

ABSTRACT
Patent expiration of first-generation biologics and the high cost of innovative biologics are 2 drivers for the development of biosimilar products. There are, however, technical challenges to the production of exact copies of such large molecules. In this study, we performed a head-to-head comparison between the originator anti-VEGF-A Fab product LUCENTIS® (ranibizumab) and an intended copy product using an integrated analytical approach. While no differences could be observed using size-exclusion chromatography, capillary electrophoresis-sodium dodecyl sulfate and potency assays, different acidic peaks were identified with cation ion exchange chromatography and capillary zone electrophoresis. Further investigation of the intact Fab, subunits and primary sequence with mass spectrometry demonstrated the presence of a modified light chain variant in the intended copy product batches. This variant was characterized with a mass increase of 27.01 Da compared to the originator sequence and its abundance was estimated in the range of 6–9% of the intended copy product light chain. MS/MS spectra interrogation confirmed that this modification relates to a serine to asparagine sequence variant found in the intended copy product light chain. We demonstrated that the integration of high-resolution and sensitive orthogonal technologies was beneficial to assess the similarity of an originator and an intended copy product.
KEYWORDS: RAZUMAB, LUCENTIS®, biosimilar, intended copy product, sequence variant, amino acid substitution, misincorporation, error-tolerant search, time-resolved deconvolution, mass spectrometry
Abbreviations
- 2D
2-dimensional
- 3D
3-dimensional
- ACN
acetonitrile
- AMD
age-related macular degeneration
- CDR
complementarity determining region
- CE-SDS
capillary electrophoresis sodium dodecyl sulfate
- CEX
cation ion exchange chromatography
- CZE
capillary zone electrophoresis
- CHO
Chinese hamster ovary
- ESI-MS
electrospray mass spectrometry
- DNA
DNA
- Fab
fragment antigen-binding
- Fc
fragment crystallizable
- FDA
US. Food and Drug Administration
- GuHCl
Guanidine-HCl
- HC
heavy chain
- HMW
high molecular weight
- IPA
isopropyl alcohol
- LC
light chain
- LC-MS
liquid chromatography – mass spectrometry
- mAb
monoclonal antibody
- MES
2-(N-morpholino)ethanesulfonic acid
- mRNA
messenger ribonucleic acid
- MS
mass spectrometry
- m/z
mass-to-charge ratio
- PTMs
post-translational modifications
- QTPP
quality target product profile
- RT
retention time
- SDS
sodium dodecyl sulfate
- SEC
size-exclusion chromatography
- TFA
trifluoroacetic acid
- tRNA
transfer ribonucleic acid
- UV
ultraviolet
- VEGF-A
vascular endothelial growth factor A
- WHO
World Health Organization
Introduction
More than 50 monoclonal antibodies (mAbs) have been approved in the European Union (EU) or United States (US) for the treatment of multiple indications since the 1980s.1-3 Six novel antibody therapeutics were approved for commercial use in the EU or US in 2016 and another 10 are currently planned for regulatory reviews in cancer and non-cancer indications in 2017.4 The number of mAbs currently under evaluation in Phase 3 clinical studies doubled since 2010, with 52 candidates as of early 2017, while over 230 molecules were in Phase 2 clinical development.4
In parallel, the high cost of innovative biological medicines is being challenged by health regulators worldwide and patents of several first-generation biological products will sequentially expire, promoting the development of biosimilars. The European Medicines Agency has been authorizing biosimilar products since 2006, with the first biosimilar mAb authorized in 2013, and there are now 19 valid biosimilar product marketing authorizations.3 The first US biosimilar was licensed in 2015.5,6 As defined by regulators, biosimilars are biological products highly similar to their respective reference products and for which similarity has been demonstrated in terms of quality, safety and effectiveness, although clinically inactive minor variations may still exist. The development of such products is attractive for healthcare systems worldwide, as they may lower prices for products losing commercial exclusive rights and increase patient access to a bioequivalent treatment. EU and US have established guidelines for the authorization and licensing of biosimilars, while WHO provides guidelines for other regions so that safe and effective similar biotherapeutic products can be made available to patients.7-11
During the development of biosimilars, analytical characterization is required to demonstrate similarity of the biosimilar compared to the originator molecule. Evidence of high similarity to the quality target product profile (QTPP), as defined by the range of batch-to-batch variability of the originator batches, may substantially shorten development timelines and reduce the costs associated with clinical programs.6,12 Side-by-side similarity assessment of multiple batches is generally performed throughout the entire biosimilar development. Producing “generic” versions of large molecules is not as straightforward as for making small molecule generic drugs due to their size, their complexity, their heterologous expression in living cells, and their sensitivity to upstream and downstream processes-relevant parameters such as pH, temperature and shear stresses. Overall, the manufacturing of biologics in mammalian or microbial cells is more complex and generates more variability than the synthesis of chemical drugs. Proteins, either native or recombinant, are amenable to post-translational modifications (PTMs), including signal peptide processing, glycosylation, phosphorylation, C-terminal lysine truncation. Additionally, chemical modifications (e.g., oxidation, deamidation, isomerization, modification by reactive metabolites in cell culture), clipping and aggregation can arise during expression, recovery, purification and storage in the final formulation.13, 14
Besides PTMs, sequence variants can be introduced during the translation step of the protein in the host system, as unintended amino acid substitution, and can lead to increased product microheterogeneity.15,16 Sequence variations can also result from differences in the coding DNA.17-20 As a result, the degree of PTMs and variants for these large molecules may differ between host cells, culture systems, downstream processes and formulations. Examples of such differences between an originator and a biosimilar product have been reported in literature. Differences in methionine oxidation, in addition to different glycosylation patterns, were previously observed in a candidate biosimilar of trastuzumab.21 A recent study of filgrastim modifications, including misincorporations, amino acid extension, and truncations, estimated their levels ranging between 0.1 and 2% across different manufacturers.22 Although these variations may not be clinically relevant at these low levels, the implementation and the integration of sensitive orthogonal analytical approaches (for a review see ref.13) are required to uncover true and relevant differences between an originator and intended copy product, when one single technique may fail in that exercise (e.g., comparison of UV chromatograms).
LUCENTIS® (ranibizumab, Novartis Pharma AG as a licensee of Genentech, Inc.) is a humanized mAb antigen-binding fragment (Fab) that targets the human vascular endothelial growth factor A (VEGF-A) for the treatment of neovascular age-related macular degeneration (wet AMD), retinal vein occlusion (RVO), and diabetic macular edema (DME). Wet AMD is a progressive condition in which central vision is affected, resulting in gradual vision loss to blindness. The LUCENTIS® 48 kDa Fab, expressed in E.coli, is composed of a 214-residue light chain connected via one inter-chain disulfide bond to a 231-residue heavy chain fragment. In this study, we performed a head-to-head comparison of LUCENTIS® and RAZUMAB using a panel of analytical techniques. RAZUMAB is an intended copy product of LUCENTIS® licensed in India, but currently not licensed or authorized in other regions such as US or EU. Therefore, the term intended copy product is used in this article, rather than biosimilar, in accordance with the published terminology.23-25 While no differences in terms of size variants and potency were observed between these products using size-exclusion chromatography (SEC), capillary electrophoresis under denaturing conditions with sodium dodecyl sulfate (CE-SDS) and potency assays, differences in charge variants were observed using cation ion exchange chromatography (CEX) and capillary zone electrophoresis (CZE). Using mass spectrometry (MS), the RAZUMAB light chain exhibited a relatively high amount (6–9%) of a + 27 Da additional peak, which was further confirmed and characterized by peptide mapping at multiple sites as being a substitution of serine by asparagine (+ 27.01 Da as monoisotopic mass shift). We demonstrated that state-of-the-art technologies such as MS are required to assess analytical similarity of biological products to their originators.
