

ABSTRACT
Conventionally, mouse hybridomas or well-plate screening are used to identify therapeutic monoclonal antibody candidates. In this study, we present an alternative to hybridoma-based discovery that combines microfluidics, yeast single-chain variable fragment (scFv) display, and deep sequencing to rapidly interrogate and screen mouse antibody repertoires. We used our approach on six wild-type mice to identify 269 molecules that bind to programmed cell death protein 1 (PD-1), which were present at an average of 1 in 2,000 in the pre-sort scFv libraries. Two rounds of fluorescence-activated cell sorting (FACS) produced populations of PD-1-binding scFv with a mean enrichment of 800-fold, whereas most scFv present in the pre-sort mouse repertoires were de-enriched. Therefore, our work suggests that most of the antibodies present in the repertoires of immunized mice are not strong binders to PD-1. We observed clusters of related antibody sequences in each mouse following FACS, suggesting evolution of clonal lineages. In the pre-sort repertoires, these putative clonal lineages varied in both the complementary-determining region (CDR)3K and CDR3H, while the FACS-selected PD-1-binding subsets varied primarily in the CDR3H. PD-1 binders were generally not highly diverged from germline, showing 98% identity on average with germline V-genes. Some CDR3 sequences were discovered in more than one animal, even across different mouse strains, suggesting convergent evolution. We synthesized 17 of the anti-PD-1 binders as full-length monoclonal antibodies. All 17 full-length antibodies bound recombinant PD-1 with KD < 500 nM (average = 62 nM). Fifteen of the 17 full-length antibodies specifically bound surface-expressed PD-1 in a FACS assay, and nine of the antibodies functioned as checkpoint inhibitors in a cellular assay. We conclude that our method is a viable alternative to hybridomas, with key advantages in comprehensiveness and turnaround time.
KEYWORDS: checkpoint inhibitors, deep sequencing, mouse repertoire, microfluidics, PD-1, yeast display
Introduction
Most monoclonal antibodies (mAbs) for cancer approved by the US Food and Drug Administration were discovered through mouse immunization followed by hybridoma screening.1 In this process, wild-type or humanized mice are immunized with a clinically relevant antigen over the course of several weeks, and then hybridomas are generated by fusing primary B cells with myeloma cells.2 Mouse hybridoma generation is cheap and efficient, but the process of screening hybridomas for antigen binders remains expensive, inefficient, and time consuming. To increase hybridoma screening throughput, large-scale discovery groups often invest in costly robotic workflows. We propose that the field would benefit from antibody discovery methods that reduce costs and improve timelines.
Alternative methods for mouse antibody repertoire mining are now available through recent advances in molecular genomic technologies. Molecular genomics uses primer pools that amplify rearranged V(D)J sequences to generate diverse libraries that are subjected to next-generation sequencing techniques that acquire millions of antibody sequences.3 This “deep” sequencing approach provides a far more comprehensive representation of mammalian antibody repertoires than hybridoma screening because RNA from millions of B cells can be analyzed in a single reaction. However, the difficulty of linking such an enormous number of sequences to function is a substantial detraction to most deep sequencing methods. For example, using conventional methods to express hundreds of thousands of antibody sequences as mAbs is currently cost prohibitive.
Prior work has described technologies that retain heavy and light chain pairing by using well plate sorting4-6 or microfluidic devices.7 The importance of such retained pairing for functional antibody discovery has also been demonstrated by combining 96-well plate sorting, yeast display, and deep sequencing for discovery of mouse mAbs.8 We are aware of no published reports of a molecular genomic method that retains native single B cell pairing of heavy and light chains and enables affinity screening of mouse repertoires for rare antibodies. Here, we describe an emulsion droplet microfluidics method to capture paired heavy and light chain libraries from millions of single B cells (Fig. 1). The libraries are then expressed as yeast scFv and screened for high-affinity binders.
Figure 1.
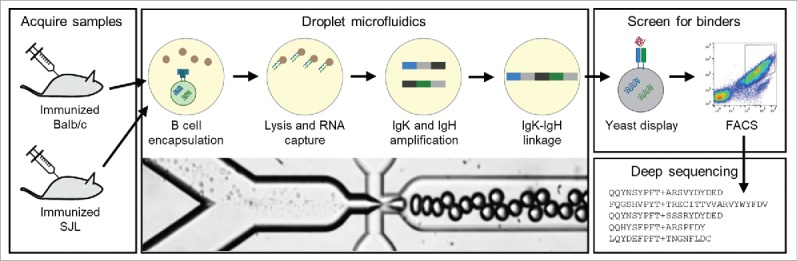
Overview of the workflow used to generate the scFv libraries from B cells isolated from Balb/c and SJL mice. B cells are isolated from lymph nodes and encapsulated into droplets with oligo-dT beads and a lysis solution. mRNA-bound beads are purified from the droplets, and then injected into a second emulsion with an OE-RT-PCR amplification mix that generates DNA amplicons that encode scFv with native pairing of heavy and light chain Ig. Libraries of natively paired amplicons are then electroporated into yeast for scFv display. FACS is used to identify high affinity scFv. Finally, deep antibody sequencing is used to identify all clones in the pre- and post-sort scFv libraries.
PD-1 is a cell surface receptor that suppresses T cell inflammatory activity, and is expressed by immune cells including T cells, B cells, and macrophages. PD-L1, the primary ligand for PD-1, is also expressed by these immune cells. The interaction of PD-1 and PD-L1 is vitally important for establishing peripheral tolerance.9 Tumor cells hijack the PD-1/PD-L1 pathway by up-regulating PD-L1 to suppress the anti-tumor immune response. Recently, therapeutic checkpoint inhibitor mAbs have been commercialized that antagonize PD-1/PD-L1 binding, thereby inhibiting tumor immune escape.10 In this study, we leveraged our novel screening method to rapidly discover an array of new anti-PD-1 antibodies from the repertoires of six mice. We found that our method is an attractive alternative to hybridoma-based antibody discovery.
Results
Overview of the experimental approach
First, we immunized five Balb/c and five SJL mice with soluble PD-1 immunogen. All five SJL mice had a positive serum titer against PD-1, whereas only one of the Balb/c mice had a positive serum titer (Fig. 2A, Fig. 3A). We made single cell suspensions from lymph nodes surgically removed from the six responsive animals, and B cells were isolated using a magnetic bead kit. We ran approximately one million B cells from each of the six animals through our emulsion droplet microfluidics platform (Supplementary Table S1). The final product for each sample was a DNA library encoding scFv from RNA of single cells, with native heavy-light Ig pairing intact.
Figure 2.
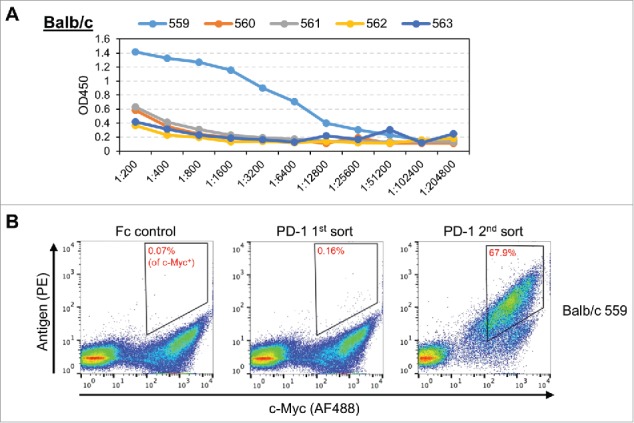
(A) Serum ELISA titers for five immunized Balb/c mice. (B) The Balb/c scFv library from mouse 559, subjected to FACS selection for an Fc control or PD-1. Staining for c-Myc (AF488) is used to differentiate yeast cells that express scFv from yeast cells that do not express scFv (x-axis). Staining for biotinylated antigen (PE) is used to identify yeast that express scFv binders. The Fc negative control (left) is used to set gates to capture yeast cells that express scFv and bind antigen (upper right corner of the FACS plot). Gates for yeast selection are indicated by the quadrangle in the upper right corner of each FACS plot. The percentage in each quadrangle (red text) indicates the proportion of c-Myc positive yeast that fell within the gate. The yeast from the first PD-1 sort (middle) are expanded and then subjected to a second PD-1 sort (right).
