

ABSTRACT
Affinity-matured, functional anti-pathogen antibodies are present at low frequencies in natural human repertoires. These antibodies are often excellent candidates for therapeutic monoclonal antibodies. However, mining natural human antibody repertoires is a challenge. In this study, we demonstrate a new method that uses microfluidics, yeast display, and deep sequencing to identify 247 natively paired anti-pathogen single-chain variable fragments (scFvs), which were initially as rare as 1 in 100,000 in the human repertoires. Influenza A vaccination increased the frequency of influenza A antigen-binding scFv within the peripheral B cell repertoire from <0.1% in non-vaccinated donors to 0.3-0.4% in vaccinated donors, whereas pneumococcus vaccination did not increase the frequency of antigen-binding scFv. However, the pneumococcus scFv binders from the vaccinated library had higher heavy and light chain Replacement/Silent mutation (R/S) ratios, a measure of affinity maturation, than the pneumococcus binders from the corresponding non-vaccinated library. Thus, pneumococcus vaccination may increase the frequency of affinity-matured antibodies in human repertoires. We synthesized 10 anti-influenza A and nine anti-pneumococcus full-length antibodies that were highly abundant among antigen-binding scFv. All 10 anti-influenza A antibodies bound the appropriate antigen at KD<10 nM and neutralized virus in cellular assays. All nine anti-pneumococcus full-length antibodies bound at least one polysaccharide serotype, and 71% of the anti-pneumococcus antibodies that we tested were functional in cell killing assays. Our approach has future application in a variety of fields, including the development of therapeutic antibodies for emerging viral diseases, autoimmune disorders, and cancer.
KEYWORDS: antibody repertoire, deep sequencing, Influenza A, microfluidics, pneumococcus
Introduction
Human antibody repertoires are a rich resource for anti-pathogen antibodies. Naturally occurring human antibodies are often excellent candidates for therapeutics because they are affinity-matured through normal B cell selection processes within a person. Human antibody repertoires also provide insight into antigenic targets for humoral immunity.1,2 Naturally paired human antibodies have been identified using various technologies,3 including human hybridomas,4,5 clonal expansion of primary B cells,6 and Epstein-Barr Virus transformation.7,8 However, the field of human antibody discovery remains constrained by the technical challenges of efficiently capturing rare B cell clones using these methods.
The advent of modern genomic technologies now provides alternative workflows for human antibody repertoire mining. These methods use multiplexed PCR or RT-PCR to amplify rearranged V(D)J sequences to generate diverse libraries of antibody sequences, which are then subjected to “deep” sequencing to acquire millions of antibody sequences.9,10 Because millions of B cells are assayed in a single reaction, deep sequencing provides a far more comprehensive representation of human antibody repertoires than human hybridoma screening. However, there are complications to most deep sequencing methods, including: 1) determining native heavy and light chain pairing and 2) linking such an enormous number of antibody sequences to function.
Pioneering work has described methods that retain pairing of heavy and light chain through 96-well plate sorting11-14 and with microfluidic devices.15 The utility of heavy and light chain pairing for functional analysis has also been demonstrated by combining 96-well plate sorting, yeast single chain variable fragment (scFv) display, and deep sequencing for discovery of mouse monoclonal antibodies (mAbs).16 However, we are not aware of any published reports of a genomic method that retains native single B cell pairing of heavy and light chain and enables high-throughput affinity screening of massively diverse human repertoires for rare antibodies. In this study, we use modern emulsion droplet microfluidics to capture paired heavy and light chain libraries from millions of single human B cells. The libraries are then expressed as yeast scFv libraries and screened for binders.
We used our new method to mine human repertoires to identify and characterize natural human mAbs reactive against influenza A virus and pneumococcus bacteria. These common infections are leading causes of morbidity and mortality in the United States, and provide an ideal system to test our methodology. In addition, deep sequencing of the humoral response to influenza A virus17-18 and pneumococcus19 have already been studied in detail, providing a useful reference for our methods. We isolated and functionally tested 10 human mAbs that bind influenza A, all of which neutralized viral infection. In addition, we identified, characterized, and functionally tested nine human mAbs that bind pneumococcal polysaccharides, five of which induced bacterial killing opsonization.
Results
Overview of the experimental approach
Prior work has mined human repertoires for mAbs present in the memory B cell (Bmem) compartment,1,2 or peripheral B cells from recently vaccinated donors.17,18 For our work, we used microfluidic methods to build a natively paired heavy and light chain library from peripheral B cells derived from a pool of 52 healthy non-vaccinated donors (Fig. 1). We also built natively paired heavy and light chain libraries from peripheral B cells derived from donors vaccinated for influenza A virus (Fluvirin; three donors) or pneumococcus (Pneumovax-23; three donors). Each non-vaccinated donor and vaccinated donor library was built from at least 1 million cells, with a maximum of 2.2 million cells (Supplementary Table S1).
Figure 1.

Overview of the workflow used to generate the scFv libraries from B cells isolated from human donors. Peripheral B cells are isolated from either vaccinated or non-vaccinated human donors. B cells are then encapsulated into droplets with oligo-dT beads and a lysis solution. The throughput of microfluidic cell encapsulation is ∼ 3 million cells per hour. mRNA-bound beads are purified from the droplets, and then injected into a second emulsion with an OE-RT-PCR amplification mix that generates DNA amplicons that encode scFv with native pairing of heavy and light chain Ig. Libraries of natively paired amplicons are then electroporated into yeast for scFv display. FACS is used to identify high affinity scFv. Finally, deep antibody sequencing is used to identify all clones in the pre- and post-sort scFv libraries. The complete workflow, from isolated B cells to sequenced scFv candidates, requires only three weeks.
Library generation was divided into three steps: 1) poly(A)+ mRNA capture, 2) multiplexed overlap extension reverse transcriptase polymerase chain reaction (OE-RT-PCR), and 3) nested PCR to remove artifacts and add adapters for deep sequencing or yeast display libraries. For poly(A)+ mRNA capture, we used a custom designed co-flow emulsion droplet microfluidic chip fabricated from glass. The microfluidic chip had two input channels for fluorocarbon oil, one input channel for the cell suspension mix, and one input channel for oligo-dT beads in cell lysis buffer. After cell lysis and capture of the mRNA onto the dT beads, mRNA-bound beads were extracted from the droplets. The beads were then re-suspended into OE-RT-PCR mix and injected into a second co-flow microfluidic chip with a mineral oil-based surfactant mix. The OE-RT-PCR mix contained a mixture of primers directed against the IgK C region, the IgG C region, and all V regions.11 The overlap region was a DNA sequence that encodes a Gly-Ser-rich scFv linker sequence.20 After isolation of the DNA fragments from droplets, nested PCR was performed to add adapters for Illumina sequencing or yeast display.
We queried the heavy and light chain V(D)J sequence content for each library using deep sequencing. The primary goal of deep sequencing was to estimate the size and diversity of each library, not to fully assess the “long tail” of rare sequences. The three libraries were transformed into yeast for scFv display. After expansion and induction, the scFv libraries were subjected to fluorescence-activated cell sorting (FACS) to identify antigen-reactive clones. The vaccinated donor libraries were analyzed for binders against their cognate antigens, which included two influenza A virus antigens (Fig. 2A) and a pool of 22 pneumococcal polysaccharide antigens (Fig. 3A). The 52-donor non-vaccinated library was analyzed for binders to the same antigens (Figs 2B, 3B). After two (flu) or three (pneumococcal) rounds of FACS, we purified plasmid from the recovered yeast and queried the heavy and light chain V(D)J sequence content for each library using deep sequencing.
Figure 2.

scFv libraries subjected to FACS for influenza A virus antigens. Staining for c-Myc (AF488) is used to differentiate yeast cells that express scFv from yeast cells that do not express scFv (x-axis). Staining for biotinylated antigen (PE) is used to identify yeast that express scFv that bind to the antigen (y-axis). The Fc negative control is used to set gates that are used to capture yeast cells that express scFv and bind antigen (upper right corner of the FACS plot). Gates for yeast selection are indicated by the quadrangle in the upper right corner of each FACS plot. The percentage in each quadrangle (red text) indicates the proportion of c-Myc-positive yeast that fell within the gate. A vertical dotted line (black) indicates the gate used to determine the number of yeast that express scFv (c-Myc+). (A) 1st and 2nd sort FACS data for H3N2 and H1N1, using an scFv library generated from B cells from donors vaccinated with the influenza A vaccine Fluvirin. (B) 1st and 2nd sort FACS data for H3N2 and H1N1, using an scFv library generated from B cells from non-vaccinated donors.
Figure 3.

scFv libraries subjected to FACS for pneumococcus polysaccharides. Staining for c-Myc (AF488) is used to differentiate yeast cells that express scFv from yeast cells that do not express scFv (x-axis). Staining for biotinylated antigen (PE) is used to identify yeast that express scFv that bind to the antigen (y-axis). The Fc negative control is used to set gates that are used to capture yeast cells that express scFv and bind antigen (upper right corner of the FACS plot). Gates for yeast selection are indicated by the quadrangle in the upper right corner of each FACS plot. The percentage in each quadrangle (red text) indicates the proportion of c-Myc positive yeast that fell within the gate. A vertical dotted line (black) indicates the gate used to determine the number of yeast that express scFv (c-Myc+). (A) 1st, 2nd, and 3rd sort FACS data for a pool of pneumococcus polysaccharides, using an scFv library generated from B cells from donors vaccinated with the pneumococcus vaccine Pneumovax-23. (B) 1st, 2nd, and 3rd sort FACS data for a pool of pneumococcus polysaccharides, using an scFv library generated from B cells from non-vaccinated donors.