Results
Potency assays
Relative potency was determined for 2 RAZUMAB batches. The potency assay analyzing binding to VEGF showed 96% and 97% relative binding activity versus the LUCENTIS® reference standard. The cell-based functional potency assay revealed 99% and 100% relative potency in comparison with the LUCENTIS® reference standard, respectively (Table 1).
Table 1.
Analytical and bioanalytical comparison of RAZUMAB batches and LUCENTIS®. Percentages of size variants as determined by SEC and CE-SDS in denaturing conditions are indicated. Percentages of charge variants were determined by CEX and CZE. HMW: High molecular weight. Potency was determined relative to the LUCENTIS®reference standard with target binding assay and cell-based functional assay. Individual determinations for each reportable result had been determined on independently prepared 96-well assay plates. Each result is provided ± standard deviation from duplicate measurements where applicable. n.d.: not detected. LOQ: limit of quantification at 0.3%. *: defined potency of LUCENTIS®reference standard.
| Analytics | Attributes | RAZUMAB #1 | RAZUMAB #2 | LUCENTIS® |
|---|---|---|---|---|
| SEC | Aggregates (%) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 |
| Main peak (%) | 100.0 ± 0.0 | 100.0 ± 0.0 | 99.9 ± 0.0 | |
| Fragments (%) | n.d. | n.d. | n.d. | |
| CE-SDS | Fragment peak 1 (%) | 0.5 ± 0.0 | 0.6 ± 0.0 | 0.6 ± 0.1 |
| Fragment peak 2 (%) | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.2 ± 0.0 | |
| Main peak (%) | 99.2 ± 0.1 | 99.1 ± 0.0 | 99.0 ± 0.1 | |
| HMW peak (%) | 0.2 ± 0.0 | 0.3 ± 0.0 | 0.3 ± 0.0 | |
| CEX | Acidic variants (%) | 0.9 ± 0.0 | 1.1 ± 0.0 | 0.7 ± 0.0 |
| Main peak (%) | 98.0 ± 0.0 | 97.9 ± 0.0 | 97.9 ± 0.0 | |
| Basic variants (%) | 1.1 ± 0.0 | 1.0 ± 0.0 | 1.4 ± 0.0 | |
| CZE | Acidic variants (%) | 2.5 | 3.1 | 1.5 |
| Main peak (%) | 97.1 | 97.4 | 97.8 | |
| Basic variants (%) | < LOQ | < LOQ | < LOQ | |
| Potency assays | Binding to VEGF (% potency) | 96 ± 2 | 97 ± 4 | 100* |
| Cell-based functional assay (% potency) | 99 ± 1 | 100 ± 5 | 100* |
The observed differences from 100% relative potency were within the expected assay variability; no differences in VEGF binding, nor in inhibition of VEGF-induced proliferation were found between the LUCENTIS® reference standard and the intended copy product RAZUMAB.
Analysis of size and charge variants by separation techniques
Two RAZUMAB batches were analyzed in parallel with the LUCENTIS® reference standard using separation techniques to investigate size and charge variants of the intended copy product relative to LUCENTIS®. SEC was performed to exhibit fragments and aggregates in these samples (Fig. 1A). The results revealed no major difference between the samples, and the LUCENTIS® reference standard contained only a slightly higher content of high molecular weight species at a level of 0.1% of total UV area compared to 0.0% for RAZUMAB (Table 1). CE-SDS was performed to investigate potential differences in terms of covalent species. Comparison of electropherograms of LUCENTIS® reference standard and RAZUMAB batches did not reveal any major difference (Fig. 1B), and similar levels below 1% for fragments, and below 0.5% for high molecular weight species, were observed in all samples (Table 1). Potential differences in charge product-related variants were investigated with CEX. Although the pattern of basic variants was highly similar between RAZUMAB and LUCENTIS® reference standard, differences could be identified in the acidic region of the chromatographic profiles, showing a minor 0.2–0.4% increase of acidic species in RAZUMAB batches with a new peak not present in LUCENTIS® reference standard (Fig. 1C). A different profile for acidic variants between RAZUMAB batches compared to LUCENTIS® reference standard was also observed using CZE as an orthogonal method, with one new acidic peak of 0.4% (Fig. 1D). A shoulder of the main peak was also detected in the acidic region of RAZUMAB batches. Results are compiled in Table 1.
Figure 1.

Analysis of size and charge variants of RAZUMAB batches and LUCENTIS® by separation techniques. (A) UV chromatogram overlay of SEC analysis. Absorbance was recorded at 280 nm. High molecular weight species (HMW) and the intact Fab peak (main peak) are shown. (B) Overlay of CE-SDS electropherograms. Fragments, the intact Fab peak (main peak) and HMW species are annotated. (C) Analysis of charge variants by CEX. Main peak, acidic and basic variants are displayed on the overlay of UV chromatograms at 280 nm. RAZUMAB and LUCENTIS® exhibit different acidic variant profiles. (D) Charge variant profiles with CZE. UV electropherograms at 214 nm are shown for RAZUMAB batches and LUCENTIS®. Main peak, acidic and basic variants are displayed. Different acidic variants are present in RAZUMAB and LUCENTIS®. *: main peak shoulder in the acidic region of RAZUMAB batches. Insert: zoomed-in view from 10 to 11 min. For all chromatograms, a signal offset of 10% has been applied between samples.
Fab intact/subunit mass analyses by LC-UV/ESI-MS
To further investigate the potential biochemical differences between RAZUMAB and LUCENTIS® reference standard causing the different acidic variants, the masses of intact Fab samples and Fab subunits after reduction/carbamidomethylation were measured without any prior enrichment. Samples were analyzed by online LC-UV/ESI-MS as intact in denaturing organic solvents, containing trifluoroacetic acid (TFA) as ion-pairing agent, to improve peak resolution. An overlay of UV chromatograms between batches of RAZUMAB and LUCENTIS® reference standard showed comparable UV profiles (Fig. 2A). Standard MS spectrum analysis usually involves averaging or summation of MS scans within a delimited peak range, and then “manual” deconvolution with user-defined settings. For this study, we performed unbiased MS data analysis and performed time-resolved deconvolution of each MS scan within a defined timeframe and created 3D heat maps of product-related variants (Fig. 2B).26 In brief, this process enables the deconvolution of each 1s MS scan in parallel for all samples processed altogether. Such display retains for each species the connection between deconvoluted mass and respective retention time. This enabled us to quickly distinguish co-eluting conventional artifacts generated in-source, e.g., water loss and TFA adduct, from real variants separated in time. Two forms were observed in the LUCENTIS® reference standard, corresponding to the main Fab at 48380 Da (4 ppm mass accuracy) and an additional signal (+16 Da) eluting slightly earlier than the main one, marked with the white cross in Fig. 2B, which most likely corresponds to the oxidized form of the LUCENTIS® reference standard. The same species could also be monitored in the 2 batches of RAZUMAB. However, an additional species with a mass increase of ∼25 Da was identified in the 2 RAZUMAB samples. Such a mass increment was not detected in LUCENTIS® reference standard intact 3D heat map.
Figure 2.
