

ABSTRACT
The abundant cell surface asialoglycoprotein receptor (ASGPR) is a highly selective receptor found on hepatocytes that potentially can be exploited as a selective shuttle for delivery. Various nucleic acid therapeutics that bind ASGPR are already in clinical development, but this receptor-mediated delivery mechanism can be saturated, which will likely result in reduced selectivity for the liver and therefore increase the likelihood for systemic adverse effects. Therefore, when aiming to utilize this mechanism, it is important to optimize both the administration protocol and the molecular properties. We here present a study using a novel ASGPR-targeted antibody to estimate ASGPR expression, turnover and internalization rates in vivo in mice. Using pharmacokinetic data (intravenous and subcutaneous dosing) and an in-silico target-mediated drug disposition (TMDD) model, we estimate an ASGPR expression level of 1.8 million molecules per hepatocyte. The half-life of the degradation of the receptor was found to be equal to 15 hours and the formed ligand-receptor complex is internalized with a half-life of 5 days. A biodistribution study was performed and confirmed the accuracy of the TMDD model predictions. The kinetics of the ASGPR shows that saturation of the shuttle at therapeutic concentrations is possible; however, simulation allows the dosing schedule to be optimized. The developed TMDD model can be used to support the development of therapies that use the ASGPR as a shuttle into hepatocytes.
KEYWORDS: TMDD, ASGPR, mAb, pharmacokinetics, modeling and simulation, liver-targeted delivery, drug targeting
Introduction
Targeted delivery of novel therapeutics has been an attractive research area because it allows the drug to concentrate in specific tissues and cell types and minimizes non-target related toxicity.1,2 This is particularly important for molecules that cannot enter cells on their own, such as RNAi or gene editing technologies mediated by CRISPR,3 or accumulate in tissues that are not targeted, such as nucleic acid therapeutics,4 leading to safety liabilities.
For targeted delivery to the liver, and more specifically to hepatocytes, the use of the asialoglycoprotein receptor (ASGPR) as a shuttle has drawn particular attention.5 This hepatic receptor has been extensively studied since its discovery in the mid-1970s by Ashwell and Morell and their co-workers.6,7 ASGPR, a transmembrane C-type lectin, recognizes a wide variety of ligands that contain either terminal galactose (Gal) or N-acetylgalactosamine (GalNAc) residues.8 Although the ASGPR native function has not been completely clarified, it is primarily cited for desialylated serum glycoproteins removal. However, the hepatic receptor is also thought to be involved in the clearance of lipoproteins,9 hepatic fibronectin,10 apoptotic cells,11 immunoglobulin A12 or involved in cell-cell interactions.13 ASGPR expression has been reported to be abundant and, at the same time, highly specific to hepatocytes.14,15 The ASGPR density has been estimated under in-vitro conditions (0.5 to 1.5 million of copies/cell)14,16,17 and in humans (1.09 million/hepatocyte).18 However, the uptake through the ASGPR is not only dependent on the receptor numbers, but also on the kinetics of the receptor, e.g., receptor turnover rate.
To investigate the in-vivo drug delivery capacity of the ASGPR, we aimed to exploit the target-mediated drug disposition (TMDD) of a newly developed anti-ASGPR antibody (ASGPR Ab) in mice.19 Therapeutic proteins often show a characteristic non-linear concentration-time profile in the plasma due to saturation of binding, distribution or elimination pathways. Thus, TMDD can be used a tool for inferring the properties of the shuttle from the plasma concentrations,20 such as the cell surface expression level, the binding constant and the internalization rate.19 These factors are all crucial to quantify the potential of the receptor for drug delivery.
Once the model is fully parametrized, it can be used to optimize a dosing protocol in order to maximize the delivery efficiency, i.e., maximizing the internalization through the receptor, and therefore reducing the off-target distribution and unspecific clearance. The model also allows simulation of the influence of the different drug properties on the delivery efficiency. To validate our model predictions, a biodistribution study was performed comparing the liver uptake of the ASGPR Ab to a non-targeting antibody.
Results
Generation and in-vitro characterization of an ASGPR-targeting antibody
The generation of the human ASGPR Ab is fully described in the supplementary material (Appendix 1). In brief, the identification of the ASGPR Ab was performed by phage display. The whole extracellular domain (ECD) or the stalk region of the ASGPR or closest homologous protein CLEC10A were fused to human IgG1 Fc (Appendix 1, Supplementary Fig. 1A, B). Binding of the ASGPR Ab was tested against these proteins (SupplementaryFig. 2A-D). Based on criteria outlined in the materials and methods section (Appendix 1, supplementary material), one clone binding to the stalk region (but not to the C-type lectin domain) of the ASGPR was selected. No cross-reactivity with the CLEC10A stalk or ECD regions was detected, underlining the specificity of the selected clone for ASGPR.
In order to assess the cross-reactivity of the ASGPR antibody among different species, a TagLite assay (CisBio) was performed. The TagLite assay is a time-resolved fluorescence resonance energy transfer (FRET)-based interaction assay, which allows antibody binding to cell surface bound receptors to be measured and quantified. In order to quantify and compare its binding to either human or murine ASGPR1, HEK293 EBNA cells were transiently transfected with the respective ASGPR1-SNAP tag fusion constructs and labeled with terbium (Tb). Interestingly, despite the rather low homology between human and murine stalk region (77%), the calculated binding curves for the ASGPR Ab to both human and murine ASGPR1 almost overlapped, clearly demonstrating that this antibody is cross-reactive to murine ASGPR1 (Fig. 1A). In addition, both calculated dissociation constants were in the low single-digit nanomolar range, supporting the finding that the ASGPR Ab binding intensity to murine ASGPR1 is comparable to the human protein (Fig. 1B).
Figure 1.
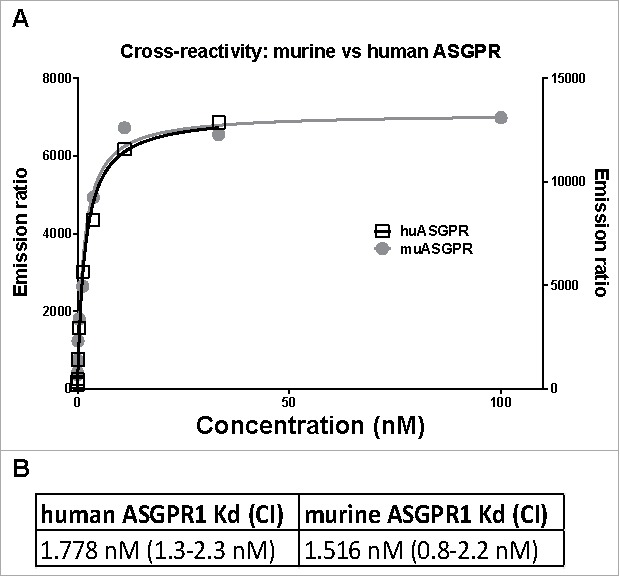
TagLite analysis of the ASGPR Ab to human and mouse ASGPR1. (A) HEK293 EBNA cells were transiently transfected with either a human or a mouse ASGPR1-SNAP tag fusion construct and the SNAP tag was labeled with Tb. A three-fold dilution series of the ASGPR Ab ranging from 100 to 0.005 nM in combination with a d2-labeled anti-human Fc antibody was incubated with the cells. After 3 hours of incubation, FRET signals were measured. Emission ratio (665 nm/620 nm) data is plotted in black (right y axis) or in grey (left y axis) for human or murine ASGPR1, respectively. Lines are the respective fitted binding curves. (B) calculated dissociation constants (Kd) and their confidence interval (CI).