Figure 3.
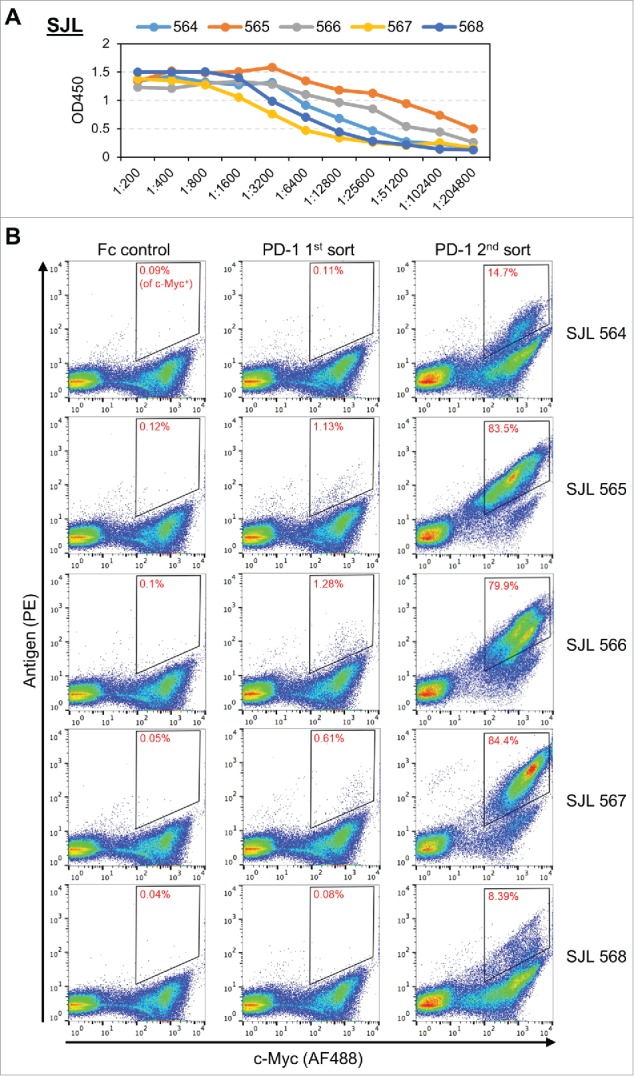
(A) Serum ELISA titers for five immunized SJL mice. (B) The SJL scFv libraries from 564, 565, 566, 567, and 568, subjected to FACS selection for an Fc control or PD-1. Staining for c-Myc (AF488) is used to differentiate yeast cells that express scFv from yeast cells that do not express scFv (x-axis). Staining for biotinylated antigen (PE) is used to identify yeast that express scFv binders. The Fc negative control (left) is used to set gates to capture yeast cells that express scFv and bind antigen (upper right corner of the FACS plot). Gates for yeast selection are indicated by the quadrangle in the upper right corner of each FACS plot. The percentage in each quadrangle (red text) indicates the proportion of c-Myc positive yeast that fell within the gate. The yeast from the first PD-1 sort (middle) are expanded and then subjected to a second PD-1 sort (right).
Next, the six DNA libraries were expressed as surface scFv in yeast (Fig. 2B, Fig. 3B). The yeast display libraries were stained with anti-c-Myc (to mark scFv-expressing cells) and biotinylated PD-1 followed by phycoerythrin (PE)-streptavidin, and then subjected to fluorescence-activated cell sorting (FACS). PD-1-binding clones were recovered and subjected to deep repertoire sequencing. Deep repertoire sequencing determines the sequences of all paired V(D)J regions of both heavy and light chain sequences. The sorted yeast cells were expanded, and then another round of staining, FACS, and deep sequencing was performed to increase confidence in the scFv binders. Seventeen scFv sequences that were present at low frequency in pre-sort libraries and became high frequency in post-sort libraries were then synthesized as full-length mAbs in Chinese hamster ovary (CHO) cells. The full-length mAbs were validated for binding kinetics through bio-layer interferometry (BLI) and checkpoint inhibition through in vitro cellular assays.
Characterization of antibody repertoires post-immunization
We define antibody “clones” as the consensus of closely related groups of sequences with ≤2 amino acid differences in their CDR3 sequences. The overall diversity of antibody clones in the pre-sort scFv libraries ranged from 4,000–13,300 antibody clones (Supplementary Table S1). The overall abundance distributions of the antibody clones were similar between animals, with several higher abundance clones followed by a long, low abundance tail (Supplementary Figure S1). However, there was variance in the level of the highest abundance clone sequences among the mice, ranging from 1.8% of reads for SJL 565 to 8.9% of reads for SJL 568.
Divergence from germline V sequences is a common metric to identify antigen-driven antibody evolution. To this end, we compared the percent identities between the V region nucleotide sequence for each antibody and its putative germline nucleotide sequence for the 500 most abundant clones from each pre-sort library. We found that the average divergence was similar across the animals: 97.9% (range 95.7%–98.7%) for Ig heavy chain and 97.8% (range 96.3%–98.4%) for Ig kappa light chain.
Isolation of PD-1 binders by yeast display
In a typical FACS dot plot, the upper right quadrant contains yeast that stain for both antigen binding and scFv expression (identified by a C-terminal c-Myc tag). The lower left quadrant contains yeast that do not stain for either the antigen or scFv expression. The lower right quadrant contains yeast that express the scFv but do not bind the antigen. We estimate the frequency of binders in each repertoire by dividing the count of yeast that double stain for antigen and scFv expression by the count of yeast that express an scFv (Fig. 2B, Fig. 3B).
Libraries generated from immunized mice yielded low percentages of scFv binders (ranging from 0.08%–1.28%) when sorted at 7 nM final antigen concentration. There was no clear association between serum titer and the frequency of binders in a repertoire. Following expansion of these sorted cells, a second round of FACS at 7 nM final antigen concentration was used to increase the specificity of the screen. The frequency of binders in the second FACS was always substantially higher than the first FACS, ranging from 8.39%–84.4%. Generally, lower frequency of binders in the first sort yielded lower frequency of binders in the second sort. Presumably, this is due to lower gating specificity for samples that have fewer bona fide binders in the original repertoire.
We used deep repertoire sequencing to assess clonal diversity of each population of binders post second FACS sort. Deep repertoire sequencing determines the sequences of all paired V(D)J regions of both heavy and light chain sequences. The population of PD-1 binders was oligoclonal, with the five most common paired, variable region amino acid sequences comprising >60% of the sequence counts for each of the six mice. Each animal yielded 38–50 unique scFv sequences present at 0.1% frequency or greater after the second FACS selection, for a total of 269 unique scFv candidate binders (Supplementary Tables S2-S3). These PD-1-binding scFvs were present at an average frequency of 0.05% in the pre-sort libraries. Using a cumulative binomial probability function, we computed that we would have had to screen >100,000 hybridoma clones to have a >90% probability of finding 40 anti-PD-1 binders that were each present at 0.05% frequency in the pre-sort libraries. In our method, the two rounds of FACS resulted in an average enrichment of 800-fold for the PD-1-binding scFvs, and the largest enrichment was nearly 29,000-fold (scFv Balb-3). In addition, many scFv were not detected in the sequencing data from the initial population of B cells from the immunized mice. This indicates that our method can enrich for rare nM-affinity binders from the initial population of B cells from immunized mice.