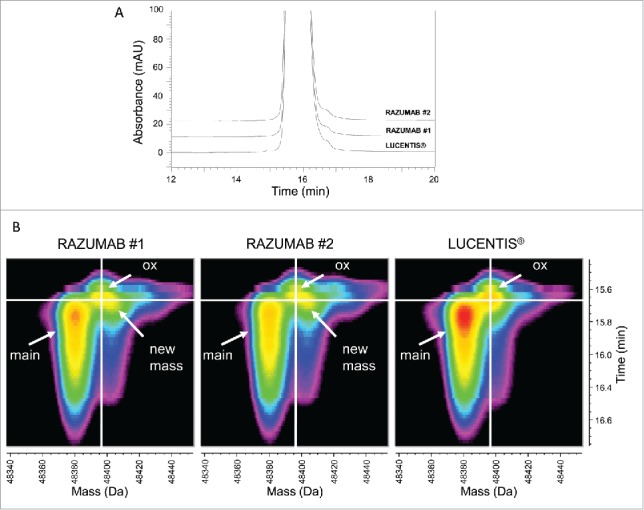
Intact Fab LC-UV/ESI-MS analysis. (A) Overlay of UV chromatograms of intact LUCENTIS® and RAZUMAB batches 1 and 2. Absorbance was recorded at 280 nm. Signal offset: 10%. (B) Time-resolved deconvolution of intact LUCENTIS® and intact RAZUMAB MS data for batches 1 and 2. Heat maps were generated from time-resolved deconvolution performed in parallel for all samples. Intensity is color-coded ranging from less intense (black) to most intense (red). The unmodified Fab (main peak) and an oxidized variant (ox) are annotated. A new mass (+ ∼25 Da) is found exclusively in RAZUMAB samples.
Analysis of the Fab light and heavy chains was performed using reduction and carbamidomethylation. An overlay of the UV chromatograms revealed a slight front shoulder in the UV chromatograms of the light chain of RAZUMAB batches only as the main difference across samples (Fig. 3A). Spectra of the Fab light and heavy chains were acquired and deconvoluted with time-resolved deconvolution in an unbiased manner as described above (Fig. 3B-C). Deconvoluted masses fit with expected masses of the light and heavy chains, at 23718.3 Da and 25242.3 Da with mass accuracies of 8 ppm and 14 ppm, respectively. A new signal with a mass of light chain + 27 Da present in RAZUMAB batches, but absent in the LUCENTIS® reference standard, matches the mass increment observed previously in the intact Fab analysis of RAZUMAB samples (Fig. 3B). This new species was also detected in a third RAZUMAB batch (Fig. S1), and was estimated in the range of 6–9% of the light chain across the 3 RAZUMAB batches (Table 2; Fig. S2). No major difference could be detected from the 3D heat map of the Fab heavy chain, containing the oxidized form as the major variant and pyroglutamate from glutamate as a minor variant (Fig. 3C). Overalkylation was observed at similar levels in these samples as a sample preparation artifact.
Figure 3.

Fab subunit LC-UV/ESI-MS analysis. (A) Overlay of UV chromatograms at 214 nm of light and heavy chains of LUCENTIS® and RAZUMAB batches 1 and 2 after reduction and carbamidomethylation. Signal offset: 10%. (B) Time-resolved deconvolution for the light chain (LC). LC, and LC + 27 Da (in RAZUMAB samples), are annotated. ║: In-source dehydration, ◊: Guanidine adduct, §: Possible LC + (2 × 27 Da) in RAZUMAB batches, ‡: Sodium adduct. (C) Time-resolved deconvolution for the heavy chain (HC) species. HC, oxidized HC (HCox) and N-terminal pyroglutamate formation (HC(pE)) are annotated. *major sample preparation artifact is overalkylation with iodoacetamide as shown by the addition of +57 Da. Heat maps were generated from time-resolved deconvolution performed in parallel for all samples. Intensity is color-coded ranging from less intense (black) to most intense (red).
Table 2.
Estimation of modified light chain (+ 27 Da) in RAZUMAB batches. Estimation was calculated as the percentage of the modified light chain (+ 27 Da) volume compared to the total volume of RAZUMAB light chain, summing up light chain volume and modified light chain (+ 27 Da) volume in 3 different batches. To reduce the impact of the possible sodium adduct noise inside the defined peak boundaries on the estimation of the modified light chain (+ 27 Da), the volume of the sodium adduct noise observed in LUCENTIS®inside the same boundaries was subtracted from the volume of the modified light chain (+ 27 Da) in RAZUMAB batches (Fig. S2). Batches 1 and 2 or batches 1 and 5 were analyzed together as independent experiments. Coefficient of variation (CV) for batch 1 across experiments 1 and 2 is provided.
| Estimation (% of total LC volume) |
|||||
| Experiment 1 |
Experiment 2 |
||||
| Species |
batch 1 |
batch 2 |
batch 1 |
batch 5 |
CV batch 1 (%) |
| LC + 27 Da | 6.7 | 8.8 | 6.1 | 8.3 | 7 |
Primary sequence analysis by LC-UV/ESI-MS/MS
The identification of a new species found at substantial levels in RAZUMAB batches was complemented by a more comprehensive characterization of the RAZUMAB primary structure. Lys-C peptide mapping analyses of RAZUMAB and LUCENTIS® reference standard were performed in parallel using online LC-UV/ESI-MS/MS acquisition with high mass accuracy and high resolution on an orbitrap mass analyzer. TFA was used as ion-pairing agent to generate well-resolved peptide peaks. Stacked UV chromatograms of RAZUMAB and LUCENTIS® reference standard are shown in Fig. 4A. No major differences were observed using chromatographic analysis. ESI-MS data were explored quantitatively in a consistent manner by first delineating peaks and clusters for all 3 samples, then comparing the corresponding cluster volumes. Clusters are composed of peaks that share the same monoisotope and retention time (thus defining a unique peptide charge state), in each sample chromatogram plane (RT-m/z) (Fig. 4B). A specific data evaluation was performed to search for clusters present or absent in each respective sample. Such analysis enabled the identification of 57 clusters found in both RAZUMAB batches but absent from LUCENTIS® reference standard (Fig. 4C). Selection of these clusters enabled the identification of these differences in the sample chromatogram planes and validation of relevant differences. Strikingly, all serine-containing peptides apart from L-L14 (SFNRGEC) of RAZUMAB light chain were present with a modified version with an additional 27.01 Da. Such modification was clearly absent in the LUCENTIS® reference standard, as shown for L-L10 (Fig. 4D). Threonine to glutamine substitution as well as serine to asparagine substitution can lead to a shift in monoisotopic mass of + 27.01 Da. The threonine to lysine sequence variation (+ 27.05 Da) could be ruled out from possible modifications due to the high mass accuracy of the orbitrap analyzer used for this study, and because this sequence variation would introduce a new Lys-C cleavage site, resulting in a shorter peptide. Manual MS/MS spectra interpretation confirmed the occurrence of a serine to asparagine sequence variation for all peptides of RAZUMAB light chain excluding SFNRGEC. Of note, several isobaric variant species with the addition of 27.01 Da were separated by time for L-L1 and L-L10 peptides, for which serine to asparagine substitution was localized at different sites. Annotated MS/MS spectra of variant and native L-L10 peptides are shown in Figure 5. Site confirmation of the substitution was also possible for other sites (Fig. S3-S4). Table 3 compiles the identification and estimation of the variant peptides of RAZUMAB light chain. In contrast, none of the Fab heavy chain peptides of RAZUMAB were affected by such sequence variation.
Figure 4.