TMDD model after intravenous administration and estimation of the ASGPR shuttle capacity and uptake kinetic parameters
A mouse PK study was designed to determine the capacity of the ASGPR shuttle using the ASGPR Ab. After intravenous (IV) dosing, the PK profiles show non-linear concentration-time profiles that are consistent with TMDD (Fig. 2). At low dose (1 mg/kg), a biphasic PK profile can be observed with an initial rapid decline in plasma concentration within the first hour followed by a slow decline. By contrast, at high dose (30 mg/kg) the decay in concentration follows 4 phases, which is also indicative of a TMDD profile when a saturating dose is administered (Fig. 2).21
Figure 2.
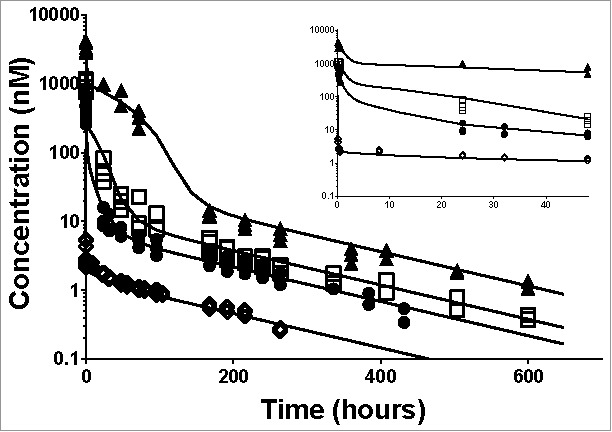
Individual plasma total anti-ASGPR antibody concentrations over time and model prediction in mice after IV bolus dosing. Markers present individual measurements for 1 mg/kg (◊), 5 mg/kg (•), 10 mg/kg (□) and 30 mg/kg (▴). Lines represent the developed model predicted values for the tested doses. The inset represents a detailed zoom-in of the data and predictions from 0 to 48 hours. The model parameters used for simulations are presented in Table 1.
The antibody-target binding processes are usually fast, in the order of minutes, in comparison with distribution or elimination processes. Therefore, the initial drop in antibody concentrations at 1 mg/kg can be attributed to the binding of the ASGPR. However, the free antibody concentrations in the first measurements (5 minutes after IV dosing) do not increase in a dose-dependent manner (see Supplementary Fig. 3). This phenomenon indicates that the binding to the receptors gets saturated at higher doses and contribute less and less to the initial decay in concentrations as the dose increases. Therefore, both non-linear PK profiles and saturable binding suggest a TMDD-like PK.
To quantify the ASGPR expression and the uptake kinetic parameters, all IV dose groups were fitted simultaneously to the TMDD model (Fig. 3). The observed data and model-fit profiles (Fig. 2) show good agreement. Relative standard errors were lower than 18% indicating that the fitted parameters have been accurately estimated (see Table 1). Individual fits and goodness of fit plots are provided in Supplementary Fig. 4 and Supplementary Fig. 5.
Figure 3.
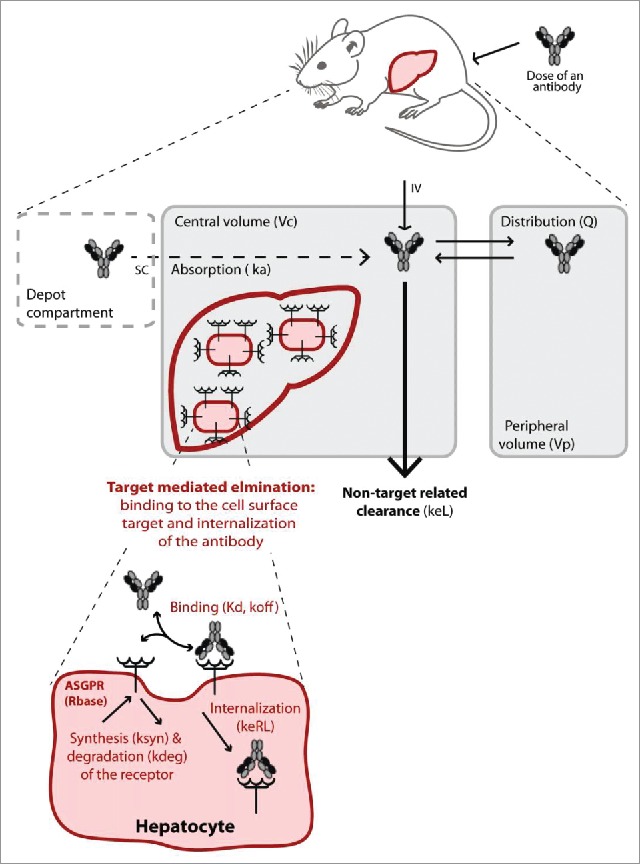
Schematic illustration of the target-mediated drug disposition model. The non-target related PK is represented by a two-compartment model (in grey) including linear distribution and clearance processes. The dashed compartment represents the depot compartment from which the antibodies are absorbed for simulating subcutaneous (SC) injection. The liver is represented in the central compartment where the ASGPR is expressed at the surface of the hepatocytes. The interaction of the antibodies with the receptors results in a nonlinear target-mediated elimination pathway (in red). Binding to the ASGPR leads to the complex formation and endocytosis. The receptor turn-over is also represented with a synthesis and degradation rate. All parameters used in the model are given in brackets.
Table 1.
Parameter estimates of the TMDD model after IV bolus.
| Population mean parameter |
Intersubject variance |
|||||
|---|---|---|---|---|---|---|
| Parameter | Description | Units | Estimate | RSE (%) | Estimate | RSE (%) |
| Vc | Volume of central compartment | mL | 1.12 | 5 | 0.115 | 26 |
| Kd | constant of dissociation | nM | 4.13 | 5 | 0.145 | 25 |
| koff | dissociation rate (binding) | 1/day | 12.31 | 2 | 0.0662 | 26 |
| Rbase | baseline target concentration | nM | 647 | 4 | — | — |
| Number of receptors/hepatocytes* | — | 1.8 million | — | — | — | |
| keRL | rate of internalization | 1/day | 0.139 | 3 | 0.0599 | 39 |
| kdeg | turnover rate of the target | 1/day | 1.06 | 13 | 0.413 | 23 |
| keL | Rate of elimination from central compartment | 1/day | 0.449 | 13 | 0.302 | 37 |
| Q | exchange between central and peripheral compartment | mL/day | 20.28 | 13 | 0.296 | 40 |
| Vp | volume of the peripheral compartment | mL | 3.79 | 18 | 0.58 | 24 |
| ksyn | Synthesis rate (secondary parameter) | nM/day | 715.84 | — | — | — |
| b | proportional error | — | 0.114 | 8 | — | — |
In support of the parameter fits, the volume of the central compartment (Vc) (estimated as 1.12 mL) is in agreement with mouse physiological plasma volume of 1 mL,22 which is where antibodies are usually distributed.23 The unspecific (non-target related) clearance of the antibody has been estimated as 25 ml/kg/day, which is relatively fast for an antibody in mice,24 but not inconsistent with previous studies for other antibodies (2.4 to 75 ml/day/kg).25-27
Regarding the target-mediated processes, the free ASGPR concentration has been estimated as 647 nM (36 nmol/kg). Assuming that the ASGPR is mainly expressed at the cell surface of hepatocytes8 and using the reported value of hepatocellularity number in mice,22,28 the binding capacity can be translated into a surface expression of 1.8 million receptors per hepatocyte (see Table 1). In addition, the half-life of the degradation of the receptor was found to be equal to 15 hours and the formed antibody-receptor complex is internalized with a half-life of 5 days. Both kinetics parameters are important to quantify and predict the time course of the receptor concentration and antibody-receptor complex internalization after dosing.