We identified 247 anti-pathogen scFv sequences that were present at ≥0.1% frequency among populations of clones selected by FACS from vaccinated and non-vaccinated donor libraries (Supplementary Tables S2-S7). For each scFv, we sequenced the full-length V(D)J regions, including complementary-determining region 3 (CDR3), and computed various sequence analysis metrics (V and J gene usage, divergence from germline, R/S ratio, and IgG isotype). We then selected a subset of the highest frequency sequences and re-engineered them as full-length IgG1 mAbs (Tables 1–2). The mAbs were subjected to a panel of functional assays, including enzyme-linked immunosorbent assays (ELISA), bio-layer interferometry (BLI), serotype-specific dot blots, and viral neutralization or multiplex opsonophagocytic killing assays, which are further described below.
Table 1.
Characteristics of 10 purified antibodies for influenza A.
| Vaccinated donors? | Sorted antigen | Ab # | CDR3K + CDR3H | Pre-sort (%) | Post-sort (%) | Affinity to H3N2 (KD) | H3N2 kon (1/Ms) | H3N2 kdis (1/s) | Affinity to H1N1 (KD) | H1N1 kon (1/Ms) | H1N1 kdis (1/s) | H3N2 neutralization (μg/ml) | H1N1 neutralization (μg/ml) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yes | H3N2 | fVax-H3-1 | QQSHTIRWT+ARTGPSGTYSGFDS | 0.086 | 61 | 0.92nM | 390,000 | 0.00036 | Does not bind | — | — | 0.781 | Not tested |
| Yes | H3N2 | fVax-H3-2 | LQESTYPLT+AREEGYYSGSGSFLPESDS | 0.02 | 5.6 | 0.073nM | 220,000 | 0.000016 | Does not bind | — | — | 3.125 | Not tested |
| Yes | H3N2 | fVax-H3-3 | LQENSYPLT+AREEGYYDGSGTWHPESDY | 0.014 | 4 | 0.19nM | 280,000 | 0.000052 | Does not bind | — | — | 3.125 | Not tested |
| Yes | H1N1 | fVax-H1-1 | QQYVNSGLFI+AREIFKGEIDYYAMDV | 0.27 | 34 | Does not bind | — | — | 1.3nM | 250,000 | 0.00032 | Not tested | 0.049 |
| Yes | H1N1 | fVax-H1-2 | QQYYSYPPIT+AKESRDSSGFYYISPEYFHH | 0.023 | 21 | Does not bind | — | — | 8.8nM | 280,000 | 0.00025 | Not tested | 0.195 |
| Yes | H1N1 | fVax-H1-3 | QQYISFGGGT!+ASYYYGSGSPSGAFDI | 0.037 | 13 | Does not bind | — | — | 0.34nM | 190,000 | 0.000063 | Not tested | 0.049 |
| No | H3N2 | NonVax-H3-1 | QQSYSISYT+TTQDVIREFEKDAFDI | 0.01 | 67 | 1.3nM | 360,000 | 0.00047 | Does not bind | — | — | 0.012 | Not tested |
| No | H3N2 | NonVax-H3-2 | QNYNTAPPLT+ARTHCYDGSGYFGVSDF | 0.00018 | 4.4 | 5.8nM | 480,000 | 0.0028 | Does not bind | — | — | 0.012 | Not tested |
| No | H1N1 | NonVax-H1-1 | QQYENLPLT+AKANFPGIAAAGSVKGQITEYYFDF | 0 | 44 | Does not bind | — | — | 0.54nM | 440,000 | 0.00024 | Not tested | 50.0 |
| No | H1N1 | NonVax-H1-2 | MQTTHWPLT+ARGPYYSGSGSYSFGDS | 0 | 22 | Does not bind | — | — | 0.46nM | 510,000 | 0.00023 | Not tested | 50.0 |
Table 2.
Characteristics of nine purified antibodies for pneumococcus.
| Ab # | CDR3K + CDR3H | Pre-sort (%) | Post-sort (%) | ELISA binding (EC50) | Serotypes bound | Serotypes killed (opsonic index) | Killing assay notes |
|---|---|---|---|---|---|---|---|
| pVax-Pn-1 | MQATHWPRT+VKGVISLDY | 0.032 | 26 | 0.03 nM | 1 | 1 (OI = 20) | No activity on serotypes 5, 7F, 19A |
| pVax-Pn-2 | LQTYRTPYT+ARLHGSGLGRWLDP | 0.28 | 20 | 0.03 nM | 18C | 18C (OI = 1284) | No activity on serotypes 6A, 9V and 19F |
| pVax-Pn-3 | LQDYNHVGT+GSRRPYFDS | 0.19 | 13 | 5.3 nM | 2, 14 | 14 (OI = 971) | Serotype 2 test not available; no activity on serotypes 4, 6B and 23F |
| pVax-Pn-4 | QQSYSTPST+ARWFSRSSTGVSYMDV | 0.11 | 10 | 0.01 nM | 1 | None | No activity on serotypes 1, 5, 7F, 19A |
| pVax-Pn-5 | QQYNSYPWT+ARIVARKPDDY | 0.038 | 7.7 | 1.5 nM | 6B | 6B (OI = 297) | No activity on serotypes 4, 14 and 23F |
| pVax-Pn-13 | MQGTHWPAWT+VTWVESNTGWLDY | 0.019 | 0.36 | 0.7 nM | non-specific | 6B (OI = 147), 7F (OI = 239) | No activity on serotypes 1,4,5,14,19A,23F |
| NonVax-Pn-1 | QQFTAYPRT+SRFWTYGRV | 0.00018 | 31 | 0.001 nM | 2 | NA | Serotype 2 test not available |
| NonVax-Pn-2 | LQHNSYPLT+ARAAYSTSYTPFDY | 0.0063 | 31 | 0.7 nM | 15B | NA | Serotype 15B test not available |
| NonVax-Pn-3 | HQYGDSPLT+ARDHFCWESPCPLRGIFDF | 0.004 | 6.4 | 0.02 nM | non-specific | None | No activity on serotypes 1,4,5,6B,7F,14,19A,23F |
Characterization of antibody repertoires post vaccination
We define antibody “clones” as the consensus of closely related groups of sequences with ≤2 amino acid differences in their CDR3 sequences. The overall diversity of antibody clones in the pre-sort libraries (non-vaccinated donors, influenza A vaccinated donors, and pneumococcus vaccinated donors) were similar, ranging from 10,000-17,500 antibody clones (Supplementary Table S1). As expected, the frequency distributions of the antibodies were different (Supplementary Figure S1). The non-vaccinated library (built from 52 donors) had a single clone with >0.2% abundance, then a very long tail of low abundance antibodies. In contrast, both vaccinated libraries (built from 3 donors each) had many highly abundant antibody sequences, and the drop-off in abundance in the tail was more gradual. The low abundance tail was similar across libraries, where the 200th most abundant antibody from each library was 0.03%-0.06% of reads.
We determined the IgG isotype of these antibodies by sequencing into the constant domain region to capture isotype-specific sequence. The non-vaccinated donors were 46% IgG1, 30% IgG2, 22% IgG3, and 2% IgG4 (Supplementary Table S1). Upon flu vaccination, IgG1 abundance increased to 57%, whereas IgG2 abundance decreased to 21% (with little to no change in IgG3/4 abundance). Upon pneumococcal vaccination, IgG2 abundance increased to 81% and all other isotypes decreased, consistent with prior observations that IgG2 is the main isotype used for pneumococcal antibodies.21 All flu binding mAbs from vaccinated donors were IgG1, and consistently ∼90% of the flu binding mAbs from non-vaccinated donors were also IgG1. For pneumococcal binding mAbs, 92% from vaccinated donors were IgG2, and 79% from non-vaccinated donors were IgG2.
Assessment of Replacement (R) as opposed to Silent (S) nucleotide mutations is often used to identify antigen-driven affinity selection of antibody sequences.22 A higher R/S ratio implies somatic hypermutation. We tabulated the R and S values across the V(D)J region, exclusive of CDR3 and framework 4 (FR4), for a random sample of ∼ 1,000 antibodies each from the pre-sort libraries (Supplementary Table S8). The R/S ratio for IgH in the pneumococcal vaccinated library (3.26) was significantly higher than the non-vaccinated library (2.92; t-test, p<0.01), whereas the R/S ratio for IgK in the pneumococcal vaccinated library (2.57) was significantly lower than the non-vaccinated library (3.75; t-test, p<0.01). It is unknown why the IgH R/S ratio was higher in the pneumococcal vaccinated library while the IgK R/S ratio was lower. There was no significant difference between the R/S ratios of the influenza A vaccinated donors and the non-vaccinated donors.
Overall divergence from germline is another metric to identify antigen-driven antibody evolution. To this end, we compared the nucleotide percent identity (%ID) between the V region for each antibody and its putative germline sequence for a random sample of ∼1,000 antibodies each from the pre-sort non-vaccinated donor library, influenza A vaccinated library, and pneumococcal vaccinated library (Supplementary Table S8). The non-vaccinated donors had IgHV and IgKV %IDs of 92.5% and 95.0%, respectively. There were no significant differences between the %IDs of the pneumococcal-vaccinated IgHV and IgKV compared to the non-vaccinated library. However, influenza A vaccination generated a repertoire with significantly higher IgHV %ID (92.9%) than the non-vaccinated library (92.5%; t-test, p<0.01), but the IgKV %ID was not significantly different between vaccinated and non-vaccinated libraries.