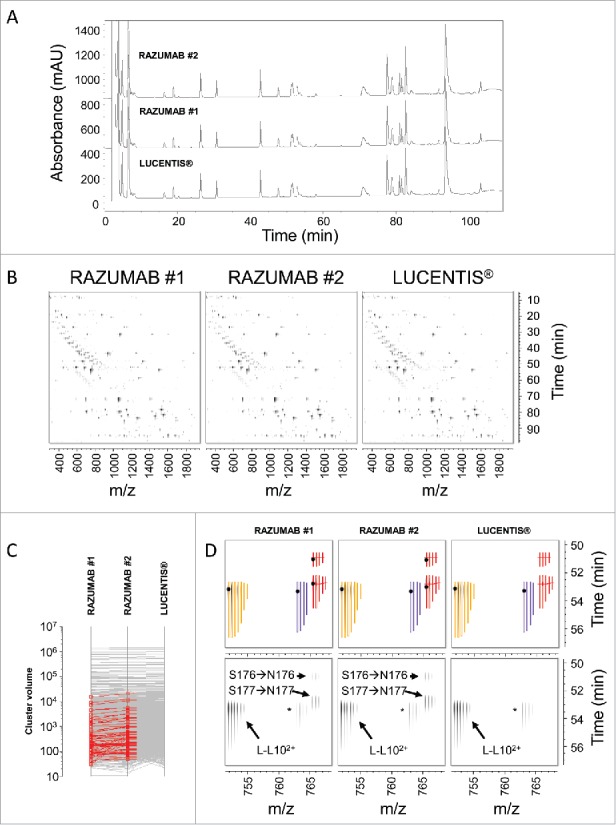
Differential analysis of RAZUMAB and LUCENTIS® peptide mapping signals. (A) Overlay of UV chromatograms of Lys-C peptide mapping of LUCENTIS® and RAZUMAB batches 1 and 2 after reduction and carbamidomethylation. Absorbance was acquired at 214 nm. No major differences were observed using UV chromatogram analysis. Signal offset: 10%. (B) Differential analysis of Lys-C peptide mapping MS data. A chromatogram plane (RT-m/z) of the MS and MS/MS data is built for each sample, in which MS peaks detected in one or more samples are clustered according to their monoisotopic mass and RT. The volume of each cluster is calculated and compared across all samples. (C) Identification of RAZUMAB-specific MS signals. Differential cluster analysis retrieved 57 clusters (shown in red) present in both RAZUMAB batches and absent in LUCENTIS®. (D) Mapping RAZUMAB-specific MS signals with a mass increment of 27.01 Da. An example is shown for L-L102+ peptide using the chromatogram planes of RAZUMAB batches and LUCENTIS®. Top panels: Clusters with isotope peak boundaries for all isotopes are shown for L-L102+ species across samples. Native L-L102+ peptide (yellow cluster) was detected in RAZUMAB batches along with variant species (red clusters), absent from LUCENTIS® sample. These variant peptides were detected with a mass increment of 27.01 Da and were assigned by MS/MS interpretation to S176→N176 and S177→N177 substitutions in RAZUMAB. A black dot represents the acquisition of MS/MS data for the specific cluster in the respective sample. The purple cluster corresponds to L-L102+ sodium adduct. Bottom panels: same chromatogram planes as in top panels without cluster isotopic peak boundaries.* L-L102+ sodium adduct.
Figure 5.

MS/MS spectra of native and variant L-L10 peptides. (A) MS/MS spectrum of L-L102+ at 765.39 m/z and retention time 50.9 min with serine to asparagine substitution localized at position 176 (marked in red). Signature ions used for Ser → Asn substitution localization are shown in red. (B) MS/MS spectrum of L-L102+ at 765.39 m/z and retention time 52.8 min with serine to asparagine substitution localized at position 177. Position and signature ions are shown in red. (C) MS/MS spectrum of native L-L102+ peptide at 751.88 m/z and retention time 53.4 min.
Table 3.
Light chain peptides identified with possible serine to asparagine substitution in RAZUMAB batches. Variant peptides of RAZUMAB light chain are shown along with the possible position of the serine to asparagine (Ser → Asn) substitution. The LC peptide number after LysC cleavage (#), the corresponding amino acid range, and the peptide sequence are shown for each entry. The possible positions of the Ser → Asn substitution are indicated in red italic and in red bold underlined when position is confirmed. Additional peptide properties are included such as retention time (RT), mass-to-charge ratio (m/z), charge, observed mass (Da), and mass delta calculated as the difference between the observed and expected precursor masses divided by the expected precursor mass multiplied by 106 (ppm). The percentage of Ser → Asn misincorporation is determined for each variant peptide as the volume of the variant peptide cluster divided by the sum of the volumes of the variant and its corresponding native peptide m/z. Results for 2 independent experiments are shown in which batch 1 was analyzed altogether with batch 2 or batch 5. Batches 2 and 5 were analyzed only in separate experiments. Values for batch 1 are reported for both experiments to assess inter-experiment reproducibility. Coefficient of variation (CV) is calculated for each ion based on the estimates obtained for batch 1 in these 2 independent experiments. *potential overestimation due to the presence of substantial sodium adduct. ‡ Signature ions for partial Ser → Asn substitution were detected for each of the 4 serine residues of peptide L-L9, indicating that the variant ion at 1081.49 m/z is a mixture of several isobaric peptides with partial misintegration of Asn for Ser at all 4 sites as observed in the chimeric MS/MS spectrum.
| Misincorporation (%) |
|||||||||||||
| Experiment 1 |
Experiment 2 |
||||||||||||
| # |
Range |
Sequence |
Ser→Asn [position] |
RT [min] |
m/z [Th] |
Charge |
Mass [Da] |
Mass Delta [ppm] |
batch 1 |
batch 2 |
batch 1 |
batch 5 |
CV batch 1 (%) |
| L-L1 | 1–39 | DIQLTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQK | 30 | 80.1 | 1463.38 | 3 | 4387.12 | 3.0 | 1.5 | 2.0 | 1.2 | 1.7 | 15 |
| L-L1 | 1–39 | DIQLTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQK | 14 | 81.1 | 1463.37 | 3 | 4387.09 | −2.9 | 1.5* | 2.1* | 2.4* | 3.2* | 33 |
| L-L4 | 46–103 | VLYIFTSSLHSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTK | 76 or 77 | 93.5 | 1608.51 | 4 | 6430.03 | −3.5 | 5.0 | 6.6 | 5.4 | 7.2 | 6 |
| L-L6 | 108–126 | RTVAAPSVFIFPPSDEQLK | 114 | 70.8 | 1065.08 | 2 | 2128.14 | 3.3 | 0.4 | 0.5 | 0.4 | 0.5 | 10 |
| L-L7 | 127–145 | SGTASVVCLLNNFYPREAK | 131 | 78.3 | 1077.05 | 2 | 2152.08 | 1.2 | 0.5 | 0.7 | 0.6 | 0.7 | 12 |
| L-L9 | 150–169 | VDNALQSGNSQESVTEQDSK | 156, 159, 162, and 168‡ | 30.8 | 1081.49 | 2 | 2160.96 | −6.8 | 2.2 | 3.1 | 1.9 | 1.9 | 11 |
| L-L10 | 170–183 | DSTYSLSSTLTLSK | 176 | 50.9 | 765.39 | 2 | 1528.76 | 0.7 | 0.6 | 0.7 | 0.5 | 0.7 | 7 |
| L-L10 | 170–183 | DSTYSLSSTLTLSK | 177 | 52.8 | 765.39 | 2 | 1528.76 | −1.1 | 2.9 | 4.0 | 2.6 | 3.5 | 6 |
| L-L13 | 191–207 | VYACEVTHQGLSSPVTK | 202 or 203 | 42.6 | 951.97 | 2 | 1901.93 | −0.7 | 0.7 | 0.9 | 0.6 | 0.7 | 18 |
Discussion
The development of generic versions of large molecules is a challenging task, owing to their size, their complex PTMs profiles, their sensitivity to chemical modifications during manufacturing steps and storage, as well as their sequence microheterogeneity, as shown in previous reports.17-22 Therefore, in-depth characterization with sensitive and specific analytical technologies is required to demonstrate high similarity between an intended copy product and its originator.12,13,27
In this study, we comprehensively compared different batches of RAZUMAB to LUCENTIS® reference standard using several state-of-the-art analytical methods. While no differences were observed between these products in terms of size variants and potency, some differences were observed in the CEX and CZE chromatographic profiles of RAZUMAB and LUCENTIS® reference standard, indicating different acidic species. A more detailed characterization using intact and subunit MS measurements enabled the identification of substantial differences in the RAZUMAB light chain. Robust sample comparison was enabled using in parallel time-resolved deconvolution of each acquired MS scan in an unbiased manner.26 This feature improved the identification of product-related variants by pattern recognition. Additionally, statistical head-to-head comparison of peptide maps using searches for absent and present signals enabled the identification of differences in RAZUMAB primary sequence at multiple sites (+ 27.01 Da as monoisotopic mass shift), which were confirmed as serine to asparagine substitution.