Validity of ASGPR shuttle model for SC administration
The shuttle capacity of the ASGPR was also tested after subcutaneous (SC) administration. SC dosing leads to a slow absorption of the antibody into the bloodstream and subsequent binding of the antibody. We investigated if the developed model of the ASGPR shuttle based on the IV data allowed the correct description of the influence of the shuttle even in the case of SC dosing. While fitting only the absorption rate parameter (all other parameters fixed from IV data), the established ASGPR shuttle model appropriately described the concentration-time profile after SC dosing (Fig. 4) for both a saturating dose of 30 mg/kg and a low dose of 1 mg/kg. This increases the confidence in the estimation of the underlying TMDD parameters. The SC injection resulted in higher inter-individual variability, especially for the largest dose, which is commonly seen for protein therapeutics.29
Figure 4.
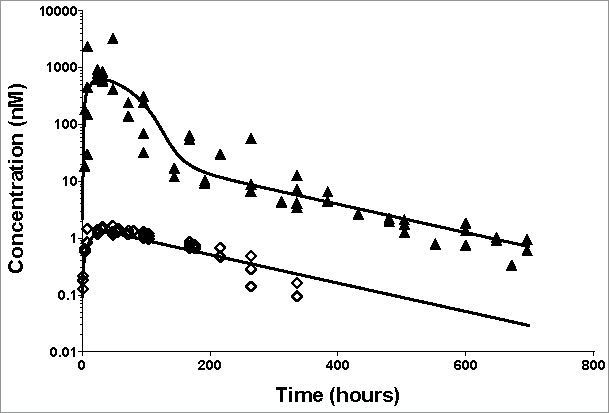
Application of the developed TMDD model for SC dosing of the anti-ASGPR antibody. Markers present individual measurements for 1 mg/kg (◊) and 30 mg/kg (▴). Lines represent model predicted values. The model parameters used for simulations are presented in Supplementary Table 2.
Parameter estimation gave good levels of relative standard errors (rse), which are displayed in Supplementary Table 1. Individual fits and goodness of fit plots are provided in Supplementary Fig. 6 and Supplementary Fig. 7.
Liver distribution of the ASGPR antibody
We performed a separate biodistribution study with the ASGPR antibody to assess the accuracy of the TMDD model in predicting the liver uptake. It is of note, however, that the liver has been found to be a major elimination organ for monoclonal antibodies.30 Therefore, a radiolabeled non-targeting antibody (IL17 Ab) was used for comparison. Both compounds were radiolabeled with 111Indium-DOTA complex. The radiochemical yields of the 111In-labeled antibodies were 45.0% for ASGPR Ab, and 50.8% for IL17 Ab. The specific activities of the 111In-labeled antibodies used in the study were 3.51 μCi/μg for ASGPR Ab and 3.02 μCi/μg for the IL17 Ab.
111Indium-DOTA complex is residualizing in tissues when antibodies are degraded.31 Due to the trapping of the radiolabel marker in the tissues, radioactive counts account for the antibodies accumulation in tissue to the extent of the marker physical decay (2.8 half-life decay). Liver samples were terminally harvested and liver content is displayed in Fig. 5A. At 8 hours post dose, 42% of the dose accumulates in the liver following a 10 mg/kg IV dose of the ASGPR Ab, compared to 4.8% of the non-targeting antibody. Therefore, the target-mediated process concentrates the ASGPR-specific antibody in the liver in comparison to the IL17-targeting antibody. In addition, we compared the liver content observations with the predictive values from the model. However, the liver content observation is not a direct measurement of the amount processed by the ASGPR. In the harvested liver, the antibody can be measured in different forms, i.e., internalized, bound or free in the residual plasma volume. Therefore, the actual observations were compared to the sum of these different entities in Fig. 5B. The TMDD accurately predicted the liver content with a maximum difference of 24%. Therefore, the accumulation in liver can be attributed to the ASGPR-mediated endocytosis, and the model is able to predict accurately the liver uptake.
Figure 5.
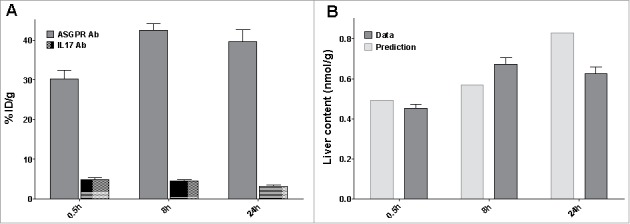
Distribution study (A) Percentage of injected dose in liver after an IV bolus dose of 10 mg/kg of the ASGPR Ab (grey) or the non-targeting Ab (dotted area) (B) Predicted (grey) versus observed (light grey) liver content of the ASGPR Ab.
Optimization of administration protocol using TMDD model
To be successful as a targeted therapy, the protocol of administration should be selected such as to maximize internalization into hepatocytes via the ASGPR route and minimize saturation that would lead to off-target distribution and unspecific clearance. The developed TMDD model includes key mechanisms of interaction between the antibody and the ASGPR, and can therefore be used to predict the uptake into hepatocytes. The TMDD model allows the calculation of how much antibody is processed through the ASGPR shuttle and how much is distributed elsewhere or via unspecific clearance (see Fig. 6A). At low dose (1 mg/kg), most of the antibody is internalized through the ASGPR and 96% of the total dose is cleared via the target pathway. However, the delivery efficiency (i.e., the percentage of the dose being internalized) decreases with increasing dose due to receptor saturation. At a high dose of 30 mg/kg, only 76% of the administered antibody is taken up by ASGPR, even though a larger total amount of antibody is taken up.
Figure 6.
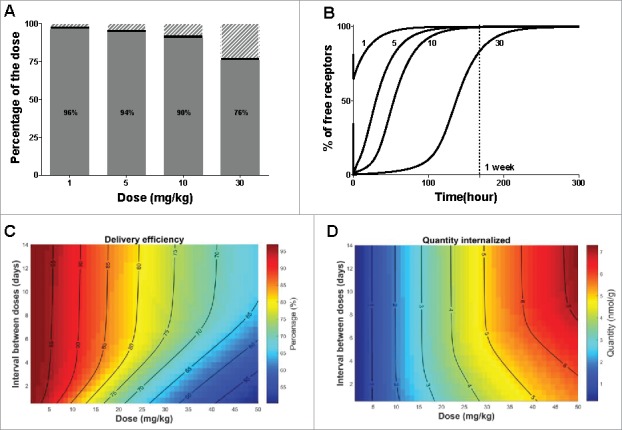
Optimization of protocol administration for maximizing delivery efficiency. (A) Delivery efficiency (grey), percentage of dose cleared via unspecific (non-target related) mechanisms (dashed area) or still bound to the receptor at 720 hours (30 days) post dose (black) after single IV bolus. (B) Simulated free available receptor concentrations over time in mice after IV bolus for all tested doses. The doses are indicated in mg/kg beside each simulation. (C) Delivery efficiency and (D) Quantity of the dose internalized via the target-mediated clearance pathway after two consecutive doses IV, varying the total amount and the interval between doses. The interval between the two consecutive doses is expressed in days, the dose in mg/kg and the quantity in nmol/g of liver. In both graphs, contour lines represent equivalent percentage or quantity values, respectively.