Isolation of pathogen antigen binders by yeast display
Yeast scFv libraries were stained for scFv expression (using a C-terminal c-Myc tag) and antigen binding. In a typical FACS dot plot, the upper right corner contains double-positive yeast that stain for both antigen binding and scFv expression. The lower left corner contains yeast that do not stain for either the antigen or scFv expression. The upper left corner contains yeast that stain for antigen but do not stain for scFv expression, presumably due to non-specific binding of antigen to yeast cell surfaces. For unknown reasons, such non-specific binding events were particularly evident in the pneumococcal antigen experiments. The lower right corner contains yeast that express the scFv but do not bind the antigen. We estimate the frequency of binders in each repertoire by dividing the count of yeast that double stain by the count of yeast that express an scFv.
The yeast scFv library from influenza A vaccinated donor peripheral B cells was typically 0.3-0.4% double positive, whereas the non-vaccinated donor scFv library was typically <0.1% double positive for two different influenza A antigens (Fig. 2). The H3N2 antigen (from the A/Sydney/5/1997 strain) and the H1N1 antigen (from the A/California/07/2009 strain) yielded similar frequencies of binders. In contrast, pneumococcal vaccination did not increase the frequency of binders in peripheral B cell repertoires, with vaccinated donors showing 0.1% and non-vaccinated donors showing 0.16% double-positive yeast (Fig. 3). Negative (Fc) controls always yielded <0.04% double-positive yeast, consistent with low non-specific staining. To increase antigen specificity, we expanded the FACS-sorted yeast from each scFv display library, and then performed a second round of FACS. The frequency of binders in the second sort was as low as 4% (for pneumococcal antigen with vaccinated donors) or as high as 76% (for H3N2 with vaccinated donors). For pneumococcal antigen, because the frequency of binders in the second sort was still relatively low, we performed a third sort that increased the frequency of binders to ∼70%.
We used antibody repertoire sequencing to assess the clonal diversity of each population of binders. As might be expected, the enrichment from the original repertoire to the population of binders was more pronounced in the non-vaccinated samples than the vaccinated samples. For example, pneumococcus binding antibody NonVax-Pn-1 was detected in the initial non-vaccinated repertoire at 0.00018%, and was present at 31% frequency after 3 rounds of FACS (Supplementary Table S7). Similarly, H3N2 binder NonVax-H3-1 was present in the un-vaccinated repertoire at 0.01%, and was enriched to 67% frequency after 2 rounds of FACS (Supplementary Table S5). Some binders were highly enriched in the post-sort data, but were apparently so rare in the original repertoire that they were not detected at all. For example, H1N1 binder NonVax-H1-1 was present in the post-sort data at 44% frequency, but was not detected in the original 52-donor non-vaccinated, pre-sort library (Supplementary Table S4).
We did not observe oligoclonality in the vaccinated or non-vaccinated libraries before sorting. However, we observed that FACS selection resulted in a significant increase in oligoclonality. For example, in each post-sort library, the top four scFv sequences accounted for >60% of the scFv sequences in the deep sequencing data (versus 4.7% on average for the input repertoires). Despite this oligoclonality, the post-sort libraries maintained substantial diversity; the library with the highest number of scFv sequences with frequencies of 0.1% or greater was the H1N1 non-vaccinated sample, with 59 scFv sequences. The library with the fewest scFv sequences at frequencies of 0.1% or greater was the H1N1 vaccinated sample, with 31 scFv sequences.
Some of the FACS plots, for example the pneumococcal antigen 2nd and 3rd sorts, showed at least two distinct populations of antigen binders. To investigate this phenomenon, we sorted the two distinct populations of antigen binders for both the vaccinated and non-vaccinated donor libraries (Supplementary Figure S2). Sequence analysis of each population revealed non-overlapping, oligoclonal sets of scFv sequences. We postulate that these non-overlapping sets of scFv sequences bind to different kinds of pneumococcal epitopes, resulting in distinct populations on the FACS plots. For example, certain scFv could bind to multiple epitopes on a given pneumococcal polysaccharide, which would increase the antigen-positive signal on the FACS plot. Another possibility is that some polysaccharide serotypes are labeled with more biotin molecules than other polysaccharide serotypes, leading to shifts in signal along the y-axis of the FACS plots.
Sequence characteristics of pathogen antigen binders
Affinity maturation and selection in vivo generates clonal lineages of similar antibody sequences. To investigate clonal lineages among FACS-sorted scFv binders, we clustered cognate-paired V(D)J sequences with at most 9 amino acid differences (Fig. 4). We define a “large clonal cluster” as clusters of three or more cognate-paired V(D)J sequences. We identified two large clonal clusters of H3N2 scFv binders and four large clonal clusters of H1N1 binders. Only one of the six anti-influenza four large clonal clusters was a mixture of scFv from both the vaccinated library and the non-vaccinated library. Seven large clonal clusters were identified among pneumococcal antigen scFv binders. None of these clusters was a mixture of scFv from both the vaccinated library and the non-vaccinated library. The clustering analysis suggests that we identified anti-influenza A scFv from at least 44 clonal clusters and anti-pneumococcus scFv from at least 32 clonal clusters.
Figure 4.
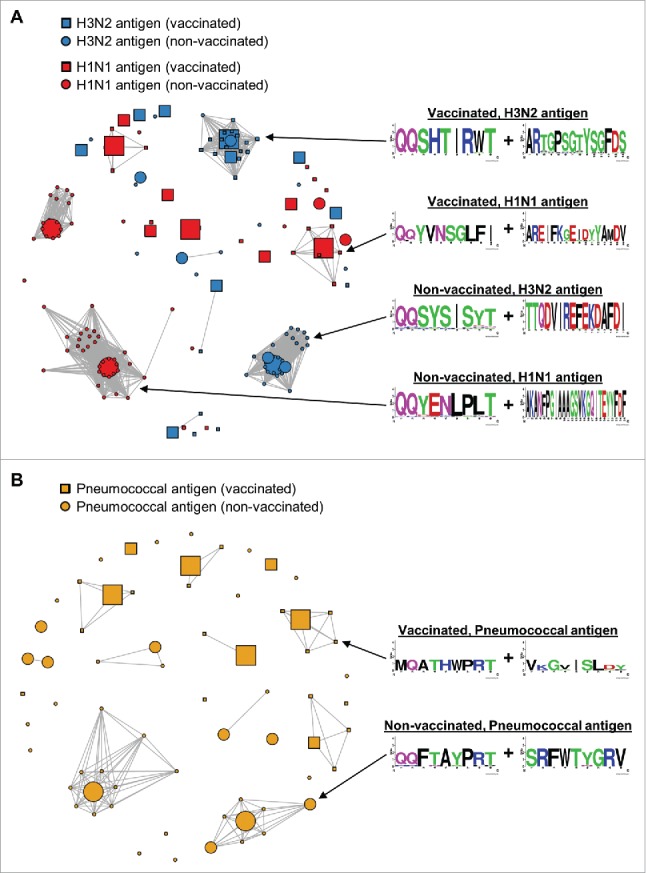
Clonal cluster analysis for FACS-sorted scFv binders. We computed the total number of amino acid differences between each pairwise alignment of FACS-sorted scFv. Edges were drawn only for pairwise alignments with ≤9 amino acid differences. The node for each scFv sequence was sized based on frequency in the FACS-sorted population: small (<1% frequency), medium (1-10% frequency), and large (>10% frequency). Web logos of the CDR3K + CDR3H amino acid sequences of the clusters used in Figure 5 are on the right. (A) Clonal clusters for anti-H3N2 scFv (blue) and anti-H1N1 scFv (red). scFv isolated from vaccinated donors are indicated with squares, and scFv isolated from non-vaccinated donors are indicated with circles. Note that two of the 19 sequences in the Vaccinated, H3N2 antigen logo were from non-vaccinated donors. (B) Clonal clusters for anti-pneumococcal antigen scFv. scFv isolated from vaccinated donors are indicated with squares, and scFv isolated from non-vaccinated donors are indicated with circles.
Understanding the amino acid residues that are conserved and variable during affinity maturation may help guide subsequent directed in vitro affinity maturation. Therefore, we aligned the full-length amino acid sequences for several clonal clusters (Fig. 4, 5). We found that most of the sequence variation within a clonal cluster was localized to the CDR3 regions, with minimal variation in CDR1, CDR2, and framework regions (54 variations in CDR3 regions of 90 total variations). We found that clonal cluster members that were enriched from the vaccinated libraries showed variation in the CDR3H with minimal variation in the light chain CDR3 (Fig. 5A), whereas the opposite was true for family members enriched from the non-vaccinated libraries, with most of the variation in the light chain CDR3 (Fig. 5B). We note that if the sequence variation within the clonal clusters were due to sequencing error alone, it is unlikely that most substitutions would occur within the CDR3.
Figure 5.

Amino acid sequence logos for groups of evolutionarily related clones. Though 1–4 such groups were present after the 2nd (influenza A) or 3rd (pneumococcus) FACS sort, we displayed a single representative dominant clonal expansion from each. Full V(D)J amino acid sequences are shown. Variant amino acids are emphasized with red arrows (the first seven amino acids are omitted due to the high probability of mis-priming during OE-RT-PCR). (A) Clonal groups for vaccinated donors: H3N2 antigen (top), H1N1 antigen (middle), and pneumococcal polysaccharides (bottom). Note that two of the 19 sequences in the H3N2 logo were from non-vaccinated donors. (B) Clonal groups for non-vaccinated donors: H3N2 antigen (top), H1N1 antigen (middle), and pneumococcal polysaccharides (bottom).