Evidence and mechanisms of amino acid misincorporation in various recombinant proteins expressed in E. coli and Chinese hamster ovary (CHO) cell lines have been extensively investigated.28-34 Sequence variants can be caused by DNA mutations or mRNA mistranslation, via tRNA mischarging with an incorrect amino acid, or through codon misreading with the use of an incorrect tRNA.28 Both the cell line history and the type of product might influence the rate of mistranslation.35,36 Furthermore, it has been previously demonstrated that limitation for a specific amino acid in the culture broth leads to the reversible substitution of this amino acid for homologs through tRNA mischarging.29,36,37 The identification of a serine to asparagine variant in CHO, NS0 and E. coli was previously reported by other groups who discussed its occurrence as the result of a possible codon-specific misreading or mischarging.38-40 In this study, most peptides of the light chain were detected along with one or more variant counterparts, both in the variable and constant region. This observation suggests the occurrence of systematic mistranslation errors in the light chain, rather than point mutations at the DNA level.36,39,40 Importantly, sequence variation exclusively occurred in the light chain of RAZUMAB, as we did not detect variant peptides of RAZUMAB heavy chain. Of note, serine is the most represented amino acid in both LUCENTIS® light chain and heavy chain. The light chain contains 34 serine residues, corresponding to nearly 16% of all amino acids of the light chain, while the heavy chain contains 30 serine residues, corresponding to 13% of all amino acids of the heavy chain. As the authors do not have access to the manufacturing process of RAZUMAB, any extensive discussion of the occurrence of the sequence variation in RAZUMAB may remain speculative and not appropriate for this analytical paper. One can only hypothesize that upstream processes, including codon optimization or expression systems, may have been different for RAZUMAB light and heavy chain or that serine concentration can be a critical process parameter for the expression of the light chain.
Among analytical technologies, MS offers unmatched resolution, specificity and sensitivity to explore the primary structure of biopharmaceuticals and maps relevant low-level differences between samples. Lower limit of detection is achieved at the peptide level compared to intact mAb analysis, so routine peptide mapping analyses using MS may be performed to mitigate the risk of sequence variation to be transferred to the final clone during technical development.36, 41 Enrichment from a separation technique, e.g., CEX, may be necessary to detect and confirm low-level sequence variants (< 0.5%).41 However, data analysis remains a bottleneck in enabling the detection and identification of such species through optimization of raw data processing and database searches in an unbiased manner. New strategies in data analysis and experimental procedures have been investigated to focus on low-level sequence variants as reported by several groups.36,40,42-44 Although an error-tolerant search is a powerful tool to identify previously uninterpreted peaks, the identification of multiple mutations in the same peptide lead to increased false positive assignments, which usually requires time-consuming manual assessment for confirmation. Also, all substitutions may not be taken into account in the error-tolerant search and some common modifications may be isobaric to sequence variants.42, 43 In parallel to the error-tolerant search, we analyzed all detected MS signals without any bias toward peptide identification parameters and we searched for absent and present signals in all samples. This methodology was extremely useful to first spot relevant differences in peptide mapping data, which needed further manual confirmation.
Nevertheless, the detection of sequence variants in a single sample or variants present at similar low levels may still be a challenging task.44 State-of-the art MS instrumentation supported with powerful search engines will undoubtedly enable examination of extremely low-level sequence variants (< 10 ppm) in an unbiased manner.40 Importantly, such sequence variations are inevitable and are commonly observed in human native proteins, at levels below 100 ppm, as shown for human serum albumin across individuals.40,45 It remains to be explored at which level above this background such modifications may affect the quality of a recombinant product (originator and intended copy product).28
In this study, we report a serine to asparagine sequence variation in the light chain of RAZUMAB that accounted for ∼6–9% in relative amount. As pI values for serine and asparagine are 5.68 and 5.41, respectively, it is unlikely that a serine to asparagine substitution would result in a major pI shift and lead to the different profiles of acidic variants observed with CEX and CZE.46 It is rather more likely that this variant may correspond to the main peak shoulder revealed with CZE analysis. The difference of 0.2–0.4% of acidic variants observed in the CEX profiles of RAZUMAB and LUCENTIS® reference standard may be due, at least in part, to the increase of the deamidated version of L-L7 (SGTASVVCLL(N→D)NFYPREAK) to the same extent in RAZUMAB batches (data not shown). We demonstrated that the implementation and the integration of sensitive orthogonal analytical approaches were required to assess analytical similarity, and investigate relevant differences between an originator and its intended copy product.
Materials and methods
Samples
Reference standard (01.REF) of LUCENTIS® (Novartis Pharma AG) was analyzed head-to-head with 2 different batches of RAZUMAB (18020001, and 18020002) from Intas Pharmaceuticals Ltd. A third RAZUMAB batch (18020005) from Intas Pharmaceuticals was also used for complementary analysis.
Reagents
Lys-C (MS grade, 125–05061) was purchased from Wako, 8 M guanidine hydrochloride (24115) was purchased from Pierce, ethylenediaminetetraacetic acid (EDTA) disodium salt, dehydrate (E5134–250G) was purchased from Sigma, 1 M Tris-hydrochloride pH8 solution (Ultrapure, 15568–025) was purchased from Gibco, dithiothreitol (43815–1G) was purchased from Sigma, iodoacetamide (Bioultra, I1149–5G) was purchased from Sigma, acetonitrile (ACN; LC-MS grade, 9821) was purchased from Biosolve, water (LC-MS grade, 9823–02) was purchased from J.T.Baker, isopropyl alcohol (IPA; LC-MS grade, 34965) from Fluka, and TFA ampoules (MS grade, 28904) were purchased from Pierce.
Potency assays
Two different methods were used for potency determinations, a target binding assay and a cell-based functional assay. An ELISA assay was used to measure binding of RAZUMAB to recombinant human VEGF immobilized on microtiter plates. Binding was quantified via an anti-human F(ab')2 antibody coupled to horseradish peroxidase from Jackson ImmunoResearch Laboratories, Inc. (109–035–097) followed by the addition of the substrate tetramethylbenzidine. The cell-based functional bioassay determines inhibition of VEGF-induced proliferation of human umbilical vein endothelial cells.