Evolutionary characteristics of PD-1 binders
Next, we studied the evolutionary characteristics of the anti-PD-1 antibody sequences. Consistent with a short immunization protocol, PD-1 binders generally showed little nucleotide divergence from germline sequences. For example, the average IgKV identity to germline was 98.3%, and the average IgHV identity to germline was 98.0% (Supplementary Tables S2-S3), similar to the average identities of the pre-sort immunized libraries (97.8% and 97.9%, respectively). Assessment of Replacement (R) versus Silent (S) nucleotide mutations is another metric that is often used to identify antigen-driven affinity selection of antibody sequences.11 The R/S ratio was computed by first determining V(D)J nucleotide variations from germline sequence, excluding the FR4 and CDR3 domains. We then determined whether each mutation leads to a different amino acid (R; replacement mutation) or the same amino acid (S; silent mutation). R/S ratios that exceed 3 imply directed somatic hypermutation and affinity maturation.12 We determined the R and S values across the V region for the FACS-selected antibody sequences. Consistent with minimal divergence from germline, the average R/S ratio for all PD-1 binders was only 1.8 for Ig heavy chain and 1.5 for Ig light chain. These data suggest that most of the antigen specificity of individual binders was conferred through V(D)J recombination rather than affinity maturation.
To further investigate clonal evolution, we looked for clusters of similar antibody sequences among the PD-1 binders. For this study, we defined a cluster as a group of scFv composed of four or more distinct protein sequences that share IgKV, IgKJ, IgHV, and IgHJ genes, and have the same length CDR3K and CDR3H. We observed at least one cluster of similar scFv sequences in each of the six animals. The dominant cluster (i.e., the cluster comprising the largest number of unique scFv clones) for each animal consistently included the most abundant scFv sequence from the FACS-selected cell population.
Next, we analyzed the amino acid locations that accounted for antibody sequence divergence within each clonal cluster. We aligned the full-length antibody sequences from the dominant cluster of anti-PD-1 binders from each mouse (Fig. 4). Sequence divergence was most common in the CDR3H, accounting for 51 of 151 total variant amino acid positions across the six exemplary post-sort clusters (note that variations in the N-terminus of the clusters are likely the result of mis-hybridization by 5’ V-gene primers during the polymerase chain reaction (PCR), and were thus excluded from analysis). CDR3K was far more invariant than CDR3H in the post-sort clonal clusters. In contrast, the six corresponding clonal clusters generated from pre-sort libraries revealed variation across both CDR3H and CDR3K regions (Fig. 4). These pre-sort clonal clusters were generally dominated by the consensus amino acid sequence, and all but one amino acid position showed variation. Though the CDR3K consensus sequences were identical for each of the six pre- and post-sort clonal cluster pairs, variation across the individual CDR3K sequences was lower in the post-sort clusters, with most positions filled only by the consensus amino acid. Post-sort CDR3H sequences also had positions filled only by the consensus amino acid; however, there was more amino acid heterogeneity at specific positions than in the pre-sort clusters. We conclude that FACS selection favored subsets of antibody sequences from the original clonal clusters generated by the mice.
Figure 4.

Amino acid sequence logos for groups of evolutionarily related clones, i.e., putative clonal lineages. CDR3 amino acid sequences from the pre-sort libraries are on the left. Full V(D)J amino acid sequences are shown on the right for post-sort libraries, and an expanded view of the CDR3 amino acid sequences is shown in the middle. Variant amino acids are emphasized with red arrows (the first seven amino acids are omitted due to the high probability of mis-priming during OE-RT-PCR). Though 2–3 such groups were present after each 2nd FACS, we display only a single representative dominant clonal expansion from each mouse: (A) Balb/c 559, (B) SJL 564, (C) SJL 565, (D) SJL 566, (E) SJL 567, and (F) SJL 568.
Though deep sequencing error rates are as high as 1%,13 we took several steps to curate high-quality sequences, including error filtering, merging variant sequences into “clones”, and merging overlapping forward and reverse reads. If the variant amino acids were mostly sequencing errors, mutations would have been more randomly distributed across the sequences, rather than clustered in CDR3H regions. Heavy chain sequences contained more variant amino acid positions than light chain sequences (108 versus 43 variant positions, respectively). Two of the clusters (from animals SJL 566 and SJL 567) also had a sizable number of amino acid substitutions in the CDR1 and CDR2 regions of both heavy and light chain (Fig. 4).
We also made alignments of antibody sequences that appear to be examples of convergent evolution across the two mouse strains or from independent SJL mice. An alignment between scFv Balb-2, Balb-3, and SJL-565-4 shows that SJL-565-4 shares all but the IgKJ gene used in the Balb/c animals (Supplementary Figure S2). However, the SJL 565 animal produced an antibody with a CDR3K that is nearly identical to the CDR3K found in the Balb/c animal. Another cross-strain alignment between Balb-9 and SJL-565-2 revealed identical Ig heavy chain protein sequences (with one silent nucleotide difference) and Ig light chain protein sequences that differ by only one amino acid in a framework region (Supplementary Figure S3). Antibodies SJL-564-2 and SJL-566-3 from two different SJL mice are identical across the entire IgKV gene with the same CDR3K sequences, but were generated with different IgKJ genes (Supplementary Figure S4). These antibodies share the same IgHV gene and CDR3H protein sequence, but have diverged at four amino acid positions, including changes in the CDR1 and CDR2 regions. Finally, antibodies SJL-566-1 and SJL-566-2, discovered in the same mouse, are an example of members from the same cluster, with some changes within and outside of the CDR3 regions (Supplementary Figure S5).
Functional validation of select mAbs
Assessing the function of all 269 FACS-selected scFv candidates was beyond the scope of this study. However, to validate the utility of the FACS approach, we chose 17 scFvs to re-engineer as purified, full-length mAbs for further validation. These mAbs comprised the 2–3 most abundant sequences in the second round of FACS for each animal. In addition, we selected antibody sequences that suggested convergent evolution between the SJL and Balb/c mouse strains: SJL-565-4, which is similar to Balb-2 and Balb-3; Balb-4, which shares an Ig light chain sequence with several scFv binders from both Balb/c and SJL mice; and Balb-9, which differs with SJL-565-2 Ig light chain by only two amino acids and has an identical Ig heavy chain protein sequence.
The binding specificity and affinity of each full-length antibody towards PD-1 was determined using BLI. We found that all 17 antibodies bound to PD-1, with affinities (KD) ranging from 0.25–430 nM (Tables 1–2; Supplementary Figure S6). Additionally, one mAb (SJL-568-1) registered at KD < 1 pM due to its inability to disassociate from PD-1. The SJL mice generally produced tighter binders, with an average KD of 20 nM, versus an average KD of 165 nM for the Balb/c mouse. Additionally, we expressed PD-1 recombinantly on the surface of CHO cells, and then used a FACS-based assay to detect mAb binding. We found that 15 of 17 antibodies specifically bound cells with surface-expressed PD-1, but they did not bind to cells expressing the unrelated surface-expressed protein OX40 (Tables 1–2; Supplementary Figure S7).
Table 1.