To better understand the dynamics of the ASGPR as a hepatocyte shuttle, we used the model to investigate how quickly the ASGPR system recovers from a dose challenge. The rate at which free receptors become available again is critical to optimize a protocol of administration in the case of multiple dosing regimens. The estimated concentration-time profile of the free receptor (expressed as the percentage compared to its baseline level) is shown in Fig. 6B. At 5 mg/kg, almost all of the receptors are bound to the antibody immediately after IV bolus and form a receptor-antibody complex, while at 1 mg/kg antibody concentrations do not saturate all free receptors. It is of note that the recovery time of free receptors is also dose dependent. At higher saturating doses (i.e., 5, 10, 30 mg/kg) unspecific clearance is not sufficiently high to prevent antibodies remaining longer in the system, and which therefore are free to bind newly synthetized receptors, thereby leading to longer depletion time of the free receptor. Hence, at 5 mg/kg for example, the free receptors return to 90% of the baseline level in 3 days, while at 30 mg/kg it takes more than a week.
The receptor time profiles suggest that dosing frequency must be adapted to the dose level. We therefore used the model to simulate the delivery efficiency after two consecutive IV doses, varying the dose level and the interval between doses (Fig. 6C). At non-saturating doses, lower than 5 mg/kg, almost all of the antibody is taken up by the shuttle and little is removed by unspecific clearance. The dosing interval thus has little effect on delivery efficiency at low doses. However, at 30 mg/kg dose, only 76% of antibody is taken up by the ASGPR, and hence the scheduling makes a larger difference. The implications of this are that an equal percentage of the dose could be delivered to the hepatocytes by using, for instance, an initial dose of 20 mg/kg and a the second dose after 48 hours (protocol 1) or an initial dose of 30 mg/kg and a second one after 7 days (protocol 2). While these two doses allow equally high delivery efficiency to be obtained, it is of interest to know how much drug eventually is delivered to the hepatocytes using the respective dosing regimens. The respective amounts delivered by the target-mediated pathway are displayed in Fig. 6D. With protocol 1 versus 2, 6 or 9 nmol would be delivered, respectively. Using the model, one can optimize the dosing protocol to maximize the delivery efficiency, and therefore reduce the off-target distribution and unspecific clearance, but also the quantity delivered to reach an efficacy threshold for instance.
While we have investigated the ASGPR capacity to transport a targeted antibody from the blood to the hepatocytes, the ASGPR is regarded as a promising shuttle for different ligand-conjugated or nanocarrier therapeutic modalities. The PK model used in this study is composed of two distinct elimination pathways, one being the non-target related clearance, with the other representing the mechanistic description of the specific clearance by the ASGPR (Fig. 3). Because the two elimination pathways are distinct, the non-target related part of the model can be adjusted while keeping the capacity estimates of the ASGPR system fixed. The longer the compound circulates in the blood stream, the higher the chance that the ASGPR clears the compound from the circulation. With regards to long-lasting PK, antibodies are very good candidates and it is likely that other ligand-conjugated drugs may have faster unspecific clearance. Therefore, we examined the effect of a faster clearance rate on the delivery efficiency. Multiplying the clearance rate by 10 decreases the delivery efficiency (Fig. 7) compared to the ASGPR antibody (Fig. 6A). Therefore, the outcome of the delivery strategy is also dependent on the unspecific clearance of the compound.
Figure 7.
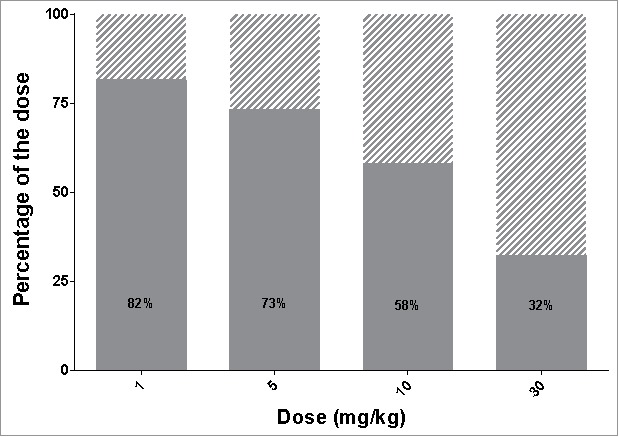
Influence of plasma sustainability on delivery efficiency. Delivery efficiency (grey area) and percentage of dose cleared via unspecific (non-target related) mechanisms (dashed area) in case of increased clearance rate (keL multiplied by a factor of 10).
Discussion
The primary goal for this PK study was to quantify the ASGPR-mediated uptake key parameters in vivo to assess the potential of the receptor for targeting delivery and allow optimization of protocol of administration. However, inferring the properties of the receptor from the plasma concentrations over time requires that all other processes (e.g., unspecific binding and clearance) are slower than the target-mediated processes.23 For this reason, a dedicated antibody is needed because targeted nucleic acids or natural ligands have rapid unspecific clearance and distribution processes in comparison. A human IgG1 specific antibody was therefore generated and selected for its specific binding to the stalk region of the sub-unit 1 of the ASGPR (ASGPR1). This antibody was also found to be non-specific to the closest homologue protein (CLEC10A), but cross reactive to the murine ASGPR1 protein, allowing study of its interaction with the ASGPR in mice.
This study describes how an adapted TMDD model was used to describe the antibody distribution and unspecific clearance, but also its interaction to the receptor and consequent degradation. However, in-silico TMDD models consist of a number of parameters that require fitting; therefore, this parameter estimation using experimental methods can be an issue and needs careful planning. Hence, we a priori selected sampling times that would help describe PK phenomena where time scales vary significantly between fast binding kinetics versus much slower distribution and elimination kinetics.21,32 The experimentally observed plasma concentrations suggest the antibody interaction with the target. First, a fast drop in concentrations was observed readily after a low IV dosing due to target binding. Additionally, saturation at the high dose is apparent by a transitory plateau in the PK profile. The dataset showed low inter-individual variability, allowing an accurate estimation of all parameters of the full TMDD model, including mean population and inter-individual variance.