As expected,21 most of the pneumococcus binders (83.5%) are IgG2, whereas most of the H1N1 and H3N2 binders are IgG1. Only two IgG3 scFv were present at 0.1% or greater frequency across all the FACS-sorted binders. One of the IgG3 scFv was found in the population sorted with H1N1 (NonVax-H1-14), and the other in the population sorted with H3N2 (NonVax-H3-12). Both IgG3 antibodies were captured from the non-vaccinated donor library. The scFv are identical in amino acid sequence across the heavy chain VDJ. However, the light chain VJ sequences are only 80% identical; they share J-region IgKJ5*01 but differ in their IgKV sequences (Supplementary Tables S4-S5). The shared heavy chain VDJ shows signs of affinity maturation (R/S ratio: 5.25), which is consistent with class-switched IgG3 antibodies. We did not observe any IgG4 scFv among the FACS-sorted binders.
We calculated the average IgH and IgK R/S ratios across FACS-sorted scFv binders (Supplementary Table S9). For pneumococcal antigen, R/S ratios for scFv binders from the vaccinated library were significantly higher (t-test, p<0.01) than for scFv binders from the corresponding non-vaccinated libraries (3.61 vs. 2.35 for IgH and 2.57 vs. 1.46 for IgK). This suggests that Pneumovax increases the frequency of the most highly affinity-matured pneumococcal antibodies in human repertoires. Similarly, the average IgK R/S ratios for H3N2 and H1N1 scFv binders were also significantly higher (t-test, p<0.01) from the influenza A vaccinated libraries (2.35 vs. 1.86 for H3N2 and 2.01 vs. 1.70 for H1N1). In contrast, the average IgH R/S ratios for H3N2 and H1N1 scFv binders from the influenza A vaccinated libraries were either no different or significantly lower, respectively, compared with the non-vaccinated libraries (2.50 vs. 2.86 for H3N2 and 3.04 vs. 5.41 for H1N1). This suggests that Fluvirin may not increase the frequency of the most highly affinity-matured anti-influenza A antibodies in human repertoires, especially in the heavy chain.
The average nucleotide %ID to germline V gene across all FACS-sorted clones was 94.2% for IgK and 94.6% for IgG. This level of divergence corresponds to an average of 6–7 amino acid substitutions per V gene. The most divergent IgK was NonVax-Pn-25 (88.1% ID), and the most divergent IgG was NonVax-H1-4 (85.0% ID). We compared the %ID values from the FACS-sorted clones from the vaccinated and non-vaccinated libraries and found that most of the populations were not significantly different (Supplementary Table S9). Only the set of IgH scFv FACS-sorted with H1N1 from the Fluvirin vaccinated library had a significantly lower V-gene %ID than the corresponding set of scFv isolated from the non-vaccinated library (92.4% vs. 96.9% ID, respectively; t-test, p<0.01).
Functional validation of select mAbs
We selected several of the highest frequency mAbs from each FACS-sorted library to make as full-length antibodies with an IgG1 constant region (Tables 1–2). The full-length antibodies were produced in Chinese hamster ovary cells and purified by Protein A chromatography. The binding specificity and affinity of each flu-targeted antibody towards each of the two antigens was determined using BLI. Antibodies sorted with either H1N1 or H3N2 only bound to their respective antigen, indicating high specificity towards the sorted antigen (Table 1, Supplementary Figures S3-S4). Each antibody bound with high affinity, with KD ranging from 73pM–8.8nM.
Influenza virus neutralization assays were performed to determine whether these anti-flu antigen antibodies can neutralize the infectivity of specific influenza strains. For this, influenza viral strains A/Sydney/5/1997 (for H3N2 binders) or A/California/07/2009 (for H1N1 binders) were incubated with the appropriate antibodies at multiple dilutions. The virus/antibody mixtures were then added to Madin-Darby canine kidney (MDCK) cells and cytopathic effects were measured. If the antibody is not capable of neutralizing, the influenza virus will lead to visible cytopathic effects; if the antibody is neutralizing, then the MDCK cells will survive. Each of the 10 mAbs that we tested neutralized the matched influenza A strain, with specific activities ranging from 0.012 to 50 μg/ml (Supplementary Figure S5, Table 1).
Pneumococcal antigen-binding antibodies were first validated for their ability to bind a pool of pneumococcal polysaccharides by ELISA. We found that all purified antibodies bound to the pneumococcal antigen pool with EC50s ranging from 0.001 nM to 5.3 nM (Supplementary Figure S6; Table 2). We then performed serotype-specific dot blots to determine the specificity of each mAb. Most of the mAbs bound to one or two serotypes, whereas two antibodies (one from vaccinated donors and one from non-vaccinated donors) bound non-specifically to most of the serotypes (Supplementary Figure S7; Table 2).
To determine if the pneumococcus-binding antibodies are functional, multiplex opsonophagocytic killing assays were performed. These assays measure antibody-mediated bacterial killing opsonization, which is the ability of the antibody to mark serotype-specific bacteria strains for ingestion/phagocytosis and subsequent killing. Most of the serotype-specific binders induced bacterial killing against the appropriate serotype (Table 2). Additional serotypes were tested to determine specificity, and we generally found that the mAbs were serotype-specific in their killing activity. One exception was antibody pVax-Pn-4, which bound to serotype 1 and had strong ELISA binding, but did not induce killing of serotype 1 bacteria. Across all mAbs that we tested, there was no association between the antibody dilution required for ELISA binding and the antibody dilution required for bacterial killing. We also tested the mAbs that non-specifically bound to many serotypes against several serotype strains. We found that the non-specific binder from vaccinated donors (pVax-Pn-13) could cause specific killing against two of the eight serotypes tested, whereas the other non-specific binder (NonVax-Pn-3) did not show any activity against any of the tested serotypes.
Discussion
In this study, we identified 247 anti-pathogen scFv binders that were initially as rare as 1 in 100,000 in human repertoires. These scFv sequences represented at least 76 distinct clonal clusters. To our knowledge, this is among the largest existing datasets of antigen-specific, naturally occurring human antibody sequences. This is also the first known success at generating natural cognate-paired scFv libraries from >1,000,000 single cells, and then screening the scFv libraries for antigen binders. For example, prior work with 96-well plates generated natural cognate-paired scFv libraries from thousands of single cells,11 and prior work with microfluidics did not screen scFv libraries for antigen binders.16 We validated several of the scFv as full-length antibodies. All 10 full-length anti-influenza A antibodies tested specifically bound the appropriate antigen at KD<10 nM and neutralized the appropriate viral strain in cellular assays. Affinities and neutralization potencies were similar to prior work on influenza A.12,14 All nine anti-pneumococcus full-length antibodies tested bound at least one polysaccharide serotype, and 71% of the anti-pneumococcus antibodies that we tested were functional in a cell killing assay. With further development, these selected mAbs could be candidates for passive immunization against their respective pathogens/serotypes. Our method is primarily limited by microfluidic and FACS throughput, whereas other methods are limited by efficiency of B cell transformation or expansion. For example, B cell transformation is typically <1%.23 Therefore, our methodology complements more established methods.
Previous studies have also successfully identified antigen-specific sequences among diverse antibody repertoires.9 One study used bioinformatics to search a diverse repertoire for antibodies similar in sequence to a known human immunodeficiency virus binder.24 In another study, the researchers vaccinated donors with seasonal influenza vaccine, paired heavy and light chain Ig through 96-well plate cloning and sequencing, and then identified binders by focusing specifically on clonal expansions.14 Another interesting method is to identify convergent antibodies, for example, antibodies shared by identical twins after a recent vaccination.25 In contrast, our method does not require convergence, a priori knowledge of antigen-specific sequences, or clonal expansions to identify reactive antibodies. This has enabled us to discover rare binders in highly diverse, non-vaccinated repertoires, and in vaccinated samples without the need for identical twins or pre-vaccination repertoires. For expedience and because functional mAb discovery was the primary goal of this study, we built libraries from pools of donors rather than individual donors. To answer questions regarding differences and similarities between donor immune repertoires, future work could build and study libraries from individual donors.
Our study also contributes to a growing body of work on the sequence characteristics of anti-pathogen immune repertoires. We found an average of 6–7 amino acid substitutions per V gene in the anti-pathogen scFvs. Prior work on influenza A14 and pneumococcus26-30 found similar rates of divergence from germline. In our work, each collection of FACS-sorted binders contained 1–4 clusters of evolutionarily related clones, i.e., putative clonal lineages. Prior work used very different experimental approaches, so it would be difficult to directly compare the nature of our clonal lineages with prior data. For example, other studies separated donors into age groups17 or looked exclusively at plasmablasts.14 Though the humoral response to pneumococcus is primarily IgG2,27 prior work has shown that intravenous immunoglobulin (IVIg) contains a significant component of anti-pneumococcus IgG1.31 Accordingly, we found that 16.4% of pneumococcus scFv binders were IgG1. One limitation to our current protocol is that we are unable to trace individual antibody sequences back to single cells. Therefore, the frequency of antibody sequences in our libraries is a function of: 1) the number of cells expressing the antibody and 2) the mRNA expression levels of those cells. To trace back antibody sequences to single cells, future work could consider adding nucleic acid barcodes to the scFv amplicons.32,33 Such a method would enable more precise measurements of clones and clonal lineages.