Characteristics of five purified full-length monoclonal antibodies from the Balb/c mouse.
| Ab # | CDR3K + CDR3H | Pre-sort (%) | Post-sort (%) | Affinity to PD-1 (KD) | kon (1/Ms) | kdis (1/s) | Binds surface PD-1? | PD-1/PD-L1 blockage (IC50; μg/ml) |
|---|---|---|---|---|---|---|---|---|
| Balb-1 | QQSNSWPLT + ASNYYWYFDV | 1 | 27.09 | 430 nM | 42,000 | 0.018 | Yes | No blocking |
| Balb-2 | QQHYSTPFT + TRSPFDY | 0.088 | 17.12 | 37 nM | 64,000 | 0.0023 | Yes | No blocking |
| Balb-3 | QQHYSTPFT + ARSPFDY | 0.00042 | 12.15 | 300 nM | 55,000 | 0.016 | Yes | No blocking |
| Balb-4 | FQGSHVPYT + TREKFDGYWYFDV | 0.0035 | 3.86 | 42 nM | 84,000 | 0.0035 | Yes | 0.98 |
| Balb-9 | FQGSHVPYT + TRECITTVVARVYWYFDV | 0 | 1 | 15 nM | 26,000 | 0.00039 | Yes | 1.9 |
Table 2.
Characteristics of the 12 purified full-length monoclonal antibodies from the SJL mice.
| Ab # | CDR3K + CDR3H | 564 pre-sort (%) | 565 pre-sort (%) | 566 pre-sort (%) | 567 pre-sort (%) | 568 pre-sort (%) | 564 post-sort (%) | 565 post-sort (%) | 566 post-sort (%) | 567 post-sort (%) | 568 post-sort (%) | Affinity to PD-1 (KD) | kon (1/Ms) | kdis (1/s) | Binds surface PD-1? | PD-1/PD-L1 blockage (IC50; μg/ml) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SJL-564-1 | FQGSHVPYT+ARSTYSNYVYAMDY | 0.08 | 0 | 0 | 0 | 0 | 41.32 | 0.0043 | 0 | 0.002 | 0.0033 | 30 nM | 110,000 | 0.0034 | No | No blocking |
| SJL-564-2 | QQWSSYPPT+AKDYYYGSSYRYYFDY | 0.0012 | 0 | 1.59 | 0 | 0 | 19.99 | 0.0028 | 17.02 | 0.002 | 0.0098 | 12 nM | 31,000 | 0.00036 | Yes | No blocking |
| SJL-565-1 | QQYNSYPFT+ARSVYDYDED | 0 | 0.15 | 0.00041 | 0.015 | 0 | 0.0065 | 41.39 | 0 | 0.002 | 0.044 | 0.25 nM | 25,000 | 0.0000063 | Yes | 2.29 |
| SJL-565-2 | FQGSHVPYT+TRECITTVVARVYWYFDV | 0 | 0.41 | 0.00062 | 0.0053 | 0 | 0.0086 | 24.65 | 0.0039 | 0.002 | 0.013 | 7.6 nM | 44,000 | 0.00034 | Yes | 0.65 |
| SJL-565-4 | QQHYSFPFT+ARSPFDY | 0 | 0.09 | 0.00072 | 0.0017 | 0 | 0 | 3.72 | 0 | 0 | 0.0049 | 8 nM | 44,000 | 0.00070 | Yes | No blocking |
| SJL-566-1 | QQHYTTPYT+ARGGLPVLDH | 0 | 0 | 0.25 | 0 | 0 | 0.0086 | 0.034 | 21.89 | 0.006 | 0.0016 | 7.2 nM | 43,000 | 0.00031 | Yes | 1.43 |
| SJL-566-2 | QQHYSTPYT+ARGGLPVLDY | 0.0003 | 0 | 2.11 | 0 | 0 | 1.93 | 0 | 18.13 | 0 | 0.0016 | 25 nM | 39,000 | 0.00095 | Yes | 1.84 |
| SJL-566-3 | QQWSSYPPT+AKDYYYGSSYRYYFDY | 0.0012 | 0 | 1.59 | 0 | 0 | 19.99 | 0.0028 | 17.02 | 0.002 | 0.0098 | 65 nM | 21,000 | 0.0014 | Yes | No blocking |
| SJL-567-1 | QQYNSYPYT+ASSNYDYGED | 0 | 0.0001 | 0 | 0.34 | 0 | 0 | 0.0028 | 0.0058 | 39 | 0.013 | 15 nM | 130,000 | 0.0019 | Yes | 0.71 |
| SJL-567-2 | QQYNSYPYT+ASSNYDYDED | 0 | 0 | 0 | 0.56 | 0.011 | 0.0022 | 0.0085 | 0.0019 | 22.42 | 0.2 | 28 nM | 94,000 | 0.0027 | Yes | 3.39 |
| SJL-568-1 | LQYDEFPYT+TNGYFVDC | 0 | 0 | 0 | 0 | 0.5 | 0.0065 | 0.03 | 0 | 0 | 31.23 | <0.001 nM | 20,000 | <0.0000001 | No | No blocking |
| SJL-568-2 | QQSIEDPWT+TRGGYGNDY | 0.0021 | 0 | 0 | 0 | 0.5 | 0.05 | 0.02 | 0.012 | 0 | 11 | 36 nM | 140,000 | 0.0051 | Yes | 1.09 |
Binding of PD-1 to PD-L1 leads to inhibition of T cell signaling.9 Antibodies that bind PD-1 and antagonize PD-1/PD-L1 interactions can therefore remove this inhibition, allowing T cells to be activated. We tested PD-1/PD-L1 checkpoint blockade through an in vitro cellular Nuclear Factor of Activated T cells (NFAT) luciferase reporter assay. In this assay, antibodies whose anti-PD-1 epitopes fall inside the PD-L1 binding domain antagonize PD-1/PD-L1 interactions, resulting in an increase of the NFAT-luciferase reporter. We assayed the 17 full-length mAb candidates that bound PD-1 expressed in CHO cells. To generate an IC50 value for each mAb, we made measurements across several concentrations (Supplementary Figure S8). We found that nine of 17 full-length mAbs were functional in checkpoint blockade in a dose dependent manner (Tables 1–2).
Finally, our examples of convergent evolution revealed that minor sequence mutations can lead to changes in binding kinetics and checkpoint blockade. For example, though Balb-2, Balb-3, and SJL-565-4 have very similar CDR3 sequences, variations throughout the heavy and light chain V regions led to different KD values of 37 nM, 300 nM, and 8 nM, respectively (Supplementary Figure S2). None of the three antibodies functioned in checkpoint blockade. Balb-9 and SJL-565-2, which only have a single amino acid difference, led to a modest but consistent shift for both affinity (15 nM and 7.6 nM, respectively) and checkpoint blockade IC50 (1.9 μg/ml and 0.65 μg/ml, respectively; Supplementary Figure S3). SJL-564-2 and SJL-566-3 have identical CDR3H amino acid sequences, but a different IgKJ gene and two amino acid differences in the CDR1H and CDR2H regions. These differences were sufficient to decrease affinity from 12 nM for SJL-564-2 to 65 nM for SJL-566-3 (Supplementary Figure S4). Neither mAb functioned in checkpoint blockade. Finally, the closely related mAbs SJL-566-1 and SJL-566-2 from the same cluster had differences in KD (7.2 nM and 25 nM, respectively) and checkpoint blockade IC50 (1.43 μg/ml and 1.84 μg/ml, respectively; Supplementary Figure S5).
Discussion
From only six animals, we identified 269 high-confidence, unique scFv binders against human PD-1 immunogen that were present at an average of 1 in 2,000 in the initial pre-sort scFv libraries. Two rounds of FACS produced populations of scFv with an average enrichment of 800-fold. However, most of the scFv present in the pre-sort mouse repertoires were eliminated following FACS. Therefore, our work suggests that most of the antibodies present in the repertoires of immunized mice are not strong binders to the immunogen. We expressed 17 scFv candidates as full-length mAbs. All 17 antibodies bound recombinant PD-1 with KD < 500 nM, and one antibody was KD < 1 pM. Fifteen of the 17 antibodies specifically bound surface-expressed PD-1 in a cellular FACS assay, and nine of these antibodies were functional in checkpoint blockade in a cell-based assay. With further development, the functional mAbs could be engineered into candidates for cancer therapy by checkpoint blockade.