Parameters of the unspecific PK, i.e., volume of central compartment, clearance and distribution, were found to be concordant with previously published values24 describing a bi-phasic linear and long-lasting antibody PK. Regarding the target-related parameters, the capacity of the ASGPR was estimated to be 36 nmol/kg of body weight. Conversion of the shuttle capacity to the number of receptors shows that 1.8 million receptors could be found at the cell surface. This in-vivo estimation isin the upper range of the reported expression values measured in vitro, from 0.5 to 1.5 million.14,15 It seems plausible that the in-vivo estimation would give a higher number than measured in vitro since the liver perfusion and cell-culture condition might impair the protein expression at the cell surface.33 It is of note that the ASGPR Ab was designed to be specific to the sub-unit 1 of the receptor (ASGPR1). However, the ASGPR is a hetero-oligomer composed of two sub-units H1 and H2,34 and we cannot exclude the possibility that the antibody interacts with a non-oligomerized H1 subunit that would inflate the estimation of the ASGPR expression. Nevertheless, by using a radiolabeled asialoglycoprotein analog, the ASGPR capacity has been estimated in vivo in healthy patients to be between 0.25 to 0.63 µmol.18,35,36 Miki and colleagues also converted this receptor capacity into 1.09 million receptors per hepatocyte by assessing the number of hepatocytes per liver,18 which is consistent with the estimate from this study (1.8 million/hepatocyte). Binding affinity (Kd) was estimated to be 4.1 nM, which is similar to the in-vitro estimation of 1.5 nM, derived from FRET analysis on transiently transfected cells expressing a transmembrane ASGPR1 protein. The PK analysis also allowed estimation of turnover dynamics for the free receptors at the cell surface. This turnover is defined mathematically, such as at baseline, and when no ligand is present the receptor concentration is in equilibrium. The half-life of the degradation of the receptor was found to be equal to 15 hours, which is in line with previously published in vitro estimates of between 12 and 20 hours.37,38 The synthesis rate, a zero-order rate, was estimated to be 29.8 nM/hour. This synthesis rate represents different contributing processes such as protein synthesis, internalization, recycling of internalized receptors back to the cell surface and mobilization of the intracellular receptor pool. The intracellular pool has been previously estimated to be as large as the number of receptors present at the cell surface.39 The antibody-ASGPR complex is internalized with a half-life of approximately 5 days; consistent with previous reports in the literature.40 This suggests that the rate-limiting step in the ASGPR-driven uptake is the internalization of the antibody-ASGPR complex. Therefore, overall the parameters and estimates from the TMDD model show good agreement with literature findings and in-vitro measured affinity (Kd). In addition, liver content after IV bolus dose was correctly predicted by the TMDD model.
The TMDD model describes the main underlying biological processes responsible for liver uptake. Therefore, it can be used to investigate the influence of the protocol of administration on the delivery outcome. As the delivery efficiency is dependent on the extent and duration of receptor saturation, dose level and dosing frequency must be tailored to minimize it. The delivery efficiency has been investigated by varying the dose and the dosing frequency. These simulations depict the dynamic response of the receptor to multiple administrations and help with finding the best administration protocol to deliver enough material while avoiding surpassing the shuttle capacity. The duration of receptor depletion can be long when administering saturating dose of antibody. For example, during 5 days after a 30 mg/kg dose, the free receptor pool only recovers 20% of the baseline. Therefore frequency of dosing must be tailored to maintain high delivery efficiency. From another perspective, the model allows the prediction of the duration of receptor saturation, which might also be important for assessing how long the compound interferes with the endogenous function of the receptor. Therefore, model-based dose optimization is useful both to ensure delivery efficiency while avoiding excessive saturation of the receptor.
The outcome of a targeting strategy is also dependent on non-target related PK properties, especially the unspecific clearance. In this study, ASGPR Ab conferred high affinity to the target (dissociation constant in the nanomolar range), but also long plasma exposure in comparison with other modalities such as small molecules or RNAi therapeutics. Therefore, high levels of delivery efficiency can be achieved. However, increasing the unspecific clearance rate impairs the capacity of the ASGPR to reach a high percentage of internalization. The TMDD is thus useful to investigate the effect of the unspecific clearance rate on the delivery performance. To further support these findings, we used a PK model developed for an oligonucleotide (ApoB targeting 13-mer LNA gapmer)41 and compared the delivery efficiency of the oligonucleotide to the reference antibody (see supplementary Appendix 4). After single IV dose, the ASGPR Ab reaches 96% when the oligonucleotide yields 68% at maximum (see supplementary figure 9A). However, this maximum delivery efficiency only takes into account the delivery through the ASGPR, and not the liver uptake specific to the unconjugated oligonucleotide. In addition, dosing the ApoB oligonucleotide at high dose (10 mg/kg) did not yield receptor saturation for long (less than 72 hours) (see supplementary figure 9B). As a consequence, the model predicts that frequent dosing may not impair delivery efficiency at therapeutic doses. The TMDD model represents therefore a tool to optimize a dosing regimen, i.e., dose level and dosing frequency, of different therapeutic modalities.
Our studies were performed in mice, but extrapolation from preclinical species to human is of interest. The ASGPR has been well conserved during evolution and has been found in human,18,42 rat (80% homology to the respective human subunit 1),43 rabbit44 and chicken.45 However, the extent to the conservation of turnover dynamics requires further study. Furthermore, patients with liver diseases may be anticipated to show reduced ASGPR expression, or function may be impaired at the cell surface.18 While it seems that ASGPR is still expressed in diseased liver tissue, the expression can be variable between patients,35,36,46 which suggests that a companion diagnostic to determine eligibility to treatment might be needed.47 The full TMDD model robustly predicted the liver uptake and the ASGPR Ab was found very specific to the receptor; however, we cannot discount the possibility that some receptor binding was on non-parenchymal cells such as Kupffer cells or endothelial cells. Further investigations are necessary to confirm that the antibody distributes in hepatocytes, and not also in parenchymal cells.
In conclusion, this study details the generation of a novel antibody directed against the ASGPR as a tool for estimating for the first time the shuttle capacity of the ASGPR receptor in vivo in mice. The study reveals a strong TMDD due to the binding of the antibody to the highly expressed ASGPR (36 nmol/kg of body weight). The in-silico TMDD model was also used to estimate the receptor turnover dynamics, i.e., its synthesis and degradation, and the internalization rate. These key kinetic parameters allow prediction of the liver uptake. A biodistribution study was performed and confirmed the accuracy of the model predictions showing that the TMDD processes indeed happen in the liver.
We show that receptor saturation by the antibody is associated with reduced delivery efficiency. The model is thus a useful tool to better appraise what parameters need to be considered for a tailored drug dosage regimens. This optimization can also be extended to different modalities, to incorporate the different PK properties (non-ASGPR related). Our TMDD model can be used to support the development of therapies that use the ASGPR as a shuttle into hepatocytes.
Materials and methods
Generation of ASGPR Ab and in-vitro characterization
The generation of a human ASGPR-specific antibody (ASGPR Ab) is described in Appendix 1 of the supplementary material. In brief, the identification of the ASGPR Ab was performed by phage display. The binding of the ASGPR Ab was tested against the whole extracellular domain (ECD) or the stalk region of the human subunit 1 of the ASGPR (ASGPR1). Isoform 2 of C-type lectin domain family 10 member A (CLEC10A) is the closest homologue of ASGPR protein. In order to exclude cross-reactivity of ASGPR Ab to CLEC10A, its binding was also tested against the ECD and the stalk region of CLEC10A.