Similar to other groups studying natural repertoire pneumococcus antibodies,30 we identified clones that are pan-serotype binders. It is possible that these antibodies are reactive against C-polysaccharide, a common cell-wall-associated antigen present across several species of pathogenic bacteria.34 Most people have high titers of non-protective antibodies against this antigen. However, one of the non-specific antibodies (pVax-Pn-13) could induce specific killing of two of the eight serotypes tested. We speculate that pVax-Pn-13 binds C-polysaccharide as well as serotype-specific polysaccharides. These observations raise the possibility that human repertoires contain pan-serotype binders that may also provide broad protection against multiple serotypes. Such binders would be excellent therapeutic mAb candidates. Future studies might also use our technology for more precise epitope mapping of pneumococcus antibodies.35
Broadly reactive anti-influenza A antibodies36-38 are excellent therapeutic candidates because influenza A antigenic drift complicates development of passive immunotherapies. However, our FACS strategy selected for scFv that bound a single influenza A strain. Accordingly, all 10 anti-influenza A full-length antibodies that we tested bound and neutralized a single influenza A strain. In future work, we envision a modified FACS strategy that selects scFv that bind multiple influenza A strains. For example, H1N1 and H3N2 could be labeled with different fluorophores, and scFv could be selected that bind to both strains, rather than a single strain. This approach could be useful in analogous contexts, for example, selecting scFv that bind both human and simian antigens, or scFv that bind an antigen, but not a closely related paralogue with high homology.
Antibody repertoire diversity is vast, and alternative experimental designs may allow for capture of higher affinity candidates. For example, we did not separately analyze different types of antibody-producing cells, such as plasma cells, plasmablasts, and Bmems. We found that scFv isolated from non-vaccinated donors did have different sequence characteristics from scFv isolated from vaccinated donors. For example, light chains were highly conserved within clonal expansions from vaccinated donors, but not from non-vaccinated donors. We may have seen such differences because scFv isolated from non-vaccinated donors were from Bmems, whereas scFv isolated from vaccinated donors were from activated and affinity-maturing cells.12,18,39 We further speculate that bone marrow plasma cells would be a rich source of high-affinity therapeutic mAb candidates. Future work should also optimize the appropriate timing of blood draws post-vaccination to capture high-affinity antibodies, as studied elsewhere.40 However, there may be natural limits to the affinity of antibodies derived from human repertoires,31 so in vitro affinity maturation may often be required as part of subsequent development of repertoire-derived therapeutic candidates.
Our approach could be modified to leverage a variety of display and screening methods, for example, panning with phage scFv display,41,42 FACS with yeast antigen-binding fragment (Fab) display,43,44 or FACS with mammalian full-length antibody display.45,46 Our methods would most easily be adapted to phage scFv display. We chose yeast scFv because we had existing experience with yeast protocols and FACS. Though the ideal yeast display format remains controversial, prior work suggests scFv can bind with lower affinity than Fabs or full length mAbs.47-49 We chose scFv because our OE-RT-PCR protocol is optimized for amplifying amplicons <2,000 nucleotides. Adapting our protocol to Fab or full-length mAb display would require amplification of a larger segment of the IgK and IgG C regions. Finally, exploratory work has shown that engineering millions-diverse full-length mammalian libraries from OE-RT-PCR scFv amplicons is possible.50 In the future, full-length mammalian libraries could be used for massively parallel functional screens, for example, to directly discover rare neutralizing antibodies from diverse repertoires.
The entire scFv binder discovery process, non-inclusive of vaccinations and peripheral blood sample collection, was accomplished in three weeks by one technician. Though estimates of antibody diversity vary widely,18 our method analyzes three million cells per hour, allowing us to sample a large fraction of immune repertoires. Full-length mAb expression used conventional technology and required less than three weeks of additional effort. With automation, it would be straightforward to express >1,000 full-length mAbs per month. The speed and convenience of our process would be especially beneficial to programs looking to mine natural repertoires for therapeutic antibodies against emerging pathogens, e.g., Zika or Ebola viruses. We expect that the methodology will also be useful for a variety of studies, such as characterization of pathogenic autoimmune antibodies and their targets, examination of age-related differences in vaccine response,17,19 or the identification of novel immunogens for active vaccines.9
Materials and methods
Sourcing and processing human materials
To generate the 52-donor non-vaccinated library (ages 21–53), we contracted AllCells, a contract research organization (CRO), to collect whole blood from generally healthy human donors into 6ml lavender-capped tubes (EDTA anticoagulant). Tubes were shipped to GigaGen at room temperature. Peripheral blood mononuclear cells (PBMCs) were isolated from the blood using SepMate™-50 tubes with Lymphoprep™ density gradient medium, and then B cells were isolated from the PBMCs using the EasySep™ Human Pan-B Cell Enrichment Kit (Stemcell Technologies).
For influenza A vaccinated libraries, we contracted AllCells to vaccinate three donors (ages 26–34) with Fluvirin (Novartis) and perform leukapheresis 10 days later to obtain PBMCs. In parallel, plasma was isolated from a separate blood draw on the day of leukapheresis; ELISA against Influenza A (IBL America) on the plasma samples confirmed a response to the vaccine as compared to plasma from the same donors prior to vaccination. For the pneumococcus vaccinated libraries, we contracted AllCells to vaccinate five donors (ages 35–57) with Pneumovax-23 (Merck) and perform leukapheresis eight days later. In parallel, plasma was isolated from a separate blood draw on the day of leukapheresis; ELISA against a pool of pneumococcal vaccine polysaccharides (Alpha Diagnostics) on the plasma samples confirmed a response to the vaccine for three of the five donors as compared to plasma from a pool of healthy donors. The three pneumococcus-responding donors were used going forward. B cells from the PBMCs were isolated using the EasySep™ Human Pan-B Cell Enrichment Kit (Stemcell Technologies).
Sample collection protocols were approved by Institutional Review Board (IRB) protocol #7000-SOP-045 (Alpha IRB, San Clemente, CA), to AllCells. Informed consent was obtained from all participants and de-identified samples were shipped to GigaGen. After isolation, B cells were cryopreserved using CryoStor® CS10 (Stemcell Technologies). Immediately prior to generating paired heavy and light chain libraries, cells were thawed, washed in cold DPBS+0.5% bovine serum albumin, assessed for viability with Trypan blue on a Countess™ cell counter (Thermo Fisher Scientific), and then re-suspended in 12% OptiPrep™ Density Gradient Medium (Sigma) at 5,000-6,000 cells per μl. This cell mixture was used for microfluidic encapsulation as described in the next section.
Generating paired heavy and light chain libraries
For poly(A)+ mRNA capture, we used a custom-designed co-flow emulsion droplet microfluidic chip fabricated from glass (Supplementary Figure S8; Dolomite). The microfluidic chip had two input channels for fluorocarbon oil (Dolomite), one input channel for the cell suspension mix, and one input channel for oligo-dT beads (NEB) at 1.25 mg/ml in cell lysis buffer (20 mM Tris pH 7.5, 0.5 M NaCl, 1 mM EDTA, 0.5% Tween-20, and 20 mM DTT). The input channels were etched to 50 μm × 150 μm for most of the chip's length, narrowed to 55 μm at the droplet junction, and were coated with hydrophobic Pico-Glide (Dolomite). Three Mitos P-Pump pressure pumps (Dolomite) were used to pump the liquids through the chip. Droplet size depends on pressure, but typically we found that droplets of ∼45 μm diameter were optimally stable. Emulsions were collected into chilled 2ml microcentrifuge tubes and incubated at 40 °C for 30 minutes for mRNA capture. The beads were extracted from the droplets using Pico-Break (Dolomite).
For multiplex OE-RT-PCR, we used glass Telos droplet emulsion microfluidic chips (Dolomite). mRNA-bound beads were re-suspended into OE-RT-PCR mix and injected into the microfluidic chips with a proprietary mineral oil-based surfactant mix (available commercially from GigaGen) at pressures that generated 27 μm droplets. The OE-RT-PCR mix contained 2 × one-step RT-PCR buffer, 2.0 mM MgSO4, SuperScript III reverse transcriptase, and Platinum Taq (Thermo Fisher Scientific), plus a mixture of primers directed against the IgK C region, the IgG C region, and all V regions11 (Supplementary Figure S9). The overlap region was a DNA sequence that encodes a Gly-Ser-rich scFv linker sequence.20 The DNA fragments are recovered from the droplets using a proprietary droplet breaking solution (available commercially from GigaGen) and then purified using QIAquick PCR Purification Kit (Qiagen).
For nested PCR, the OE-RT-PCR product was first run on a 1.7% agarose gel for 80 minutes at 150V. A band at 1200–1500 base pair (bp) corresponding to the linked product was excised and purified using NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel). PCR was then performed to add adapters for Illumina sequencing or yeast display; for sequencing a randomer of seven nucleotides was added to increase base calling accuracy in subsequent next generation sequencing steps. Nested PCR was performed with 2 × NEBNext High-Fidelity amplification mix (NEB) with either Illumina adapter containing primers or primers for cloning into the yeast expression vector. The nested PCR product was run on a 1.2% agarose gel for 50 minutes at 150V. A band at 800–1100 bp was excised and purified using NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel).