For both assay formats, the test samples and the LUCENTIS® reference standard were normalized on the basis of protein content; dilution series of reference standard and test samples were analyzed on each assay plate. Assay data, i.e., the resulting concentration-response curves were analyzed with statistical software (PLA, Stegmann Systems) and relative potency was calculated based on the parallel line model. The final potency result was expressed as relative potency (in percent) of a RAZUMAB sample compared to the LUCENTIS® reference standard.
SEC
Samples were diluted in mobile phase (0.20 M potassium phosphate, 0.25 M potassium chloride, pH 6.2) to 1 mg/mL and injected onto a Tosoh TSK-GEL G2000SWXL column (300 × 7.8 mm, 5 μm) with an Agilent 1200 HPLC system. Peak separation was performed over a 30 min isocratic elution at a flowrate of 0.5 mL/min. Column was kept at ambient temperature and peak UV detection was performed at 280 nm.
CEX
Samples were diluted in mobile phase A (20 mM MES, pH 5.70) to 4 mg/mL and injected onto a Thermo ProPac WCX-10 (250 × 4 mm, 10 μm) with an Agilent 1100 HPLC system. Peaks were separated by applying a linear gradient from 8% mobile phase B (20 mM MES, 200 mM NaCl pH 5.70) to 66% B in 85 min at a flowrate of 0.8 mL/min. UV absorbance was measured at 280 nm.
CE-SDS
Samples were diluted to 0.5 mg/mL with labeling reaction buffer (0.1 M sodium bicarbonate pH 8.3) then 0.5 mL of this diluted sample was loaded onto a NAP-5 column to complete buffer exchange. Eluent was discarded and sample collected in 1mL of labeling reaction buffer. For labeling, 10 µL of the dye reagent working solution (5-carboxytetramethylrhodamine succinimidyl ester in dimethylsulfoxide 1 mg/mL) was added to 190 µL of buffer-exchanged sample and incubated for 30 min at 30 °C. Excess of dye reagent was removed by loading 190 µL of labeled sample onto a NAP-5 column, washing column with 0.4 mL of labeling reaction buffer and collecting sample in 0.7 mL labeling reaction buffer. The labeled sample was diluted twice with CE-SDS sample buffer (0.1 M Tris-HCl / 5% SDS, pH 8.0). The injection was performed into a bare, fused-silica capillary at 5 kV for 20 seconds using a Beckman PA800 Enhanced/Plus. The instrument separated proteins in an electric field of 484 V/cm for 35 minutes. Detection was performed via an Argon-ion laser with an excitation at 488 nm and an emission at 560 nm. Chromeleon software was used for data analysis.
CZE
Samples were diluted in sample buffer (5 mM sodium phosphate, pH 7.30) to 3 mg/mL. The injection was performed into a bare, fused-silica capillary at 0.5 psi for 4 seconds using a Beckman PA800 Enhanced/Plus. The instrument separated proteins at a voltage of 20 kV for 15 minutes in a 400 mM 6-aminocaproic acid, 2 mM triethylenetetramine pH 5.7 adjusted with acetic acid and 0.03% Tween 20. UV detection was performed at 214 nm. Chromeleon software was used for data analysis.
Intact Fab analysis by LC-UV/ESI-MS
Samples were analyzed sequentially by online LC-UV/ESI-MS using a ToF Xevo G2-S (Waters, Manchester, UK) mass spectrometer. Twenty micrograms of each sample were injected onto a PLRPS column (2.1 × 150 mm, 3 µm, 300 Å) set at 60 °C using a Waters Acquity UPLC. After sample loading and column washing for 4 min with 65% A (0.1% TFA in water) and 35% B (0.09% TFA in 70:20:10 IPA:ACN:water), separation was performed at a flowrate of 0.2 mL/min with a linear gradient ramping from 35% B to 50% B in 24 min. UV traces were recorded at 280 nm. The MS instrument was operating in positive mode with a capillary voltage of 3.8 kV, cone voltage of 160V and source offset of 120V. The source temperature was set at 120°C and the desolvation temperature at 500°C. Scans of 1 second were acquired from 500 to 5000 m/z for 36 min. Lockmass correction was applied every 20 scans from an average of 3 scans by infusion of 1 pmol/μL of Glu-1-Fibrinopeptide B.
Fab subunit analysis by LC-UV/ESI-MS
Prior to analysis, samples were denatured in parallel with 150 µL of 6 M GuHCl, 5 mM Na2EDTA in 50 mM Tris-HCl at pH 8, and incubated with 1.5 µL 1 M dithiothreitol for 1h at 37 °C to reduce disulfide bonds. Then, alkylation was performed for 1 h in the dark using 3 µL of 1 M iodoacetamide. 1 µL DTT was finally used to quench the alkylation. Samples were then analyzed sequentially by online LC-UV/ESI-MS using a ToF Xevo G2-S (Waters, Manchester, UK) mass spectrometer. Six micrograms of each sample were injected onto a PLRPS column as described above. Sample loading and column washing for 4 min was performed with 68% A (0.1% TFA in water) and 32% B (0.09% TFA in 70:20:10 IPA:ACN:Water), while separation was performed at a flowrate of 0.2 mL/min with a linear gradient ramping from 32% B to 47% B in 37 min. UV traces were recorded at 214 nm. The MS instrument was operating in positive mode with a capillary voltage of 3.8 kV, cone voltage of 120V and source offset of 60V. The source temperature was set at 120°C and the desolvation temperature at 500°C. Scans of 1 second were acquired from 500 to 3000 m/z for 50 min. Lockmass correction was applied as described above.
Intact and subunit MS data analysis
Data were imported into the Refiner MS software package (Genedata) without any prior data conversion. An adapted and application specific workflow was built and used for chemical noise reduction, RT and m/z restriction, and finally time-resolved deconvolution. Each 1s scan was deconvoluted while retaining retention time information using the so-called time-resolved deconvolution performed with the embedded harmonic suppression deconvolution method using 0.1 Da step. The peak volumes of light chain + 27 Da and light chain were calculated to estimate the percentage of light chain + 27 Da. The volumes of both peaks were defined as the integral of the intensity values inside the peak boundaries, resulting in the combination of 3 dimensions, i.e., intensity values x retention time window x mass width. The volume of the sodium adduct noise inside these boundaries in LUCENTIS® reference standard was subtracted from the peak volumes of light chain + 27 Da in RAZUMAB samples for this estimation (Fig. S2).
Lys-C peptide mapping analysis by LC-UV/ESI-MS/MS
All samples were denatured, reduced and alkylated together as described for the analysis of Fab subunits. Then samples were prepared as previously reported.26 Briefly, samples were digested with 4 µg of Lysyl endopeptidase for 1 h at 37 °C. A further 4 μg of Lysyl endopeptidase were added to the samples and incubation was prolonged for another 3 h at 37 °C. An aliquot of 100 µL of the digested samples was injected on column and analyzed by LC-UV/ES-MS/MS using an Agilent 1200 online connected to a Thermo Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Reinach, Switzerland). The mobile phases were 0.1% TFA in water as A and 0.09% TFA in 90% ACN as B and the flow rate was set at 0.2 mL/min. The column used for the separation was a Vydac C18 (2.1 × 150 mm, 5 µm, 300 Å) and was set at 40 °C. The UV traces were recorded at 214 nm. The separation was achieved over 146 min by starting at 2% B and maintaining it for 5 min then ramping to 22% B in 45 min, then to 24% B in 10 min, to 36% B in 28 min, further to 38% B in 10 min and to 90% B in 17 min and maintaining it for 10 min. The HPLC was directly coupled to the mass spectrometer. The instrument was operating in positive ion mode with a capillary voltage of 3.5 kV. The capillary temperature was set at 250 °C. The MS instrument method was built as a typical data-dependent acquisition experiment: first full scan in the Orbitrap is followed by 3 collision-induced dissociation MS/MS events of the 3 most intense ions and by 3 higher-energy collisional dissociation MS/MS of the same ions all measured in the Orbitrap Elite. Dynamic exclusion was used to prevent fragmentation of the same precursor ion multiple times.