In order to assess the cross-reactivity of the human ASGPR antibody to the murine ASGPR1, avidity-mediated binding to its epitope on ASGPR-expressing cells was determined by FRET analysis. For this analysis, the DNA sequence encoding the SNAP Tag (plasmid purchased from Cisbio) was fused to the C-terminal end of the full length human ASGPR1 sequence. The plasmid encoding the resulting fusion protein was transiently transfected into HEK293 cells. After an incubation time of 20 hours, cells were washed with phosphate-buffered saline (PBS) and incubated for 1 h at 37°C in LabMed buffer (Cisbio) containing 100 nM SNAP-Lumi4 Tb (Cisbio), leading to specific labeling of the SNAP Tag. Subsequently, cells were washed 4 times with LabMed buffer to remove unbound dye. Avidity of the ASGPR Ab was measured by adding the antibody at concentrations ranging from 0.39–50 nM to labeled cells (100 cells per well) followed by addition of anti-huFc-d2 (final 200 nM per well, Cisbio Catalog number 61HFCDAA) as acceptor molecule for the FRET. After an incubation time of three hours at room temperature, the emission of the acceptor dye (665 nm) as well as of the donor dye (620 nm) was determined using a fluorescence Reader (Victor 3, Perkin Elmer). The ratio of acceptor to donor emission was calculated and the ratio of the background control (cells with anti-huFc-d2) subtracted. The curve was analyzed in GraphPad Prism 5 and the dissociation constant (KD) was calculated.
Antibody solutions and radiochemistry
For the purpose of performing PK studies, ASGPR antibody solutions were formulated in 20 mM His/HisCl and 140 mM NaCl, at concentration of 8.5 mg/ml. A biodistribution study was also performed to assess the liver accumulation of ASGPR antibodies in comparison with a non-targeting antibody, a recombinant, humanized immunoglobulin G1 monoclonal antibody directed against interleukin IL-17A (Roche compound). Both antibodies were labeled through random modification of lysine residues followed by conjugation to 1,4,7,10-tetraazacyclododecane- N,N′,N,”N”'-tetraacetic acid (DOTA) for indium-111 (111In) complexation.31,48 The radiolabeled proteins were purified using PD-10 column (GE Healthcare Life Sciences,) pre-equilibrated in PBS. All radiolabeled antibodies were characterized by size-exclusion high performance liquid chromatography with in-line radiometric and UV detectors to compare the profiles of radioimmunoconjugates and corresponding unlabeled antibodies.
Animals and treatments
Male C57BL/6 mice were used for all in-vivo studies. In a first PK study, 12 mice (28–32g) were randomly assigned to 4 dose levels groups. The ASGPR antibody was administered to the mice via a single dose bolus IV injection in the tail vein at a dose of 1, 5, 10 or 30 mg/kg. A second PK study was conducted where mice received a single subcutaneous (SC) injection of either 1 or 30 mg/kg. Three animals were assigned in the dose group of 1 mg/kg, and 4 animals in the 30 mg/kg group, allowing a composite PK profile. PK studies were conducted according to applicable guidelines and approved by Swiss authority. The animal laboratory is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
A third study was conducted to assess liver distribution. Animal experiments were conducted at Chelatec, France, in accordance with the European Council Directive 2010/63/EU. Experimental plan was preliminary reviewed and approved by the local ethical committee “Comité d'Ethique en Expérimentation Animale des Pays de La Loire – C2EA-06”, and the authorization was then delivered by the Ministry of Research. Mice received either the ASGPR-targeting antibody (group 1) or a non-targeting antibody, a recombinant, humanized immunoglobulin G1 directed against IL-17 (group 2). Both groups received a 10 mg/kg dose bolus IV injection of the respective antibody in the tail vein. Both antibodies were radiolabeled with 111In-DOTA (30 µCi), a residualizing radiolabel (2.8 days decay life). Nine mice (24–27.6 g) were allocated to each group. For all studies, mice were fed ad libitum and kept under controlled conditions.
Sample collection and analysis
For the PK studies, after IV or SC injection and using micro-sampling technique, 16 plasma samples per animal were withdrawn from the tail vein. Sampling times were chosen to cover both the rapid initial decay phase and the long elimination of the antibody to obtain a rich data set in order to contain all necessary information for parameter estimation.21 Plasma samples were analyzed via a serial sandwich enzyme-linked immunosorbent assay using human Fc-specific derivatized capture and detection antibodies, as previously described by Stubenrauch et al.49 The limit of quantitation for the assay was defined as the lowest spiked plasma sample (7.8 ng/ml) that demonstrated acceptable accuracy and precision.
For the distribution study, liver samples were terminally harvested at 0.5, 8 and 24 hours. Liver samples were rinsed with PBS, blot-dried, weighed, and stored at +4°C until radiometric analysis. Radioactivity counts were measured in liver samples using a 2470 Wizard 2 automatic gamma counter (Perkin Elmer). Radioactive counts were converted to dose-normalized concentrations by calculating the percentage of injected dose per gram of tissue. It should be noted that no mathematical correction was applied to account for residual blood from liver samples. Liver samples were harvested at early time points, i.e., before 24 h, to prevent a bias estimate of accumulation in liver due to degradation of the antibody and marker (physical decay) in the tissue.
Target-mediated disposition model
A mechanistically based mathematical modeling approach was used for PK data analysis. The model was based on a generalized PK model for drugs exhibiting TMDD as originally described by Mager and Jusko in 2001.19 The TMDD model equations provide a framework for quantitative description of the underlying biological processes. Figure 3 illustrates the model; equations are provided in the supplementary information (Appendix 5).
Briefly, antibodies distribute in a linear process to a peripheral compartment ( and inter-compartmental clearance, ) and undergo an unspecific elimination (elimination rate, ) from the central compartment (volume, ). In addition, antibodies bind (dissociation constant, and dissociation rate, ) to free ASGPR targets in the central compartment to form a receptor-antibody complex (, concentration). It is assumed that one antibody binds a single ASGPR molecule. Once formed, the complex can either dissociate () or be internalized (, internalization rate). The unbound receptor is also synthesized () and degraded () so that the receptor is at a dynamic equilibrium before dosing (.
Software and parameters estimation
Parameters were estimated using a non-linear mixed-effect approach in MONOLIX 4.3.3. Inter-subject variability was estimated for all parameters, unless stated, and a residual proportional error model was used to describe the remaining differences between observed data and model-simulated profiles. Simulations have been performed with the final PK model and mean parameter estimates using Simbiology 5.4 in Matlab® R2016a. The model parameters used for simulation are presented in Table 1.
Simulation experiment using the TMDD model for optimization of administration protocol
A simulation experiment was performed using the TMDD model. First, we defined the delivery efficiency as the percentage of the total given dose that is internalized via the target-mediated pathway:
| (1) |
| (2) |
By contrast, the rest of the dose that is not internalized is distributed in the rest of body (off-target distribution), and subsequently eliminated via unspecific mechanisms (unspecific clearance). We also define the recovery as the percentage of free receptor, which is defined by comparison to the receptor baseline level (Rbase).
| (3) |
Delivery efficiency and percentage of free receptors have been simulated for all tested doses after IV bolus. To test the influence of the extent and duration of receptor saturation on delivery efficiency, we also performed simulations for 2 successive doses varying the dose level and time between the two doses. Finally, simulations were performed assuming a 10-fold increase clearance rate, to account for unspecific PK properties.