Preparation of antigens for FACS
Human IgG1-Fc protein (Thermo Fisher Scientific), Influenza A H3N2-His protein (A/Sydney/5/1997; Sino Biological) and Influenza A H1N1-His protein (A/California/07/2009; Sino Biological) were biotinylated using the EZ-Link Micro Sulfo-NHS-LC-Biotinylation kit (Thermo Fisher Scientific). The biotinylation reagent was resuspended to 9 mM and added to the protein at a 50-fold molar excess. The reaction was incubated on ice for 2 hours, then the biotinylation reagent was removed using Zeba desalting columns (Thermo Fisher Scientific). The final protein concentration was calculated with a Bradford assay.
Twenty-two of the 23 Pneumovax-23 polysaccharides were purchased from ATCC (serotype 8 was not available at the time of the study). For biotinylation, sugar solutions were prepared at 1 mg/ml total sugar concentration in 0.1 M sodium acetate, pH 5.5. A 20 mM solution of cold sodium meta-periodate was prepared in 0.1 M sodium acetate, pH 5.5 and stored on ice in an amber tube. The two solutions were mixed at equal volumes and incubated on ice for 30 minutes. The excess meta-periodate was removed by buffer exchange using a Zeba desalting column (Thermo Fisher Scientific) according to the manufacturer's instructions. The oxidized and desalted sugar solution was mixed with Hydrazide-LC-biotin (Thermo Fisher Scientific) at 18.55 mg/ml in dimethyl sulfoxide, at a 9:1 ratio (sugar:hydrazide-LC-biotin) to give a final concentration of 5 mM hydrazide-LC-biotin. Ten μl of 5 mM AminoLink Reductant solution (Thermo Fisher Scientific) was added per milliliter of protein conjugation mixture (50 mM AminoLink Reductant final). The reaction was incubated overnight at 4 °C. The reaction was quenched by the addition of 50 μl 1M Tris, pH 7.5 per milliliter of conjugation solution and incubation at room temperature for 30 minutes. Excess reagents were removed and the solution was buffer exchanged into phosphate-buffered saline (PBS) using a Zeba desalting column (Thermo Fisher Scientific) according to the manufacturer's instructions. The final solution was not quantified, and instead a specific volume was used for FACS screening.
Yeast library screening
We built a yeast surface display vector (pYD) that contains a GAL1/10 promoter, an Aga2 cell wall tether, and a C-terminal c-Myc tag (Supplementary Figure S10). The GAL1/10 promoter induces expression of the scFv protein in medium that contains galactose. The Aga2 cell wall tether is required to shuttle the scFv to the yeast cell surface and tether the scFv to the extracellular space. The c-Myc tag is used during the flow sort to stain for yeast cells that express in-frame scFv protein. Saccharomyces cerevisiae cells (ATCC) were electroporated (Bio-Rad Gene Pulser II; 0.54 kV, 25 uF, resistance set to infinity) with gel-purified nested PCR product and linearized pYD vector for homologous recombination in vivo. Transformed cells were expanded and induced with galactose, to generate yeast scFv display libraries.
To confirm scFv surface expression, ∼ 2 × 106 cells from the expanded scFv libraries were stained with anti-c-Myc (Thermo Fisher Scientific A21281) and an AF488-conjugated secondary antibody (Thermo Fisher Scientific A11039). To select scFv-expressing cells that bind to antigen, yeast cells were stained with biotinylated antigen (7 nM final concentration for protein antigens or 50 μl for polysaccharide antigens) and then stained with phycoerythrin (PE)-streptavidin (Thermo Fisher Scientific). Cells were then flow sorted on a BD Influx (Stanford Shared FACS Facility) for double-positive cells (AF488+/PE+). Recovered clones were then plated on SD-CAA plates with kanamycin, streptomycin, and penicillin (Teknova) and expanded. The expanded first round FACS clones were then subjected to a second (and sometimes third) round of FACS with the same antigen at the same molarity (7 nM final concentration for protein antigen) or volume (50 μl for polysaccharide antigens). Plasmid minipreps (Zymo Research) were prepared from yeast recovered from the final FACS sort. Tailed-end PCR was used to add Illumina adapters to the plasmid libraries for deep sequencing (Supplementary Figure S9).
Deep antibody repertoire sequencing
Deep antibody sequencing libraries were quantified using a quantitative PCR Illumina Library Quantification Kit (KAPA) and diluted to 17.5 pM. Libraries were sequenced on a MiSeq (Illumina) using a 500 cycle MiSeq Reagent Kit v2, according to the manufacturer's instructions. To obtain high-quality sequence reads with maintained heavy and light chain linkage, we performed sequencing in two separate runs. In the first run (“linked run”), we directly sequenced the scFv libraries to obtain forward read of 340 cycles for the light chain V-gene and CDR3, and reverse read of 162 cycles that covered the heavy chain CDR3 and part of the heavy chain V-gene (Supplementary Figure S11). In the second run (“unlinked run”), we first used the scFv library as a template for PCR to separately amplify heavy and light chain V-genes. Then, we obtained forward reads of 340 cycles and reverse reads of 162 cycles for the heavy and light chain Ig separately. This produced forward and reverse reads that overlap at the CDR3 and part of the V-gene, which increased confidence in nucleotide calls.
To remove base call errors, we used a previously published expected error filtering method.51 The expected number of errors (E) for a read was calculated from its Phred scores. By default, reads with E>1 were discarded, leaving reads for which the most probable number of base call errors is zero. As an additional quality filter, singleton nucleotide reads were discarded, because sequences found two or more times have a high probability of being correct.52 Finally, we generated high-quality, linked antibody sequences by merging filtered sequences from the linked and unlinked runs. Briefly, we wrote a series of scripts in Python that first merges forward and reverse reads from the unlinked run. We discarded any pairs of forward and reverse sequences that contain mismatches. Next, we used the nucleotide sequences from the linked run to query merged sequences in the unlinked run. The final output from the scripts was a series of full-length, high-quality V(D)J sequences, with native heavy and light chain Ig pairing.
To identify reading frame and FR/CDR junctions, we first processed a database of well-curated immunoglobulin sequences53 to generate position-specific sequence matrices (PSSMs) for each FR/CDR junction (Supplementary Figure S12). We used these PSSMs to identify FR/CDR junctions for each of the merged nucleotide sequences generated using the processes described above. This identified the protein reading frame for each of the nucleotide sequences. CDR sequences that have a low identity score to the PSSMs are indicated by an exclamation point. Python scripts were then used to translate the sequences. We required reads to have a valid predicted CDR3 sequence, so, for example, reads with a frame-shift between the V and J segments were discarded. Next, we ran UBLAST54 using the scFv nucleotide sequences as queries, and V and J gene sequences from the IMGT database53 as the reference sequences. The UBLAST alignment with the lowest E-value was used to assign V and J gene families and compute %ID to germline. R/S values were computed using V gene segments spanning the first amino acid through the FR3-CDR3 boundary. CDR3 and FR4 were not used in R/S value computation.
To estimate the diversity of the libraries, we defined “clones” conservatively (Supplementary Table S1). First, we concatenated the CDR3K and CDR3H amino acid sequences from each scFv sequence into a single contiguous amino acid sequence. Next, we used UBLAST54 to compute the total number of amino acid differences in all pairwise alignments between each concatenated sequence in each data set. Groups of sequences with ≤2 amino acid differences in the concatenated CDR3s were counted as a single clone. Finally, we used the majority amino acid identity at each residue position to generate the consensus amino acid sequence of the clone from the sequences of the members of the group.
To generate clonal cluster plots (Fig. 4), we first used UBLAST54 to compute the total number of amino acid differences between each pairwise alignment of FACS-sorted scFv sequences. Binders to influenza A antigens (H3N2 and H1N1) were combined into a single analysis. Binders to pneumococcal antigen were treated as an independent data set. Next, we used the igraph R package55 to generate clustering plots for the pairwise alignments. Antibody clones are represented by “nodes” in the plots. Each node was sized based on the frequency of the antibody clone in the FACS-sorted population: small (<1% frequency), medium (1-10% frequency), and large (>10% frequency). We define “edges” as the links between nodes. The “layout_with_kk” layout option was used to format the output. Edges were drawn only for pairwise alignments with ≤9 amino acid differences. Edge lengths are proportional to the number of amino acid differences between linked nodes, i.e., longer edges indicate more amino acid differences.
Monoclonal antibody expression and purification
The mAbs were expressed from a variant of the pCDNA5/FRT mammalian expression vector (Thermo Fisher Scientific). The vector used an EF1-alpha promoter (from pEF1a-IRES, Clontech) for the light chain, followed by a BGH polyA sequence from pCDNA5/FRT. The vector used the cytomegalovirus promoter from pCDNA5/FRT for expression of the heavy chain, followed by a BGH polyA sequence (Supplementary Figure S13). Antibody expression constructs were built using GeneBlocks (Integrated DNA Technologies) and NEBuilder® HiFi DNA Assembly Master Mix (New England BioLabs). All constructs were synthesized as human IgG1 isotype, regardless of a given antibody's IgG isotype in the original repertoire. Constructs were transformed into NEB 10-beta E. coli and purified with the ZymoPURE™ Plasmid Maxiprep Kit (Zymo Research). The purified plasmid was then used for transient transfection in the ExpiCHO system (Thermo Fisher Scientific). Transfected cells were cultured for 7–9 days in ExpiCHO medium and then antibodies were purified from filtered supernatant using protein A columns (Millipore). Antibody purity and proper size was verified by Coomassie stained SDS-PAGE gel (Thermo Fisher Scientific).