Peptide mapping data evaluation using RefinerMS and Analyst
The raw data were directly imported into the Refiner MS software. Data from all samples were processed in parallel with chemical noise subtraction, peak detection (isotope peak detection), charge assignment (isotope clustering), and MS/MS consolidation. No data normalization was performed. Clusters were searched against LUCENTIS® light and heavy chain sequences and this search retrieved 100% sequence coverage (precursor and fragment mass tolerance: 20ppm, carbamidomethylation on cysteines). All clusters were used for quantitative assessment. To identify sample-specific signals, quantitative evaluation of cluster volumes was performed in Analyst by sorting out clusters with a null volume in the LUCENTIS® reference standard and positive volumes in all RAZUMAB batches. By doing so, we were able to identify MS signals specifically present only in RAZUMAB batches and absent from the LUCENTIS® reference standard. To reduce any potential bias toward single isotope measurements, cluster volumes were calculated by summing up volumes for all isotope peaks included in the isotopic cluster. The volume of a peak is defined as the integral of the intensity values inside the peak boundaries, resulting in the combination of 3 dimensions, i.e., intensity values x retention time window x m/z width. In parallel, a complementary error-tolerant search with maximum 1 missense mutation per peptide was performed to assist the identification of the peptides with a mass increment of + 27.01 Da.
Supplementary Material
Disclosure of potential conflicts of interest
All authors were employed by Novartis Pharma AG at the time they completed work described in this publication. Novartis Pharma AG funded all of this research, and some of the authors own Novartis stocks. LUCENTIS® was developed by Genentech Inc. and Novartis. Genentech Inc. has the commercial rights to LUCENTIS® in the United States. Novartis has exclusive rights in the rest of the world. LUCENTIS® is a registered trademark of Genentech Inc.
Acknowledgments
The authors would like to thank Dr. Christoph Rösli for providing a constructive review of this work.
Funding details
This work was supported by Novartis Pharma AG.
References
- 1.Reichert JM. Antibodies to watch in 2016. Mabs. 2016;8:197-204. doi: 10.1080/19420862.2015.1125583. PMID:26651519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichert JM. Antibodies to watch in 2015. Mabs. 2015;7:1-8. doi: 10.4161/19420862.2015.988944. PMID:25484055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. Mabs. 2015;7:9-14. doi: 10.4161/19420862.2015.989042. PMID:25529996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9:167-81. doi: 10.1080/19420862.2016.1269580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holzmann J, Balser S, Windisch J. Totality of the evidence at work: The first US biosimilar. Expert Opin Biol Ther. 2016;16:137-42. doi: 10.1517/14712598.2016.1128410. PMID:26634611 [DOI] [PubMed] [Google Scholar]
- 6.Kurki P, Ekman N. Biosimilar regulation in the EU. Expert Rev Clin Pharmacol. 2015;8:649-59. doi: 10.1586/17512433.2015.1071188. PMID:26294076 [DOI] [PubMed] [Google Scholar]
- 7.WHO Guidelines on evaluation of similar Biotherapeutic Products (SBPs), Annex 2. Technical Report Series. 2013;977:51-89. URL: http://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=1. Access date: July.07.2017. [Google Scholar]
- 8.WHO Guidelines on evaluation of monoclonal antibodies as similar biotherapeutic products (SBPs), Annex 2. Technical Report Series 2017;1004:93–127. URL: http://www.who.int/biologicals/biotherapeutics/WHO_TRS_1004_web_Annex_2.pdf?ua=1. Access date: July.07.2017. [Google Scholar]
- 9.WHO Regulatory assessment of approved rDNA-derived biotherapeutics, Annex 3. Technical Report Series 2016;999:131–146. URL: http://www.who.int/biologicals/areas/biological_therapeutics/Annex_3_Regulatory_assessment_of_approved_rDNA-derived_biotherapeutics.pdf?ua=1. Access date: July.07.2017. [Google Scholar]
- 10.FDA Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product. 2015. URL: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf. Access date: July.07.2017. [Google Scholar]
- 11.EMA Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Quality issues (revision 1). 2014. URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf. Access date: July.07.2017. [Google Scholar]
- 12.Beck A, Debaene F, Diemer H, Wagner-Rousset E, Colas O, Van Dorsselaer A, Cianférani S. Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J Mass Spectrom. 2015;50:285-97. doi: 10.1002/jms.3554. PMID:25800010 [DOI] [PubMed] [Google Scholar]
- 13.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S. Characterization of Therapeutic Antibodies and Related Products. Anal Chem. 2013;85:715-36. doi: 10.1021/ac3032355. PMID:23134362 [DOI] [PubMed] [Google Scholar]
- 14.Chumsae C, Hossler P, Raharimampionona H, Zhou Y, McDermott S, Racicot C, Radziejewski C, Zhou ZS. When Good Intentions Go Awry: Modification of a Recombinant Monoclonal Antibody in Chemically Defined Cell Culture by Xylosone, an Oxidative Product of Ascorbic Acid. Anal Chem. 2015;87:7529-34. doi: 10.1021/acs.analchem.5b00801. PMID:26151084 [DOI] [PubMed] [Google Scholar]
- 15.Parker J, Pollard JW, Friesen JD, Stanners CP. Stuttering – high-level mistranslation in animal and bacterial-cells. Proc Natl Acad Sci U S A. 1978;75:1091-5. doi: 10.1073/pnas.75.3.1091. PMID:349556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos MAS, Tuite MF. New insights into messenger-rna decoding – implications for heterologous protein-synthesis. Trends Biotechnol. 1993;11:500-5. doi: 10.1016/0167-7799(93)90028-8. PMID:7764419 [DOI] [PubMed] [Google Scholar]
- 17.Tan Q, Guo Q, Fang C, Wang C, Li B, Wang H, Li J, Guo Y. Characterization and comparison of commercially available TNF receptor 2-Fc fusion protein products. Mabs. 2012;4:761-774. doi: 10.4161/mabs.22276. PMID:23032066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Fu T, Liu T, Guo H, Guo Q, Xu J, Zhang D, Qian W, Dai J, Li B, et al.. Characterization of alanine to valine sequence variants in the Fc region of nivolumab biosimilar produced in Chinese hamster ovary cells. MAbs. 2016;8:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Yang B, Zhou D, Xu J, Ke Z, Suen WC. Discovery and characterization of antibody variants using mass spectrometry-based comparative analysis for biosimilar candidates of monoclonal antibody drugs. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1025:57-67. doi: 10.1016/j.jchromb.2016.05.004. PMID:27214604 [DOI] [PubMed] [Google Scholar]
