionization mass spectrometry: a technology for studying noncovalent macromolecular complexes. Int J Mass Spectrom. 2000:175-86. doi: 10.1016/S1387-3806(00)00298-0 [DOI] [Google Scholar]
- 21.Deng B, Lento C, Wilson DJ. Hydrogen deuterium exchange mass spectrometry in biopharmaceutical discovery and development – A review. Anal Chim Acta. 2016;940:8-20. doi: 10.1016/j.aca.2016.08.006. PMID:27662755 [DOI] [PubMed] [Google Scholar]
- 22.Pirrone GF, Iacob RE, Engen JR. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal Chem. 2015;87:99-118. doi: 10.1021/ac5040242. PMID:25398026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci Publ Protein Soc. 1993;2:522-31. doi: 10.1002/pro.5560020404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konermann L, Pan J, Liu Y-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224-34. doi: 10.1039/C0CS00113A. PMID:21173980 [DOI] [PubMed] [Google Scholar]
- 25.Englander JJ, Del Mar C, Li W, Englander SW, Kim JS, Stranz DD, Hamuro Y, Woods VL. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc Natl Acad Sci U S A. 2003;100:7057-62. doi: 10.1073/pnas.1232301100. PMID:12773622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hager-Braun C, Tomer KB. Determination of protein-derived epitopes by mass spectrometry. Expert Rev Proteomics. 2005;2:745-56. doi: 10.1586/14789450.2.5.745. PMID:16209653 [DOI] [PubMed] [Google Scholar]
- 27.Konermann L. Heavy lessons in protein allostery. Nat Struct Mol Biol. 2016;23:511-2. doi: 10.1038/nsmb.3234. PMID:27273638 [DOI] [PubMed] [Google Scholar]
- 28.Resetca D, Wilson DJ. Characterizing rapid, activity-linked conformational transitions in proteins via sub-second hydrogen deuterium exchange mass spectrometry. FEBS J. 2013;280:5616-25. doi: 10.1111/febs.12332. PMID:23663649 [DOI] [PubMed] [Google Scholar]
- 29.Resetca D, Haftchenary S, Gunning PT, Wilson DJ. Changes in signal transducer and activator of transcription 3 (STAT3) dynamics induced by complexation with pharmacological inhibitors of Src homology 2 (SH2) domain dimerization. J Biol Chem. 2014;289:32538-47. doi: 10.1074/jbc.M114.595454. PMID:25288792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S, Shala A, Bezginov A, Sljoka A, Audette G, Wilson DJ. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PloS One. 2015;10:e0120416. doi: 10.1371/journal.pone.0120416. PMID:25767879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rob T, Liuni P, Gill PK, Zhu S, Balachandran N, Berti PJ, Wilson DJ. Measuring dynamics in weakly structured regions of proteins using microfluidics-enabled subsecond H/D exchange mass spectrometry. Anal Chem. 2012;84:3771-9. doi: 10.1021/ac300365u. PMID:22458633 [DOI] [PubMed] [Google Scholar]
- 32.Wilson DJ, Konermann L. A capillary mixer with adjustable reaction chamber volume for millisecond time-resolved studies by electrospray mass spectrometry. Anal Chem. 2003;75:6408-14. doi: 10.1021/ac0346757. PMID:16465695 [DOI] [PubMed] [Google Scholar]
- 33.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815-20. doi: 10.1021/ac8008862. PMID:18672890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anti-Myoglobin antibody [EP3081Y] (ab77232) | Abcam product information [accessed July 2017]. http://www.abcam.com/myoglobin-antibody-ep3081y-ab77232.html
- 35.Atassi MZ. Antigenic structure of myoglobin: the complete immunochemical anatomy of a protein and conclusions relating to antigenic structures of proteins. Immunochemistry. 1975;12:423-38. doi: 10.1016/0019-2791(75)90010-5. PMID:53189 [DOI] [PubMed] [Google Scholar]
- 36.Rob T, Gill PK, Golemi-Kotra D, Wilson DJ. An electrospray ms-coupled microfluidic device for sub-second hydrogen/deuterium exchange pulse-labelling reveals allosteric effects in enzyme inhibition. Lab Chip. 2013;13:2528-32. doi: 10.1039/c3lc00007a. PMID:23426018 [DOI] [PubMed] [Google Scholar]
- 37.Whiteley W. Counting out to the flexibility of molecules. Phys Biol. 2005;2:S116-126. doi: 10.1088/1478-3975/2/4/S06. PMID:16280617 [DOI] [PubMed] [Google Scholar]
- 38.Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Protein flexibility predictions using graph theory. Proteins. 2001;44:150-65. doi: 10.1002/prot.1081. PMID:11391777 [DOI] [PubMed] [Google Scholar]
- 39.Kim TH, Mehrabi P, Ren Z, Sljoka A, Ing C, Bezginov A, Ye L, Pomès R, Prosser RS, Pai EF. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science. 2017;355:eaag2355. doi: 10.1126/science.aag2355. PMID:28104837 [DOI] [PubMed] [Google Scholar]
- 40.Sowole MA, Simpson S, Skovpen YV, Palmer DRJ, Konermann L. Evidence of Allosteric Enzyme Regulation via Changes in Conformational Dynamics: A Hydrogen/Deuterium Exchange Investigation of Dihydrodipicolinate Synthase. Biochemistry (Mosc). 2016;55:5413-22. doi: 10.1021/acs.biochem.6b00764 [DOI] [PubMed] [Google Scholar]
- 41.Laskowski RA, Gerick F, Thornton JM. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009;583:1692-8. doi: 10.1016/j.febslet.2009.03.019. PMID:19303011 [DOI] [PubMed] [Google Scholar]
- 42.Hacisuleyman A, Erman B. Entropy Transfer between Residue Pairs and Allostery in Proteins: Quantifying Allosteric Communication in Ubiquitin. Plos Comput Biol. 2017;13:e1005319. doi: 10.1371/journal.pcbi.1005319. PMID:28095404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ceruso MA, Periole X, Weinstein H. Molecular dynamics simulations of transducin: interdomain and front to back communication in activation and nucleotide exchange. J Mol Biol. 2004;338:469-81. doi: 10.1016/j.jmb.2004.02.064. PMID:15081806 [DOI] [PubMed] [Google Scholar]
- 44.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831-8. doi: 10.1038/nsmb1132. PMID:16906160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taraska JW, Zagotta WN. Structural dynamics in the gating ring of cyclic nucleotide-gated ion channels. Nat Struct Mol Biol. 2007;14:854-60. doi: 10.1038/nsmb1281. PMID:17694071 [DOI] [PubMed] [Google Scholar]
- 46.Li J, Callaway DJE, Bu Z. Ezrin Induces Long-Range Interdomain Allostery in the Scaffolding Protein NHERF1. J Mol Biol. 2009;392:166-80. doi: 10.1016/j.jmb.2009.07.005. PMID:19591839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2016 [Google Scholar]
- 48.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinforma Oxf Engl. 2014;30:1771-3. doi: 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mashiach E, Schneidman-Duhovny D, Peri A, Shavit Y, Nussinov R, Wolfson HJ. An integrated suite of fast docking algorithms. Proteins. 2010;78:3197-204. doi: 10.1002/prot.22790. PMID:20607855 [DOI] [PMC free article] [PubMed] [Google Scholar]