Monoclonal antibody characterization
For measurement of kinetics of binding to H1N1-His and H3N2-His, antibodies were loaded onto an anti-Human IgG Fc (AHC) biosensor using the Octet Red96 system (ForteBio) by a CRO (LakePharma). Loaded biosensors were dipped into antigen dilutions beginning at 300 nM, with 6 serial dilutions at 1:3. Kinetic analysis was performed using a 1:1 binding model and global fitting (Supplementary Figures S3-S4).
A CRO (Virapur) performed the influenza microneutralization assays. Briefly, serial dilutions of mAb were mixed with titered stock influenza A/California/07/2009 (H1N1) or A/Sydney/5/1997 (H3N2) strains and incubated for 1 hour at 37 °C. The virus/antibody solution was then pipetted into a 96-well plate of MDCK cells. The plates were incubated for 4 days at 37 °C. Viral infection of the cells was quantified using a microscope. All measurements were performed in triplicate. The data are presented as the lowest concentration of antibody that displayed neutralization activity in the assay (Supplementary Figure S5).
For analysis of pneumococcal mAbs binding to a pool of 23 sugars, the human anti-S. pneumoniae vaccine (Pneumovax/CPS23) IgG ELISA kit (Alpha Diagnostics) was used in parallel with the human anti-S. pneumoniae CWPS/22F IgG ELISA kit (Alpha Diagnostics). Antibody samples were absorbed with C22 absorbent mix from the anti-S. pneumoniae CWPS/22F IgG ELISA kit for 1 hour at room temperature at the highest concentration of antibody tested to remove non-specific binding activity. After absorption, serial dilutions of antibody were performed in Low non-specific binding sample diluent (Alpha Diagnostics). Samples were then analyzed by anti-S. pneumoniae CWPS/22F IgG ELISA according to the manufacturer's instructions to confirm that non-specific binding activity was removed by the absorption step, and analyzed by human anti-S. pneumoniae vaccine (Pneumovax/CPS23) IgG ELISA to determine the anti-S. pneumoniae titer. Quantitative measurements were performed on a plate reader at 450 nm (Perkin Elmer) (Supplementary Figure S6). EC50 values were calculated using SoftMax Pro (Molecular Devices).
To determine the pneumococcus polysaccharide-binding specificity of individual mAbs, a dot blot method was used. Individual polysaccharides (ATCC), as well as the 22-polysaccharide pool, were spotted onto a 0.45 μm nitrocellulose membrane (VWR), 2 μl per spot at 2 mg/ml. Once the spots were dry, the membrane was washed in PBST (1 × PBS + 0.1% Tween-20) 3 × 5 minutes while shaking. The membrane was blocked in 5% dry milk in PBST for 20–60 minutes, then washed 3 times briefly with 1 × PBST. The membrane was then incubated with the purified mAb of interest diluted in 1 × PBST for 1 hour at room temperature while shaking. After 1 hour the membrane was washed 3 × briefly and then 3 × 5 minutes shaking in PBST. The membrane was then incubated with an HRP-conjugated secondary antibody (Thermo Fisher Scientific 31410) for 45 minutes while shaking. The membrane was washed 3 × briefly and then washed in PBST for 3 × 5 minutes while shaking. The membrane was visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) on a c300 imaging system (Azure Biosystems) (Supplementary Figure S7). Human intravenous immunoglobulin (Gammagard) was used as a positive control, with the expectation that it would bind to all polysaccharides, individually and as a pool.
Functional antibodies to pneumococcal serotypes were measured in a multiplexed opsonophagocytic assay as previously described.56,57
Supplementary Material
Disclosure statement
ASA, RAM, MJS, MSA, MAA, RCE, JL, RL, and DSJ are employees of GigaGen Inc. and receive both equity shares and salary for their work. LR, RW, and DG are employees of University College London and receive salary for their work.
Acknowledgments
We thank Everett Meyer for his useful insights during development of the technology. The staff at the Stanford Shared FACS Facility were a valuable resource for flow sorting.
Funding
This work was supported by the National Science Foundation under grant 1230150; National Institute for Allergy and Infectious Diseases under grant R44AI124901.
References
- 1.Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, Lampley R, Kose N, Ilinykh PA, Kuzmina N, et al.. Cross-Reactive and Potent Neutralizing Antibody Responses in Human Survivors of Natural Ebolavirus Infection. Cell. 2016;164(3):392-405. doi: 10.1016/j.cell.2015.12.022. PMID:26806128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilchuk I, Gilchuk P, Sapparapu G, Lampley R, Singh V, Kose N, Blum DL, Hughes LJ, Satheshkumar PS, Townsend MB, et al.. Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell. 2016;167(3):684-694.e9. doi: 10.1016/j.cell.2016.09.049. PMID:27768891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beerli RR, Rader C. Mining human antibody repertoires. MAbs. 2010;2(4):365-78. doi: 10.4161/mabs.12187. PMID:20505349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith SA, Crowe JE Jr. Use of Human Hybridoma Technology To Isolate Human Monoclonal Antibodies. Microbiol Spectr. 2015;3(1):AID-0027-2014. doi: 10.1128/microbiolspec.AID-0027-2014. PMID:26104564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495-7. doi: 10.1038/256495a0. PubMed PMID:1172191 [DOI] [PubMed] [Google Scholar]
- 6.de Wildt RM, Hoet RM. The recovery of immunoglobulin sequences from single human B cells by clonal expansion. Methods Mol Biol. 2002;178:121-31. PMID:11968481 [DOI] [PubMed] [Google Scholar]
- 7.Steinitz M, Klein G, Koskimies S, Makel O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature. 1977;269(5627):420-2. doi: 10.1038/269420a0. PMID:198669 [DOI] [PubMed] [Google Scholar]
- 8.Chiorazzi N, Wasserman RL, Kunkel HG. Use of Epstein-Barr virus-transformed B cell lines for the generation of immunoglobulin-producing human B cell hybridomas. J Exp Med. 1982;156(3):930-5. doi: 10.1084/jem.156.3.930. PMID:6286839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galson JD, Clutterbuck EA, Trück J, Ramasamy MN, Münz M, Fowler A, Cerundolo V, Pollard AJ, Lunter G, Kelly DF. BCR repertoire sequencing: different patterns of B-cell activation after two Meningococcal vaccines. Immunol Cell Biol. 2015;93(10):885-95. doi: 10.1038/icb.2015.57. PMID:25976772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd SD, Crowe JE Jr. Deep sequencing and human antibody repertoire analysis. Curr Opin Immunol. 2016;40:103-9. doi: 10.1016/j.coi.2016.03.008. PMID:27065089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meijer PJ, Nielsen LS, Lantto J, Jensen A. Human antibody repertoires. Methods Mol Biol. 2009;525:261-77, xiv. doi: 10.1007/978-1-59745-554-1_13. PMID:19252857 [DOI] [PubMed] [Google Scholar]
- 12.DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, Giesecke C, Dörner T, Andrews SF, et al.. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol 2013; 31:166-9; doi: 10.1038/nbt.2492. PMID:23334449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busse CE, Czogiel I, Braun P, Arndt PF, Wardemann H. Single-cell based high-throughput sequencing of full-length immunoglobulin heavy and light chain genes. Eur J Immunol. 2014;44(2):597-603. doi: 10.1002/eji.201343917. PMID:24114719 [DOI] [PubMed] [Google Scholar]
- 14.Tan YC, Blum LK, Kongpachith S, Ju CH, Cai X, Lindstrom TM, Sokolove J, Robinson WH. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin Immunol. 2014;151(1):55-65. doi: 10.1016/j.clim.2013.12.008. PMID:24525048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, Georgiou G. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med 2015; 21:86-91. doi: 10.1038/nm.3743. PMID:25501908 [DOI] [PubMed] [Google Scholar]
- 16.Wang B, Lee CH, Johnson EL, Kluwe CA, Cunningham JC, Tanno H, Crooks RM, Georgiou G, Ellington AD. Discovery of high affinity anti-ricin antibodies by B cell receptor sequencing and by yeast display of combinatorial VH:VL libraries from immunized animals. MAbs. 2016;8(6):1035-44. doi: 10.1080/19420862.2016.1190059. PMID:27224530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang N, He J, Weinstein JA, Penland L, Sasaki S, He XS, Dekker CL, Zheng NY, Huang M, Sullivan M, et al.. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci Transl Med. 2013;5(171):171ra19 Erratum in: Sci Transl Med. 2013 Jul 10;5(193):193er8. doi: 10.1126/scitranslmed.3004794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vollmers C, Sit RV, Weinstein JA, Dekker CL, Quake SR. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc Natl Acad Sci U S A. 2013;110(33):13463-8. doi: 10.1073/pnas.1312146110. PMID:23898164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu YC, Kipling D, Dunn-Walters DK. Age-Related Changes in Human Peripheral Blood IGH Repertoire Following Vaccination. Front Immunol. 2012;3:193. doi: 10.3389/fimmu.2012.00193. PMID:22787463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dangaj D, Lanitis E, Zhao A, Joshi S, Cheng Y, Sandaltzopoulos R, Ra HJ, Danet-Desnoyers G, Powell DJ Jr, Scholler N. Novel recombinant human b7-h4 antibodies overcome tumoral immune escape to potentiate T-cell antitumor responses. Cancer Res. 2013;73(15):4820-9. doi: 10.1158/0008-5472.CAN-12-3457. PMID:23722540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrante A, Beard LJ, Feldman RG. IgG subclass distribution of antibodies to bacterial and viral antigens. Pediatr Infect Dis J. 1990;9(8 Suppl):S16-24. PMID:2216603 [PubMed] [Google Scholar]