- 20.Li C, Rossomando A, Wu S-L, Karger BL. Comparability analysis of anti-CD20 commercial (rituximab) and RNAi-mediated fucosylated antibodies by two LC-MS approaches. Mabs. 2013;5:565-75. doi: 10.4161/mabs.24814. PMID:23751726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, et al.. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs. 2010;2:379-94. doi: 10.4161/mabs.11986. PMID:20458189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hausberger A, Lamanna WC, Hartinger M, Seidl A, Toll H, Holzmann J. Identification of Low-Level Product-Related Variants in Filgrastim Products Presently Available in Highly Regulated Markets. BioDrugs. 2016;30:233-42. doi: 10.1007/s40259-016-0169-2. PMID:27026103 [DOI] [PubMed] [Google Scholar]
- 23.Declerck P, Danesi R, Petersel D, Jacobs I. The language of biosimilars: Clarification, definitions, and regulatory aspects. Drugs. 2017;77:671-7. doi: 10.1007/s40265-017-0717-1. PMID:28258517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macdonald JC, Hartman H, Jacobs IA. Regulatory considerations in oncologic biosimilar drug development. Mabs. 2015; 7:653-61. doi: 10.1080/19420862.2015.1040973. PMID:25961747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mysler E, Pineda C, Horiuchi T, Singh E, Mahgoub E, Coindreau J, Jacobs I. Clinical and regulatory perspectives on biosimilar therapies and intended copies of biologics in rheumatology. Rheumatol Int. 2016;36:613-25. doi: 10.1007/s00296-016-3444-0. PMID:26920148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griaud F, Denefeld B, Lang M, Hensinger H, Haberl P, Berg M. Unbiased in-depth characterization of CEX fractions from a stressed monoclonal antibody by mass spectrometry. MAbs. 2017;9:820-30. doi: 10.1080/19420862.2017.1313367. PMID:28379786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beck A, Sanglier-Cianferani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 2012;84:4637-46. doi: 10.1021/ac3002885. PMID:22510259 [DOI] [PubMed] [Google Scholar]
- 28.Harris RP, Kilby PM. Amino acid misincorporation in recombinant biopharmaceutical products. Curr Opin Biotechnol. 2014;30:45-50. doi: 10.1016/j.copbio.2014.05.003. PMID:24922333 [DOI] [PubMed] [Google Scholar]
- 29.Bogosian G, Violand BN, Dorwardking EJ, Workman WE, Jung PE, Kane JF. Biosynthesis and incorporation into protein of norleucine by escherichia-coli. J Biol Chem. 1989;264:531-9. PMID:2642478 [PubMed] [Google Scholar]
- 30.Brinkmann U, Mattes RE, Buckel P. High-level expression of recombinant genes in escherichia-coli is dependent on the availability of the dnay gene-product. Gene. 1989;85:109-14. doi: 10.1016/0378-1119(89)90470-8. PMID:2515992 [DOI] [PubMed] [Google Scholar]
- 31.Calderone TL, Stevens RD, Oas TG. High-level misincorporation of lysine for arginine at AGA codons in a fusion protein expressed in Escherichia coli. J Mol Biol. 1996;262:407-12. doi: 10.1006/jmbi.1996.0524. PMID:8893852 [DOI] [PubMed] [Google Scholar]
- 32.Harris RP, Mattocks J, Green PS, Moffatt F, Kilby PM. Determination and control of low-level amino acid misincorporation in human thioredoxin protein produced in a recombinant Escherichia coli production system. Biotechnol Bioeng. 2012;109:1987-95. doi: 10.1002/bit.24462. PMID:22334292 [DOI] [PubMed] [Google Scholar]
- 33.Seetharam R, Heeren RA, Wong EY, Braford SR, Klein BK, Aykent S, et al.. Mistranslation in igf-1 during over-expression of the protein in escherichia-coli using a synthetic gene containing low-frequency codons. Biochem Biophys Res Commun. 1988;155:518-23. doi: 10.1016/S0006-291X(88)81117-3. PMID:3137938 [DOI] [PubMed] [Google Scholar]
- 34.Samaranayake H, Wirth T, Schenkwein D, Raty JK, Yla-Herttuala S. Challenges in monoclonal antibody-based therapies. Ann Med. 2009;41:322-31. doi: 10.1080/07853890802698842. PMID:19234897 [DOI] [PubMed] [Google Scholar]
- 35.Feeney L, Carvalhal V, Yu XC, Chan B, Michels DA, Wang YJ, Shen A, Ressl J, Dusel B, Laird MW. Eliminating tyrosine sequence variants in CHO cell lines producing recombinant monoclonal antibodies. Biotechnol Bioeng. 2013;110:1087-97. doi: 10.1002/bit.24759. PMID:23108857 [DOI] [PubMed] [Google Scholar]
- 36.Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, Huang Y-M, Foley SF, Garber E, Pederson N, Meier W. Discovery and Investigation of Misincorporation of Serine at Asparagine Positions in Recombinant Proteins Expressed in Chinese Hamster Ovary Cells. J Biol Chem. 2009;284:32686-94. doi: 10.1074/jbc.M109.059360. PMID:19783658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khetan A, Huang Y-m, Dolnikova J, Pederson NE, Wen D, Yusuf-Makagiansar H, Chen P, Ryll T. Control of misincorporation of serine for asparagine during antibody production using cho cells. Biotechnol Bioeng. 2010;107:116-23. doi: 10.1002/bit.22771. PMID:20506364 [DOI] [PubMed] [Google Scholar]
- 38.Guo D, Gao A, Michels DA, Feeney L, Eng M, Chan B, Laird MW, Zhang B, Yu XC, Joly J, et al.. Mechanisms of Unintended Amino Acid Sequence Changes in Recombinant Monoclonal Antibodies Expressed in Chinese Hamster Ovary (CHO) Cells. Biotechnol Bioeng. 2010;107:163-71. doi: 10.1002/bit.22780. PMID:20506532 [DOI] [PubMed] [Google Scholar]
- 39.Yu XC, Borisov OV, Alvarez M, Michels DA, Wang YJ, Ling V. Identification of Codon-Specific Serine to Asparagine Mistranslation in Recombinant Monoclonal Antibodies by High-Resolution Mass Spectrometry. Anal Chem. 2009;81:9282-90. doi: 10.1021/ac901541h. PMID:19852494 [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z, Shah B, Bondarenko PV. G/U and Certain Wobble Position Mismatches as Possible Main Causes of Amino Acid Misincorporations. Biochemistry. 2013;52:8165-76. doi: 10.1021/bi401002c. PMID:24128183 [DOI] [PubMed] [Google Scholar]
- 41.Ren D, Zhang J, Pritchett R, Liu H, Kyauk J, Luo J, Amanullah A. Detection and identification of a serine to arginine sequence variant in a therapeutic monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:2877-84. doi: 10.1016/j.jchromb.2011.08.015 [DOI] [PubMed] [Google Scholar]
- 42.Zeck A, Regula JT, Larraillet V, Mautz B, Popp O, Goepfert U, Wiegeshoff F, Vollertsen UE, Gorr IH, Koll H, et al.. Low level sequence variant analysis of recombinant proteins: An optimized approach. Plos One. 2012;7. doi: 10.1371/journal.pone.0040328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, Strahan A, Li C, Shen A, Liu H, Ouyang J, Katta V, Francissen K, Zhang B. Detecting low level sequence variants in recombinant monoclonal antibodies. Mabs. 2010;2:285-298. doi: 10.4161/mabs.2.3.11718. PMID:20400866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brady LJ, Scott RA, Balland A. An optimized approach to the rapid assessment and detection of sequence variants in recombinant protein products. Anal Bioanal Chem. 2015;407:3851-60. doi: 10.1007/s00216-015-8618-1. PMID:25795027 [DOI] [PubMed] [Google Scholar]
- 45.Gatlin CL, Eng JK, Cross ST, Detter JC, Yates JR. Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal Chem. 2000;72:757-63. doi: 10.1021/ac991025n. PMID:10701260 [DOI] [PubMed] [Google Scholar]
- 46.Haynes WM. “Properties of Amino Acids” in CRC Handbook of Chemistry and Physics. Boca Raton, FL: CRC Press/Taylor & Francis, 2017; 97th Edition (Internet Version 2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.