Other groups have successfully discovered high-affinity antibodies in the repertoires of immunized mice using deep sequencing, but without the benefit of microfluidic technology for Ig pairing.3,6,14-16 Generally, these groups first used deep sequencing followed by bioinformatics to identify the most common antibodies in the animals. Then, they inferred Ig pairings by matching frequent heavy chains with frequent light chains. These methods generally yielded high-affinity binders, achieving 0–40% false discovery rates. However, our method has two distinct advantages over most previously published deep sequencing methods: 1) we achieve native Ig pairing, and 2) our process is designed to discover rare antibodies. Though accurate false negative rates are impossible to compute without clear truth models, our method must have a lower false negative rate than prior methods, since prior methods explicitly exclude rare antibodies and do not necessarily include natively paired antibodies.
Further improvements to our protocols can be made. For example, many scFv that are <0.1% frequency in FACS-selected populations may also be high affinity binders. Future studies should more precisely determine the appropriate post-FACS frequency cut-off for identifying antibodies of the desired binding affinity. In this study, we did not try different antigen molarities for optimal FACS selection. Future work might optimize the balance between lower antigen molarities, which might yield more KD < 1 nM scFv binders, and higher antigen molarities, which might yield a greater variety of scFv sequences. Prior work suggests scFv can bind with lower affinity than full length mAbs.17-19 To address this issue, our approach could be modified to leverage a variety of display methods, for example yeast antigen-binding fragment display.20,21 Additionally, we note that it is possible to perform yeast scFv display with DNA libraries generated using randomly paired Ig sequences.22 This may generate a larger diversity of scFv binders because a given heavy or light chain may still bind antigen if paired with a non-cognate light or heavy chain, respectively.23,24 Such non-native pairings may result in additional antibodies with favorable properties, augmenting intellectual property portfolios. However, we speculate that non-natively paired antibodies may not be as developable as natively-paired antibodies. Finally, future work might explore methods that first enrich B cells using the desired antigen25-29 prior to microfluidic droplet encapsulation. Pre-enrichment might yield yeast scFv libraries with a higher proportion of binders, thereby increasing the probability of discovering rare antibodies. On the other hand, pre-enrichment might exclude useful populations of scFv binders, for example because different enrichment protocols are required for enrichment of memory B cells as opposed to plasma cells.29
FACS selection yielded clusters of similar scFv sequences, which suggests early stages of clonal divergence. Other groups have observed a similar phenomenon in the repertoires of immunized mice.6,15,16,30 We acknowledge that the high error rate of deep sequencing could produce erroneous sequences that masquerade as clonal divergence, a problem that has been addressed elsewhere with alternative molecular methods.31,32 Still, to reduce the probability of errors, we filtered out low-quality reads, required multiple identical sequencing reads, and merged overlapping forward and reverse reads to generate high-confidence consensus antibody sequences. In addition, sequencing errors would be spread out across the entire heavy and light chain regions. However, our data show that most of the sequence differences between antibody clusters reside within the CDR3 regions, suggesting true clonal divergence. Similar to our study, prior work also detected antibody sequence convergence between mouse replicates.3,16 Though some apparent convergence may result from workflow contamination,3 future work should investigate whether convergence helps to detect high-affinity antibodies.16 We are confident that some convergence does occur, since we identified large clonally related antibodies in multiple animals, rather than just a few shared reads.
Prior work on mouse plasma cell repertoires found only 2–5 amino acid substitutions per V-gene,3 roughly in line with our data on lymph node B cells and other published work on splenocytes.14,30 In the future, we hope to improve our mouse immunization protocol to increase divergence from germline and affinity maturation in vivo. One approach will focus on bone marrow plasma cells, which are <1% of all lymphoid cells but generate most antibodies in circulation. Bone marrow plasma cells may yield more evolved and higher-affinity antibodies than B cells from the lymph node or spleen.33 Another approach will focus on mouse immunization procedures that allow for improved in vivo affinity maturation, with the goal of finding a large variety of high-affinity antibodies from cohorts of <10 mice.
In this study, we performed a limited set of biochemical and cellular assays to validate the mAbs identified using our discovery methodology. We acknowledge that more exhaustive pre-clinical studies would be required to develop any of these candidate mAbs as therapeutics. For example, the mAbs should be humanized34 and subjected to in vitro affinity maturation.35 Our data on clonal lineages might simplify such antibody engineering development work, for example, by identifying highly constrained amino acid residues. Additionally, further development would require comprehensive epitope mapping36 or epitope binning37 on hundreds of our mAb candidates. Antibody development would also require larger-scale studies of specificity, for example with peptide arrays.38 Such studies would also help elucidate whether our microfluidic and molecular genomic protocols bias antibody discovery toward certain epitopes, or identify antibodies with lower specificity than alternative discovery methods.
The complete process of scFv binder discovery, not including mouse immunizations, required only three weeks and the work of a single technician. The process of scFv re-engineering and mAb expression is widely practiced and requires less than three weeks of additional effort, even without automation. To increase the throughput of scFv re-engineering, exploratory work has shown that engineering millions-diverse full-length mammalian libraries from OE-RT-PCR scFv amplicons is possible.39 In the future, full-length mammalian libraries could be used for massively parallel functional screens, for example, to directly discover rare neutralizing antibodies from diverse repertoires. In general, the speed and convenience of our methods result in a larger number of candidates for development pipelines. More mice can be screened and each mouse can be screened more exhaustively. This has substantial advantages for intellectual property because a program can choose a target and quickly identify hundreds of unique, validated binders. In theory, technologies such as ours could also lead to shorter commercialization timelines.
Materials and methods
Mouse immunization and sample preparation
All mouse work was performed at Aragen Bioscience, a contract research organization, and was overseen by a licensed veterinarian. Five SJL mice and five Balb/c mice were immunized with recombinant His-tagged PD-1 protein (R&D Systems), using TiterMax as an adjuvant. One μg of immunogen was injected into each hock and 3 μg of immunogen was administered intraperitoneally, every third day for 15 days. Titer was assessed by enzyme-linked immunosorbent assay (ELISA) on a 1:2 dilution series of each animal's serum, starting at a 1:200 dilution. A final intravenous boost of 2.5 μg/hock without adjuvant was given to each animal before harvest. Lymph nodes (popliteal, inguinal, axillary, and mesenteric) were surgically removed after sacrifice. Single cell suspensions for each animal were made by manual disruption followed by passage through a 70 μm filter. Next, we used the EasySep™ Mouse Pan-B Cell Isolation Kit (Stemcell Technologies) negative selection kit to isolate B cells from each sample. The lymph node B cell populations were quantified by counting on a C-Chip hemocytometer (Incyto) and assessed for viability using Trypan blue. The cells were then diluted to 5,000–6,000 cells/μL in phosphate-buffered saline (PBS) with 12% OptiPrep™ Density Gradient Medium (Sigma). This cell mixture was used for microfluidic encapslation as described in the next section
Generating paired heavy and light chain libraries
Library generation is divided into three steps: 1) poly(A)+ mRNA capture, 2) multiplexed overlap extension reverse transcriptase polymerase chain reaction (OE-RT-PCR), and 3) nested PCR to remove artifacts and add adapters for deep sequencing or yeast display libraries. We created scFv libraries from approximately one million B cells from each animal that achieved a positive ELISA titer.