- 22.Lossos IS, Tibshirani R, Narasimhan B, Levy R. The inference of antigen selection on Ig genes. J Immunol. 2000;165(9):5122-6. doi: 10.4049/jimmunol.165.9.5122. PMID:11046043 [DOI] [PubMed] [Google Scholar]
- 23.Yu X, McGraw PA, House FS, Crowe JE Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods. 2008;336(2):142-51. doi: 10.1016/j.jim.2008.04.008. PMID:18514220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu J, Wu X, Zhang B, McKee K, O'Dell S, Soto C, Zhou T, Casazza JP, NISC Comparative Sequencing Program, Mullikin JC, Kwong PD, et al.. De novo identification of VRC01 class HIV-1-neutralizing antibodies by next-generation sequencing of B-cell transcripts. Proc Natl Acad Sci U S A. 2013;110(43):E4088-97. doi: 10.1073/pnas.1306262110. PMID: 24106303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Liu Y, Cavanagh MM, Le Saux S, Qi Q, Roskin KM, Looney TJ, Lee JY, Dixit V, Dekker CL, et al.. B-cell repertoire responses to varicella-zoster vaccination in human identical twins. Proc Natl Acad Sci U S A. 2015;112(2):500-5. doi: 10.1073/pnas.1415875112. PMID:25535378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas AH, Moulton KD, Tang VR, Reason DC. Combinatorial library cloning of human antibodies to Streptococcus pneumoniae capsular polysaccharides: variable region primary structures and evidence for somatic mutation of Fab fragments specific for capsular serotypes 6B, 14, and 23F. Infect Immun. 2001;69(2):853-64. doi: 10.1128/IAI.69.2.853-864.2001. PMID:11159978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Lottenbach KR, Barenkamp SJ, Reason DC. Somatic hypermutation and diverse immunoglobulin gene usage in the human antibody response to the capsular polysaccharide of Streptococcus pneumoniae Type 6B. Infect Immun. 2004;72(6):3505-14. doi: 10.1128/IAI.72.6.3505-3514.2004. PMID:15155658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolibab K, Smithson SL, Rabquer B, Khuder S, Westerink MA. Immune response to pneumococcal polysaccharides 4 and 14 in elderly and young adults: analysis of the variable heavy chain repertoire. Infect Immun. 2005;73(11):7465-76. doi: 10.1128/IAI.73.11.7465-7476.2005. PMID:16239548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baxendale HE, Davis Z, White HN, Spellerberg MB, Stevenson FK, Goldblatt D. Immunogenetic analysis of the immune response to pneumococcal polysaccharide. Eur J Immunol. 2000;30(4):1214-23. doi: 10.1002/(SICI)1521-4141(200004)30:4%3c1214::AID-IMMU1214%3e3.0.CO;2-D. PMID:10760811 [DOI] [PubMed] [Google Scholar]
- 30.Smith K, Muther JJ, Duke AL, McKee E, Zheng NY, Wilson PC, James JA. Fully human monoclonal antibodies from antibody secreting cells after vaccination with Pneumovax®23 are serotype specific and facilitate opsonophagocytosis. Immunobiology. 2013;218(5):745-54. doi: 10.1016/j.imbio.2012.08.278. PMID:23084371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Gunten S, Smith DF, Cummings RD, Riedel S, Miescher S, Schaub A, Hamilton RG, Bochner BS. Intravenous immunoglobulin contains a broad repertoire of anticarbohydrate antibodies that is not restricted to the IgG2 subclass. J Allergy Clin Immunol. 2009;123(6):1268-76.e15. doi: 10.1016/j.jaci.2009.03.013. PMID:19443021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson DS, Meyer EH. (2012). WO2012083225 A3. System and methods for massively parallel analysis of nucleic acids in single cells. [Google Scholar]
- 33.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al.. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202-14. doi: 10.1016/j.cell.2015.05.002. PMID:26000488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q, Abeygunawardana C, Ng AS, Sturgess AW, Harmon BJ, Hennessey JP Jr. Characterization and quantification of C-polysaccharide in Streptococcus pneumoniae capsular polysaccharide preparations. Anal Biochem. 2005;336(2):262-72. Erratum in: Anal Biochem. 2005 Jul 15;342(2):358. doi: 10.1016/j.ab.2004.10.019. PMID:15620891 [DOI] [PubMed] [Google Scholar]
- 35.Khan N, Jan AT. Towards Identifying Protective B-Cell Epitopes: The PspA Story. Front Microbiol. 2017;8:742. doi: 10.3389/fmicb.2017.00742. PMID:28512452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krause JC, Tsibane T, Tumpey TM, Huffman CJ, Basler CF, Crowe JE Jr. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J Virol. 2011;85(20):10905-8. doi: 10.1128/JVI.00700-11. PMID:21849447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, et al.. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 2011;108(34):14216-21. doi: 10.1073/pnas.1111497108. PMID:21825125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al.. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med. 2011;208(1):181-93. Epub 2011 Jan 10. Erratum in: J Exp Med. 2011 Feb 14;208(2):411. doi: 10.1084/jem.20101352. PMID:21220454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frölich D, Giesecke C, Mei HE, Reiter K, Daridon C, Lipsky PE, Dörner T. Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J Immunol. 2010;185(5):3103-10. Epub 2010 Aug 6. Erratum in: J Immunol. 2014 Jan 1;192(1):535. doi: 10.4049/jimmunol.1000911. PMID:20693426 [DOI] [PubMed] [Google Scholar]
- 40.Poulsen TR, Jensen A, Haurum JS, Andersen PS. Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J Immunol. 2011;187(8):4229-35. doi: 10.4049/jimmunol.1000928. PMID:21930965 [DOI] [PubMed] [Google Scholar]
- 41.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348(6301):552-4. . doi: 10.1038/348552a0. PMID:2247164 [DOI] [PubMed] [Google Scholar]
- 42.Clackson T, Hoogenboom HR, Griffiths AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991;352(6336):624-8. doi: 10.1038/352624a0. PMID:1907718 [DOI] [PubMed] [Google Scholar]
- 43.van den Beucken T, Pieters H, Steukers M, van der Vaart M, Ladner RC, Hoogenboom HR, Hufton SE. Affinity maturation of Fab antibody fragments by fluorescent-activated cell sorting of yeast-displayed libraries. FEBS Lett. 2003;546(2-3):288-94. doi: 10.1016/S0014-5793(03)00602-1. PMID:12832056 [DOI] [PubMed] [Google Scholar]
- 44.Walker LM, Bowley DR, Burton DR. Efficient recovery of high-affinity antibodies from a single-chain Fab yeast display library. J Mol Biol. 2009;389(2):365-75. doi: 10.1016/j.jmb.2009.04.019. PMID:19376130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho M, Pastan I. Mammalian cell display for antibody engineering. Methods Mol Biol. 2009;525:337-52, xiv. doi: 10.1007/978-1-59745-554-1_18. PMID:19252852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowers PM, Horlick RA, Kehry MR, Neben TY, Tomlinson GL, Altobell L, Zhang X, Macomber JL, Krapf IP, Wu BF, et al.. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods. 2014;65(1):44-56. doi: 10.1016/j.ymeth.2013.06.010. PMID:23792919 [DOI] [PubMed] [Google Scholar]
- 47.Bird RE, Walker BW. Single chain antibody variable regions. Trends Biotechnol. 1991;9(4):132-7. doi: 10.1016/0167-7799(91)90044-I. PMID:1367550 [DOI] [PubMed] [Google Scholar]
- 48.Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, et al.. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003;77(19):10557-65. doi: 10.1128/JVI.77.19.10557-10565.2003. PubMed PMID:12970440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintero-Hernández V, Juárez-González VR, Ortíz-León M, Sánchez R, Possani LD, Becerril B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol. 2007;44(6):1307-15. doi: 10.1016/j.molimm.2006.05.009. PMID:16814388 [DOI] [PubMed] [Google Scholar]
- 50.Johnson DS, Adler AS, Mizrahi RM. (2016). US20160362681 A1. Recombinant fusion proteins and libraries from immune cell repertoires. http://www.necsi.edu/events/iccs6/papers/c1602a3c126ba822d0bc4293371c.pdf [Google Scholar]
- 51.Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476-82. doi: 10.1093/bioinformatics/btv401. PMID:26139637 [DOI] [PubMed] [Google Scholar]
- 52.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996-8. doi: 10.1038/nmeth.2604. PMID:23955772 [DOI] [PubMed] [Google Scholar]
- 53.Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al.. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37(Database issue):D1006-12. doi: 10.1093/nar/gkn838. PMID:18978023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460-1. doi: 10.1093/bioinformatics/btq461. PMID:20709691 [DOI] [PubMed] [Google Scholar]
- 55.Csardi G, Nepusz T. The igraph software package for complex network research, InterJournal Complex Systems 2006;1695. [Google Scholar]
- 56.Burton RL and Nahm MH. Development and validation of a fourfold multiplex opsonization assay (MOPA4) for pneumococcal antibodies. Clin Vaccine Immunol. 2006;13(9):1004-9. doi: 10.1128/CVI.00112-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rose CE, Romero-Steiner S, Burton RL, Carlone GM, Goldblatt D, Nahm MH, Ashton L, Haston M, Ekström N, Haikala R, et al.. Multilaboratory comparison of Streptococcus pneumoniae opsonophagocytic killing assays and their level of agreement for the determination of functional antibody activity in human reference sera. Clin Vaccine Immunol. 2011;18(1):135-42. doi: 10.1128/CVI.00370-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.