Prediction of biodistribution study results
To compare the predictions from the TMDD model to the biodistribution study results, we simulated the predicted liver content at all sampling times. However, the harvested liver samples contain antibodies in three different states, i.e., free, bound and internalized antibodies. Therefore, the liver content (L) was simulated as follows:
| (4) |
with being the liver weight (1.8 g),22 the free ASGPR Ab concentration in the central compartment, RL the concentration of bound antibodies, the residual blood volume in liver (1.5 mL),50 the volume of the central compartment.
Supplementary Material
Disclosure of potential conflicts of interest
The authors declare no competing interests in relation to the work described here. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgments
We sincerely thank Dr. S. Lohman from Hoffmann-La Roche for performing the bioanalytical assay of the PK studies. Furthermore, we thank Chelatec for performing the biodistribution study. Finally, we thank Dr. Hans-Peter Grimm, Miro Eigenmann and Dr. Nicolas Frances for reviewing the paper.
References
- 1.Kennedy PJ, Oliveira C, Granja PL, and Sarmento B. Antibodies and associates: Partners in targeted drug delivery. Pharmacol Ther. 2017;177:129-145. doi: 10.1016/j.pharmthera.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Park K. Controlled drug delivery systems: past forward and future back. J Control Release. 2014;190:3-8. doi: 10.1016/j.jconrel.2014.03.054. PMID:24794901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J and Shui SL. Delivery methods for site-specific nucleases: Achieving the full potential of therapeutic gene editing. J Control Release. 2016;244(Pt A):83-97. doi: 10.1016/j.jconrel.2016.11.014. PMID:27865852. [DOI] [PubMed] [Google Scholar]
- 4.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44(14):6518-6548. doi: 10.1093/nar/gkw236. PMID:27084936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Souza AA and Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release. 2015;203:126-39. doi: 10.1016/j.jconrel.2015.02.022. PMID:25701309. [DOI] [PubMed] [Google Scholar]
- 6.Ashwell G and Morell AG. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv Enzymol Relat Areas Mol Biol. 1974;41(0):99-128. PMID:4609051.2745406 [DOI] [PubMed] [Google Scholar]
- 7.Weiss P and Ashwell G. Ligand-induced modulation of the hepatic receptor for asialoglycoproteins. Evidence for the role of cell surface hyposialylation. J Biol Chem. 1989;264(20):11572-4. PMID:2745406. [PubMed] [Google Scholar]
- 8.Hubbard AL and Stukenbrok H. An electron microscope autoradiographic study of the carbohydrate recognition systems in rat liver. II. Intracellular fates of the 125I-ligands. J Cell Biol. 1979;83(1):65-81.doi: 10.1083/jcb.83.1.65. PMID:511942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Windler E, Greeve J, Levkau B, Kolb-Bachofen V, Daerr W, and Greten H. The human asialoglycoprotein receptor is a possible binding site for low-density lipoproteins and chylomicron remnants. Biochem J. 1991;276 (Pt 1):79-87.doi: 10.1042/bj2760079. PMID:1645533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rotundo RF, Vincent PA, McKeown-Longo PJ, Blumenstock FA, and Saba TM. Hepatic fibronectin matrix turnover in rats: involvement of the asialoglycoprotein receptor. Am J Physiol. 1999;277(6 Pt 1):G1189-99. PMID:10600816. [DOI] [PubMed] [Google Scholar]
- 11.Dini L, Autuori F, Lentini A, Oliverio S, and Piacentini M. The clearance of apoptotic cells in the liver is mediated by the asialoglycoprotein receptor. FEBS Lett. 1992;296(2):174-8. doi: 10.1016/0014-5793(92)80373-O. PMID:1370803. [DOI] [PubMed] [Google Scholar]
- 12.Roccatello D, Picciotto G, Torchio M, Ropolo R, Ferro M, Franceschini R, Quattrocchio G, Cacace G, Coppo R, Sena LM, et al.. Removal systems of immunoglobulin A and immunoglobulin A containing complexes in IgA nephropathy and cirrhosis patients. The role of asialoglycoprotein receptors. Lab Invest. 1993;69(6):714-23. PMID:8264233. [PubMed] [Google Scholar]
- 13.Weigel PH. Galactosyl and N-acetylgalactosaminyl homeostasis: a function for mammalian asialoglycoprotein receptors. Bioessays. 1994;16(7):519-24. doi: 10.1002/bies.950160713. PMID:7945281. [DOI] [PubMed] [Google Scholar]
- 14.Weigel PH and Oka JA. The large intracellular pool of asialoglycoprotein receptors functions during the endocytosis of asialoglycoproteins by isolated rat hepatocytes. J Biol Chem. 1983;258(8):5095-102. PMID:6300113. [PubMed] [Google Scholar]
- 15.Rice KG and Lee YC. Oligosaccharide valency and conformation in determining binding to the asialoglycoprotein receptor of rat hepatocytes. Adv Enzymol Relat Areas Mol Biol. 1993;66:41-83. PMID:8430516. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz AL, Fridovich SE, Knowles BB, and Lodish HF. Characterization of the asialoglycoprotein receptor in a continuous hepatoma line. J Biol Chem. 1981;256(17):8878-81. PMID:6267054. [PubMed] [Google Scholar]
- 17.Li Y, Huang G, Diakur J, and Wiebe LI. Targeted delivery of macromolecular drugs: asialoglycoprotein receptor (ASGPR) expression by selected hepatoma cell lines used in antiviral drug development. Curr Drug Deliv. 2008;5(4):299-302. doi: 10.2174/156720108785915069. PMID:18855599. [DOI] [PubMed] [Google Scholar]
- 18.Miki K, Kubota K, Inoue Y, Vera DR, and Makuuchi M. Receptor measurements via Tc-GSA kinetic modeling are proportional to functional hepatocellular mass. J Nucl Med. 2001;42(5):733-7. PMID:11337568. [PubMed] [Google Scholar]
- 19.Mager DE and Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28(6):507-32. doi: 10.1023/A:1014414520282. PMID:11999290. [DOI] [PubMed] [Google Scholar]
- 20.Krippendorff BF, Kuester K, Kloft C, and Huisinga W. Nonlinear pharmacokinetics of therapeutic proteins resulting from receptor mediated endocytosis. J Pharmacokinet Pharmacodyn. 2009;36(3):239-60. doi: 10.1007/s10928-009-9120-1. PMID:19554432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peletier LA and Gabrielsson J. Dynamics of target-mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn. 2012;39(5):429-51. doi: 10.1007/s10928-012-9260-6. PMID:22851162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies B and Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093-5. doi: 10.1023/A:1018943613122. PMID:8378254. [DOI] [PubMed] [Google Scholar]
- 23.Dirks NL and Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633-59. doi: 10.2165/11535960-000000000-00000. PMID:20818831. [DOI] [PubMed] [Google Scholar]