For poly(A)+ mRNA capture, we used a custom designed co-flow emulsion droplet microfluidic chip fabricated from glass (Dolomite). The microfluidic chip has two input channels for fluorocarbon oil (Dolomite), one input channel for the cell suspension mix described above, and one input channel for oligo-dT beads (NEB) at 1.25 mg/ml in cell lysis buffer (20 mM Tris pH 7.5, 0.5 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Tween-20, and 20 mM dithiothreitol). The input channels are etched to 50 μm × 150 μm for most of the chip's length, narrow to 55 μm at the droplet junction, and are coated with hydrophobic Pico-Glide (Dolomite). Three Mitos P-Pump pressure pumps (Dolomite) were used to pump the liquids through the chip. Droplet size depends on pressure, but typically we find that droplets of ∼45 μm diameter are optimally stable. Emulsions are collected into chilled 2 ml microcentrifuge tubes and incubated at 40 °C for 15 minutes for mRNA capture. The beads were extracted from the droplets using Pico-Break (Dolomite).
For multiplex OE-RT-PCR, we used glass Telos droplet emulsion microfluidic chips (Dolomite). mRNA-bound beads were re-suspended into OE-RT-PCR mix and injected into the microfluidic chips with a mineral oil-based surfactant mix (available commercially from GigaGen) at pressures that generate 27 μm droplets. The OE-RT-PCR mix contains 2 × one-step RT-PCR buffer, 2.0 mM MgSO4, SuperScript III reverse transcriptase, and Platinum Taq (Thermo Fisher Scientific), plus a mixture of primers directed against the IgK C region, the IgG C region, and all V regions (Supplementary Figure S9).22 The overlap region is a DNA sequence that encodes a Gly-Ser rich scFv linker sequence.40 The DNA fragments are recovered from the droplets using a droplet breaking solution (available commercially from GigaGen) and then purified using QIAquick PCR Purification Kit (Qiagen).
For nested PCR (Supplementary Figure S9), the purified OE-RT-PCR product was first run on a 1.7% agarose gel for 80 minutes at 150 V. A band at 1200–1500 base pair (bp) corresponding to the linked product was excised and purified using NucleoSpin Gel and PCR Clean-up Kit (Macherey Nagel). PCR was then performed to add adapters for Illumina sequencing or yeast display; for sequencing, a randomer of seven nucleotides is added to increase base calling accuracy in subsequent next-generation sequencing steps. Nested PCR was performed with 2 × NEBNext High-Fidelity amplification mix (NEB) with either Illumina adapter containing primers or primers for cloning into the yeast expression vector. The nested PCR product was run on a 1.2% agarose gel for 50 minutes at 150V. A band at 800–1100 bp was excised and purified using NucleoSpin Gel and PCR Clean-up Kit (Macherey Nagel).
Yeast library screening
Human IgG1-Fc (Thermo Fisher Scientific) and PD-1 (R&D Systems) proteins were biotinylated using the EZ-Link Micro Sulfo-NHS-LC-Biotinylation kit (Thermo Fisher Scientific). The biotinylation reagent was resuspended to 9 mM and added to the protein at a 50-fold molar excess. The reaction was incubated on ice for 2 hours and then the biotinylation reagent was removed using Zeba desalting columns (Thermo Fisher Scientific). The final protein concentration was calculated with a Bradford assay.
We built a yeast surface display vector (pYD) that contains a GAL1/10 promoter, an Aga2 cell wall tether, and a C-terminal c-Myc tag (Supplementary Figure S10). The GAL1/10 promoter induces expression of the scFv protein in medium that contains galactose. The Aga2 cell wall tether is required to shuttle the scFv to the yeast cell surface and tether the scFv to the extracellular space. The c-Myc tag is used during the flow sort to stain for yeast cells that express in-frame scFv protein. Saccharomyces cerevisiae cells (ATCC) are electroporated (Bio-Rad Gene Pulser II; 0.54 kV, 25 uF, resistance set to infinity) with gel-purified nested PCR product and linearized pYD vector for homologous recombination in vivo. Transformed cells are expanded and induced with galactose to generate yeast scFv display libraries.
Two million yeast cells from the expanded scFv libraries are stained with anti-c-Myc (Thermo Fisher Scientific A21281) and an AF488-conjugated secondary antibody (Thermo Fisher Scientific A11039). To select scFv-expressing cells that bind to antigen, biotinylated antigen is added to the yeast culture (7 nM final) during primary antibody incubation and then stained with PE-streptavidin (Thermo Fisher Scientific). Yeast cells are flow sorted on a BD Influx (Stanford Shared FACS Facility) for double-positive cells (AF488+/PE+), and recovered clones are then plated on SD-CAA plates with kanamycin, streptomycin, and penicillin (Teknova) for expansion. The expanded first round FACS clones are then subjected to a second round of FACS with the same antigen at the same molarity (7 nM final). Plasmid minipreps (Zymo Research) are prepared from yeast recovered from the final FACS sort. Tailed-end PCR is used to add Illumina adapters to the plasmid libraries for deep sequencing.
Deep antibody repertoire sequencing
Deep antibody sequencing libraries were quantified using a quantitative PCR Illumina Library Quantification Kit (KAPA) and diluted to 17.5 pM. Libraries were sequenced on a MiSeq (Illumina) using a 500 cycle MiSeq Reagent Kit v2, according to the manufacturer's instructions. To obtain high quality sequence reads with maintained heavy and light chain linkage, we performed sequencing in two separate runs (Supplementary Figure S11). In the first run (“linked run”), we directly sequenced the scFv libraries to obtain forward read of 340 cycles for the light chain V-gene and CDR3, and reverse read of 162 cycles that cover the heavy chain CDR3 and part of the heavy chain V-gene. In the second run (“unlinked run”), we first used the scFv library as a template for PCR to separately amplify heavy and light chain V-genes. Then, we obtained forward reads of 340 cycles and reverse reads of 162 cycles for the heavy and light chain Ig separately. This produces forward and reverse reads that overlap at the CDR3 and part of the V-gene, which increases confidence in nucleotide calls.
To remove base call errors, we used a previously published expected error filtering method.13 The expected number of errors (E) for a read were calculated from its Phred scores. By default, reads with E >1 were discarded, leaving reads for which the most probable number of base call errors is zero. As an additional quality filter, singleton nucleotide reads were discarded because sequences found two or more times have a high probability of being correct.41 Finally, we generated high-quality, linked antibody sequences by merging filtered sequences from the linked and unlinked runs. Briefly, we wrote a series of scripts in Python that first merged forward and reverse reads from the unlinked run. We discarded any pairs of forward and reverse sequences that contain mismatches. Next, we used the nucleotide sequences from the linked run to query merged sequences in the unlinked run. The final output from the scripts is a series of full-length, high-quality V(D)J sequences, with native heavy and light chain Ig pairing.
To identify reading frame and FR/CDR junctions, we first processed a database of well-curated immunoglobulin sequences42 to generate position-specific sequence matrices (PSSMs) for each FR/CDR junction (Supplementary Figure S12). We used these PSSMs to identify FR/CDR junctions for each of the merged nucleotide sequences generated using the processes described above. This identified the protein reading frame for each of the nucleotide sequences. CDR sequences that have a low identify score to the PSSMs are indicated by an exclamation point. Python scripts were then used to translate the sequences. We required reads to have a valid predicted CDR3 sequence, so, for example, reads with a frame-shift between the V and J segments were discarded. Next, we ran UBLAST43 using the scFv nucleotide sequences as queries and V and J gene sequences from the IMGT database42 as the reference sequences. The UBLAST alignment with the lowest E-value was used to assign V and J gene families and compute %ID to germline. R/S values were computed using V gene segments spanning the first amino acid through the FR3-CDR3 boundary. CDR3s were not used in R/S value computation.
To estimate the diversity of the libraries, we defined “clones” conservatively (Supplementary Table S1). First, we concatenated the CDR3K and CDR3H amino acid sequences from each scFv sequence into a single contiguous amino acid sequence. Next, we used UBLAST43 to compute the total number of amino acid differences in all pairwise alignments between each concatenated sequence in each data set. Groups of sequences with ≤2 amino acid differences in the concatenated CDR3s were counted as a single clone. Finally, we used the majority amino acid identity at each residue position to generate the consensus amino acid sequence of the clone from sequences of the members of the group.
Monoclonal antibody expression
The mAbs were expressed from a variant of the pCDNA5/FRT mammalian expression vector (Thermo Fisher Scientific). The vector uses an EF1-alpha promoter to drive light chain expression, followed by a BGH polyA sequence and a CMV promoter to drive expression of the heavy chain, followed by a second BGH polyA sequence (Supplementary Figure S13). Antibody expression constructs were built using GeneBlocks (Integrated DNA Technologies) and NEBuilder® HiFi DNA Assembly Master Mix (New England BioLabs). All constructs were synthesized as human IgG1, regardless of a given antibody's IgG isotype in the original mouse repertoire. Constructs were amplified in NEB 10-beta E. coli and purified with the ZymoPURE™ Plasmid Maxiprep Kit (Zymo Research). The purified plasmids were used for transient transfection in the ExpiCHO system (Thermo Fisher Scientific). Transfected cells were cultured for 7–9 days in ExpiCHO medium and antibodies were purified from filtered supernatant using protein A columns (Millipore). Antibody purity and size was verified by Coomassie stained SDS-PAGE gels (Thermo Fisher Scientific).
Monoclonal antibody characterization
For measurement of PD-1 binding kinetics, antibodies were loaded onto an Anti-Human IgG Fc (AHC) biosensor using the Octet Red96 system (ForteBio), by a contract research organization (LakePharma). Loaded biosensors were dipped into antigen dilutions beginning at 300 nM, with 6 serial dilutions at 1:3. Kinetic analysis was performed using a 1:1 binding model and global fitting (Supplementary Figure S6). Pembrolizumab produced in-house was used as a positive control.
Stable OX40 and PD-1 expressing Flp-In CHO (Thermo Fisher Scientific) cells were generated and mixed at a 50:50 ratio. One million cells were stained with 2 μg of our purified anti-PD-1 recombinant antibodies in 100 μl of MACS Buffer (DPBS with 0.5% bovine serum albumin and 2 mM EDTA) for 30 minutes at 4 °C. Cells were then co-stained with anti-human CD134 (OX40)-APC [Ber-ACT35] (BioLegend 350008) and anti-human IgG Fc-PE [M1310G05] (BioLegend 41070) antibodies for 30 minutes at 4°C. An anti-human CD279 (PD-1)-FITC [EH12.2H7] (BioLegend 329903) antibody was used as a control for these mixing experiments and cell viability was assessed with DAPI. Flow cytometry analysis was conducted on a BD Influx at the Stanford Shared FACS Facility and data was analyzed using FlowJo (Supplementary Figure S7).
For analysis of the antibodies' ability to block the PD-1/PD-L1 interaction, the PD-1/PD-L1 Blockade Bioassay (Promega) was used according to the manufacturer's instructions. On the day prior to the assay, PD-L1 aAPC/CHO-K1 cells were thawed into 90% Ham's F-12/10% fetal bovine serum (FBS) and plated into the inner 60 wells of two 96-well plates. The cells were incubated overnight at 37 °C, 5% CO2. On the day of assay, antibodies were diluted in 99% RPMI/1% FBS. The antibody dilutions were added to the wells containing the PD-L1 aAPC/CHO-K1 cells, followed by addition of PD-1 effector cells (thawed into 99% RPMI/1% FBS). The cell/antibody mixtures were incubated at 37 °C, 5% CO2 for 6 hours, after which Bio-Glo Reagent was added and luminescence was read using a Spectramax i3x plate reader (Molecular Devices). Fold-induction was plotted by calculating the ratio of [signal with antibody]/[signal with no antibody], and the plots were used to calculate the IC50 using SoftMax Pro (Molecular Devices) (Supplementary Figure S8). In-house produced pembrolizumab was used as a positive control, and an antibody binding to an irrelevant antigen was used as a negative control.
Supplementary Material
Disclosure of potential conflicts of interest
ASA, RAM, MJS, MSA, MAA, RCE, JL, RL, and DSJ are employees of GigaGen Inc. and receive both equity shares and salary for their work.
Acknowledgments
We thank Everett Meyer for his useful insights during development of the technology. The staff at the Stanford Shared FACS Facility were a valuable resource for flow sorting.
Funding
This work was supported by the National Science Foundation under grant 1230150; National Cancer Institute under grant R44CA187852.
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767-74. doi: 10.1038/nrd3229. PMID:20811384 [DOI] [PubMed] [Google Scholar]
- 2.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495-7. doi: 10.1038/256495a0. PMID:1172191 [DOI] [PubMed] [Google Scholar]
- 3.Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, Chrysostomou C, Hunicke-Smith SP, Iverson BL, Tucker PW, et al.. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol. 2010;28(9):965-9. doi: 10.1038/nbt.1673. PMID:20802495 [DOI] [PubMed] [Google Scholar]
- 4.DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, Giesecke C, Dörner T, Andrews SF, et al.. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31:166-9. doi: 10.1038/nbt.2492. PMID:23334449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busse CE, Czogiel I, Braun P, Arndt PF, Wardemann H. Single-cell based high-throughput sequencing of full-length immunoglobulin heavy and light chain genes. Eur J Immunol. 2014;44(2):597-603. doi: 10.1002/eji.201343917. PMID:24114719 [DOI] [PubMed] [Google Scholar]
- 6.Wang B, Kluwe CA, Lungu OI, DeKosky BJ, Kerr SA, Johnson EL, Jung J, Rezigh AB, Carroll SM, Reyes AN, et al.. Facile discovery of a diverse panel of anti-ebola virus antibodies by immune repertoire mining. Sci Rep. 2015;5:13926. doi: 10.1038/srep13926. PMID:26355042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, Georgiou G. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med 2015; 21:86-91. doi: 10.1038/nm.3743. PMID:25501908 [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Lee CH, Johnson EL, Kluwe CA, Cunningham JC, Tanno H, Crooks RM, Georgiou G, Ellington AD. Discovery of high affinity anti-ricin antibodies by B cell receptor sequencing and by yeast display of combinatorial VH:VL libraries from immunized animals. MAbs. 2016;8(6):1035-44. doi: 10.1080/19420862.2016.1190059. PMID:27224530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767-1778. doi: 10.1056/NEJMra1514296. PMID:27806234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56-61. doi: 10.1126/science.aaa8172. PMID:25838373 [DOI] [PubMed] [Google Scholar]
- 11.Lossos IS, Tibshirani R, Narasimhan B, Levy R. The inference of antigen selection on Ig genes. J Immunol. 2000;165(9):5122-6. doi: 10.4049/jimmunol.165.9.5122. PMID:11046043 [DOI] [PubMed] [Google Scholar]
- 12.Bose B, Sinha S. Problems in using statistical analysis of replacement and silent mutations in antibody genes for determining antigen-driven affinity selection. Immunology. 2005;116(2):172-83. doi: 10.1111/j.1365-2567.2005.02208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476-82. doi: 10.1093/bioinformatics/btv401. PMID:26139637 [DOI] [PubMed] [Google Scholar]
- 14.Saggy I, Wine Y, Shefet-Carasso L, Nahary L, Georgiou G, Benhar I. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng Des Sel. 2012;25(10):539-49. doi: 10.1093/protein/gzs060. PMID:22988130 [DOI] [PubMed] [Google Scholar]