- 24.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, and Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs. 2011;3(1):61-6. doi: 10.4161/mabs.3.1.13799. PMID:20962582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myzithras M, Bigwarfe T, Li H, Waltz E, Ahlberg J, Giragossian C, and Roberts S. Utility of immunodeficient mouse models for characterizing the preclinical pharmacokinetics of immunogenic antibody therapeutics. MAbs. 2016;8(8):1606-1611. doi: 10.1080/19420862.2016.1229721. PMID:27598372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan YY, Dowty ME, Zhang M, Zhang Y, Cheng A, et al.. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8(6):1064-78. doi: 10.1080/19420862.2016.1193660. PMID:27232760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng R, Meng YG, Hoyte K, Lutman J, Lu Y, Iyer S, DeForge LE, Theil FP, Fielder PJ, and Prabhu S. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. MAbs. 2012;4(1):101-9. doi: 10.4161/mabs.4.1.18543. PMID:22327433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sohlenius-Sternbeck AK. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro. 2006;20(8):1582-6. doi: 10.1016/j.tiv.2006.06.003. PMID:16930941. [DOI] [PubMed] [Google Scholar]
- 29.Kagan L. Pharmacokinetic modeling of the subcutaneous absorption of therapeutic proteins. Drug Metab Dispos. 2014;42(11):1890-905. doi: 10.1124/dmd.114.059121. [DOI] [PubMed] [Google Scholar]
- 30.Eigenmann MJ, Fronton L, Grimm HP, Otteneder MB, and Krippendorff BF. Quantification of IgG monoclonal antibody clearance in tissues. MAbs. 2017;9(6):1007-1015. doi: 10.1080/19420862.2017.1337619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip V, Palma E, Tesar DB, Mundo EE, Bumbaca D, Torres EK, Reyes NA, Shen BQ, Fielder PJ, Prabhu S, et al.. Quantitative cumulative biodistribution of antibodies in mice: effect of modulating binding affinity to the neonatal Fc receptor. MAbs. 2014;6(3):689-96. doi: 10.4161/mabs.28254. PMID:24572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eudy RJ, Riggs MM, and Gastonguay MR. A Priori Identifiability of Target-Mediated Drug Disposition Models and Approximations. AAPS J. 2015;17(5):1280-4. doi: 10.1208/s12248-015-9795-8. PMID:26077506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Severgnini M, Sherman J, Sehgal A, Jayaprakash NK, Aubin J, Wang G, Zhang L, Peng CG, Yucius K, Butler J, et al.. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 2012;64(2):187-95. doi: 10.1007/s10616-011-9407-0. PMID:22105762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Massarelli I, Chiellini F, Chiellini E, and Bianucci AM. Three-dimensional models of the oligomeric human asialoglycoprotein receptor (ASGP-R). Int J Mol Sci. 2010;11(10):3867-84. doi: 10.3390/ijms11103867. PMID:21152305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudo M, Todo A, Ikekubo K, Yamamoto K, Vera DR, and Stadalnik RC. Quantitative assessment of hepatocellular function through in vivo radioreceptor imaging with technetium 99m galactosyl human serum albumin. Hepatology. 1993;17(5):814-9. PMID:8491449. [PubMed] [Google Scholar]
- 36.Pimstone NR, Stadalnik RC, Vera DR, Hutak DP, and Trudeau WL. Evaluation of hepatocellular function by way of receptor-mediated uptake of a technetium-99m-labeled asialoglycoprotein analog. Hepatology. 1994;20(4 Pt 1):917-23. doi: 10.1002/hep.1840200421. PMID:7927233. [DOI] [PubMed] [Google Scholar]
- 37.Warren R and Doyle D. Turnover of the surface proteins and the receptor for serum asialoglycoproteins in primary cultures of rat hepatocytes. J Biol Chem. 1981;256(3):1346-55. PMID:6256393. [PubMed] [Google Scholar]
- 38.Bischoff J and Lodish HF. Two asialoglycoprotein receptor polypeptides in human hepatoma cells. J Biol Chem. 1987;262(24):11825-32. PMID:3040719. [PubMed] [Google Scholar]
- 39.Schwartz AL. Trafficking of asialoglycoproteins and the asialoglycoprotein receptor. Targeted Diagn Ther. 1991;4:3-39. PMID:1797161. [PubMed] [Google Scholar]
- 40.Vexler V, Yu L, Pamulapati C, Garrido R, Grimm HP, Sriraman P, Bohini S, Schraeml M, Singh U, Brandt M, et al.. Target-mediated drug disposition and prolonged liver accumulation of a novel humanized anti-CD81 monoclonal antibody in cynomolgus monkeys. MAbs. 2013;5(5):776-86. doi: 10.4161/mabs.25642. PMID:23924796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimizu R, Kitade M, Kobayashi T, Hori S, and Watanabe A. Pharmacokinetic-pharmacodynamic modeling for reduction of hepatic apolipoprotein B mRNA and plasma total cholesterol after administration of antisense oligonucleotide in mice. J Pharmacokinet Pharmacodyn. 2015;42(1):67-77. doi: 10.1007/s10928-014-9398-5. PMID:25376372. [DOI] [PubMed] [Google Scholar]
- 42.Baenziger JU and Maynard Y. Human hepatic lectin. Physiochemical properties and specificity. J Biol Chem. 1980;255(10):4607-13. PMID:7372599. [PubMed] [Google Scholar]
- 43.Spiess M and Lodish HF. Sequence of a second human asialoglycoprotein receptor: conservation of two receptor genes during evolution. Proc Natl Acad Sci U S A. 1985;82(19):6465-9.doi: 10.1073/pnas.82.19.6465. PMID:3863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardy MR, Townsend RR, Parkhurst SM, and Lee YC. Different modes of ligand binding to the hepatic galactose/N-acetylgalactosamine lectin on the surface of rabbit hepatocytes. Biochemistry. 1985;24(1):22-8. doi: 10.1021/bi00322a004. PMID:3994969. [DOI] [PubMed] [Google Scholar]
- 45.Loeb JA and Drickamer K. The chicken receptor for endocytosis of glycoproteins contains a cluster of N-acetylglucosamine-binding sites. J Biol Chem. 1987;262(7):3022-9. PMID:3818631. [PubMed] [Google Scholar]
- 46.Shi B, Abrams M, and Sepp-Lorenzino L. Expression of asialoglycoprotein receptor 1 in human hepatocellular carcinoma. J Histochem Cytochem. 2013;61(12):901-9. doi: 10.1369/0022155413503662. PMID:23979840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Witzigmann D, Quagliata L, Schenk SH, Quintavalle C, Terracciano LM, and Huwyler J. Variable asialoglycoprotein receptor 1 expression in liver disease: Implications for therapeutic intervention. Hepatol Res. 2016;46(7):686-96. doi: 10.1111/hepr.12599. PMID:26422581. [DOI] [PubMed] [Google Scholar]
- 48.Pastuskovas CV, Mundo EE, Williams SP, Nayak TK, Ho J, Ulufatu S, Clark S, Ross S, Cheng E, Parsons-Reponte K, et al.. Effects of anti-VEGF on pharmacokinetics, biodistribution, and tumor penetration of trastuzumab in a preclinical breast cancer model. Mol Cancer Ther. 2012;11(3):752-62. doi: 10.1158/1535-7163.MCT-11-0742-T. PMID:22222630. [DOI] [PubMed] [Google Scholar]
- 49.Stubenrauch K, Wessels U, and Lenz H. Evaluation of an immunoassay for human-specific quantitation of therapeutic antibodies in serum samples from non-human primates. J Pharm Biomed Anal. 2009;49(4):1003-8. doi: 10.1016/j.jpba.2009.01.030. PMID:19250787. [DOI] [PubMed] [Google Scholar]
- 50.Shah DK and Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67-86. doi: 10.1007/s10928-011-9232-2. PMID:22143261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.