- 15.Gray SA, Moore M, VandenEkart EJ, Roque RP, Bowen RA, Van Hoeven N, Wiley SR, Clegg CH. Selection of therapeutic H5N1 monoclonal antibodies following IgVH repertoire analysis in mice. Antiviral Res. 2016 Jul;131:100-8. doi: 10.1016/j.antiviral.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kono N, Sun L, Toh H, Shimizu T, Xue H, Numata O, Ato M, Ohnishi K, Itamura S. Deciphering antigen-responding antibody repertoires by using next-generation sequencing and confirming them through antibody-gene synthesis. Biochem Biophys Res Commun. 2017;487(2):300-306. doi: 10.1016/j.bbrc.2017.04.054. PMID:28412367 [DOI] [PubMed] [Google Scholar]
- 17.Bird RE, Walker BW. Single chain antibody variable regions. Trends Biotechnol. 1991;9(4):132-7. doi: 10.1016/0167-7799(91)90044-I. PMID:1367550 [DOI] [PubMed] [Google Scholar]
- 18.Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, et al.. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003;77(19):10557-65. doi: 10.1128/JVI.77.19.10557-10565.2003. PMID:12970440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quintero-Hernández V, Juárez-González VR, Ortíz-León M, Sánchez R, Possani LD, Becerril B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol. 2007;44(6):1307-15. doi: 10.1016/j.molimm.2006.05.009. PMID:16814388 [DOI] [PubMed] [Google Scholar]
- 20.van den Beucken T, Pieters H, Steukers M, van der Vaart M, Ladner RC, Hoogenboom HR, Hufton SE. Affinity maturation of Fab antibody fragments by fluorescent-activated cell sorting of yeast-displayed libraries. FEBS Lett. 2003;546(2–3):288-94. doi: 10.1016/S0014-5793(03)00602-1. PMID:12832056 [DOI] [PubMed] [Google Scholar]
- 21.Walker LM, Bowley DR, Burton DR. Efficient recovery of high-affinity antibodies from a single-chain Fab yeast display library. J Mol Biol. 2009;389(2):365-75. doi: 10.1016/j.jmb.2009.04.019. PMID:19376130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller KD, Pefaur NB, Baird CL. Construction and screening of antigen targeted immune yeast surface display antibody libraries. Curr Protoc Cytom. 2008;Chapter 4:Unit4.7. doi: 10.1002/0471142956.cy0407s45. PMID:18770649 [DOI] [PubMed] [Google Scholar]
- 23.Alzari PM, Spinelli S, Mariuzza RA, Boulot G, Poljak RJ, Jarvis JM, Milstein C. Three-dimensional structure determination of an anti-2-phenyloxazolone antibody: the role of somatic mutation and heavy/light chain pairing in the maturation of an immune response. EMBO J. 1990;9(12):3807-14 PMID:2123450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czerwinski M, Siemaszko D, Siegel DL, Spitalnik SL. Only selected light chains combine with a given heavy chain to confer specificity for a model glycopeptide antigen. J Immunol. 1998;160(9):4406-17 PMID:9574545 [PubMed] [Google Scholar]
- 25.Lagerkvist AC, Furebring C, Borrebaeck CA. Single, antigen-specific B cells used to generate Fab fragments using CD40-mediated amplification or direct PCR cloning. Biotechniques. 1995;18(5):862-9 PMID:7542459 [PubMed] [Google Scholar]
- 26.Dohmen SE, Mulder A, Verhagen OJ, Eijsink C, Franke-van Dijk ME, van der Schoot CE. Production of recombinant Ig molecules from antigen-selected single B cells and restricted usage of Ig-gene segments by anti-D antibodies. J Immunol Methods. 2005;298(1–2):9-20. doi: 10.1016/j.jim.2004.12.013. PMID:15847793 [DOI] [PubMed] [Google Scholar]
- 27.Ditzel HJ. Affinity isolation of antigen-specific circulating B cells for generation of phage display-derived human monoclonal antibodies. Methods Mol Biol. 2009;562:37-43. doi: 10.1007/978-1-60327-302-2_3. PMID:19554285 [DOI] [PubMed] [Google Scholar]
- 28.Di Niro R, Mesin L, Raki M, Zheng NY, Lund-Johansen F, Lundin KE, Charpilienne A, Poncet D, Wilson PC, Sollid LM. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. 2010;185(9):5377-83. doi: 10.4049/jimmunol.1001587. PMID:20935207 [DOI] [PubMed] [Google Scholar]
- 29.Kodituwakku AP, Jessup C, Zola H, Roberton DM. Isolation of antigen-specific B cells. Immunol Cell Biol. 2003;81(3):163-70. doi: 10.1046/j.1440-1711.2003.01152.x. PMID:12752679 [DOI] [PubMed] [Google Scholar]
- 30.Wilson JR, Tzeng WP, Spesock A, Music N, Guo Z, Barrington R, Stevens J, Donis RO, Katz JM, York IA. Diversity of the murine antibody response targeting influenza A(H1N1pdm09) hemagglutinin. Virology. 2014;458–459:114-24. doi: 10.1016/j.virol.2014.04.011. PMID:24928044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Briney B, Le K, Zhu J, Burton DR. Clonify: unseeded antibody lineage assignment from next-generation sequencing data. Sci Rep. 2016;6:23901. doi: 10.1038/srep23901. PMID:27102563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole C, Volden R, Dharmadhikari S, Scelfo-Dalbey C, Vollmers C. Highly accurate sequencing of full-length immune repertoire amplicons using Tn5-enabled and molecular identifier-guided amplicon assembly. J Immunol. 2016;196(6):2902-7. doi: 10.4049/jimmunol.1502563. PMID:26856699 [DOI] [PubMed] [Google Scholar]
- 33.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388(6638):133-4. doi: 10.1038/40540. PMID:9217150 [DOI] [PubMed] [Google Scholar]
- 34.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321(6069):522-5. doi: 10.1038/321522a0. PMID:3713831 [DOI] [PubMed] [Google Scholar]
- 35.Boder ET, Wittrup KD. Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 2000;328:430-44. doi: 10.1016/S0076-6879(00)28410-3. PMID:11075358 [DOI] [PubMed] [Google Scholar]
- 36.Davidson E, Doranz BJ. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology. 2014;143(1):13-20. doi: 10.1111/imm.12323. PMID:24854488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks BD, Miles AR, Abdiche YN. High-throughput epitope binning of therapeutic monoclonal antibodies: why you need to bin the fridge. Drug Discov Today. 2014;19(8):1040-4. doi: 10.1016/j.drudis.2014.05.011. PMID:24880105 [DOI] [PubMed] [Google Scholar]
- 38.Weber LK, Palermo A, Kügler J, Armant O, Isse A, Rentschler S, Jaenisch T, Hubbuch J, Dübel S, Nesterov-Mueller A, et al.. Single amino acid fingerprinting of the human antibody repertoire with high density peptide arrays. J Immunol Methods. 2017;443:45-54. doi: 10.1016/j.jim.2017.01.012. PMID:28167275 [DOI] [PubMed] [Google Scholar]
- 39.Johnson DS, Adler AS, Mizrahi RM. US20160362681 A1. Recombinant fusion proteins and libraries from immune cell repertoires. 2016 [Google Scholar]
- 40.Dangaj D, Lanitis E, Zhao A, Joshi S, Cheng Y, Sandaltzopoulos R, Ra HJ, Danet-Desnoyers G, Powell DJ Jr, Scholler N. Novel recombinant human b7-h4 antibodies overcome tumoral immune escape to potentiate T-cell antitumor responses. Cancer Res. 2013;73(15):4820-9. doi: 10.1158/0008-5472.CAN-12-3457. PMID:23722540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996-8. doi: 10.1038/nmeth.2604. PMID:23955772 [DOI] [PubMed] [Google Scholar]
- 42.Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al.. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37(Database issue):D1006-12. doi: 10.1093/nar/gkn838. PMID:18978023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460-1. doi: 10.1093/bioinformatics/btq461. PMID:20709691 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.