

Abstract
Hematopoietic stem/progenitor cells in the adult mammalian bone marrow ensure blood cell renewal. Their cellular microenvironment, called ‘niche’, regulates hematopoiesis both under homeostatic and immune stress conditions. In the Drosophila hematopoietic organ, the lymph gland, the posterior signaling center (PSC) acts as a niche to regulate the hematopoietic response to immune stress such as wasp parasitism. This response relies on the differentiation of lamellocytes, a cryptic cell type, dedicated to pathogen encapsulation and killing. Here, we establish that Toll/NF-κB pathway activation in the PSC in response to wasp parasitism non-cell autonomously induces the lymph gland immune response. Our data further establish a regulatory network where co-activation of Toll/NF-κB and EGFR signaling by ROS levels in the PSC/niche controls lymph gland hematopoiesis under parasitism. Whether a similar regulatory network operates in mammals to control emergency hematopoiesis is an open question.
Research organism: D. melanogaster
Introduction
In adult mammals, Hematopoietic Stem and Progenitor Cells (HSPCs) present in the bone marrow are at the origin of blood cell production throughout an individual’s life. Self-renewal, proliferation and differentiation of HSPCs are under the control of the bone marrow microenvironment called the ‘niche’ which has emerged as a key regulator of hematopoiesis (Calvi and Link, 2015; Morrison and Scadden, 2014). While considerable effort has been directed toward uncovering the mechanisms that regulate HSPC/niche communication under normal conditions, processes that take place in response to infection and inflammation remain more elusive (Kobayashi et al., 2016; Zhao and Baltimore, 2015). With the discovery that many transcriptional regulators and signaling pathways controlling blood cell development are conserved between insects and humans, Drosophila has become an important model to investigate hematopoiesis under normal conditions and in response to immune stress (Letourneau et al., 2016; Martinez-Agosto et al., 2007).
Immune cells in Drosophila, called hemocytes, contribute to innate immune defense (Lemaitre and Hoffmann, 2007). Two phases of hematopoiesis occur in Drosophila (Gold and Brückner, 2015; Holz et al., 2003). A first wave of immune cell production takes place during embryogenesis and generates two types of hemocytes, plasmatocytes and crystal cells, which are involved in phagocytosis and melanisation/wound healing, respectively (Lemaitre and Hoffmann, 2007). Both cell types increase in numbers at larval stages and either circulate in the hemolymph or aggregate into pockets under the epidermis (sessile hemocytes) (Makhijani et al., 2011; Márkus et al., 2009). A second wave of de novo hematopoiesis takes place in a larval specialized hematopoietic organ, the lymph gland, a multi-lobed organ which develops along the anterior cardiac tube, the aorta region (Jung et al., 2005; Krzemien et al., 2010a; Lanot et al., 2001). During the third larval stage, the anterior lobes of the mature lymph gland are composed of a medullary zone (MZ) containing progenitors (prohemocytes), a cortical zone (CZ) composed of differentiating plasmatocytes and crystal cells, and the posterior signaling center (PSC) (Jung et al., 2005). During metamorphosis, lymph gland rupture releases all hemocytes into circulation (Grigorian et al., 2011; Lanot et al., 2001). A third blood cell type, called lamellocyte, differentiates in emergency situations, such as wasp parasitism, from lymph gland progenitors and circulating/sessile hemocytes (Anderl et al., 2016; Crozatier et al., 2004; Honti et al., 2010; Lanot et al., 2001; Márkus et al., 2009; Stofanko et al., 2010). Lamellocytes are specialized hemocytes which mediate the encapsulation and killing of pathogens too large to be phagocytosed. Following wasp egg laying inside the Drosophila larva, the egg is recognized as a foreign body. This leads to differentiation of lamellocytes at the expense of other immune cells (Honti et al., 2010; Krzemień et al., 2007; Márkus et al., 2009; Stofanko et al., 2010), the premature dispersal of the lymph gland and the release of lamellocytes into the hemolymph (Crozatier et al., 2004; Sorrentino et al., 2002).
The PSC plays a major role to control the immune response to wasp parasitism (Benmimoun et al., 2015; Crozatier et al., 2004; Krzemień et al., 2007; Oyallon et al., 2016; Sinenko et al., 2011). In the absence of PSC, such as in collier (col/kn/ebf) mutants, or upon its genetic ablation, massive differentiation of lamellocytes fails to occur. It was recently established that wasp parasitism increases Reactive Oxygen Species (ROS) levels in PSC cells, leading to the secretion of Spitz (Spi), one ligand of the Epidermal Growth Factor Receptor (EGFR) signaling pathway (Sinenko et al., 2011). These authors proposed that Spi released from PSC cells activates the EGFR pathway in circulating hemocytes, inducing them to differentiate into lamellocytes.
Several independent studies established that the evolutionarily conserved Toll/NF-κB pathway regulates Drosophila hematopoiesis both under homeostatic and wasp parasitism conditions (Gao et al., 2016; Gueguen et al., 2013; Matova and Anderson, 2006, 2010; Qiu et al., 1998; Schmid et al., 2014; Sorrentino et al., 2004). However, how Toll/NF-κB controls wasp egg encapsulation remains unknown. Here, we show that this pathway is activated in PSC cells in response to egg laying by Leptopilina boulardi wasps and non-cell autonomously controls lamellocyte differentiation in the lymph gland. This, in turn, leads to lymph gland disruption and in fine to successful wasp egg encapsulation. We establish that a regulatory network linking ROS to Toll/NF-κB in the PSC and to EGFR/Erk signaling in lymph gland progenitors controls the lymph gland response to parasitism and the success of wasp egg neutralization.
Results
Toll/NF-κB signaling controls the lymph gland response to wasp infection
In Drosophila, Toll/NF-κB pathway activation by the ligand Spätzle (Spz) leads to the recruitment of an adaptor complex that consists of three death domain-containing proteins, Myd88, Tube, and Pelle (Pll). This complex triggers phosphorylation and degradation of the IκB factor Cactus (Cact) and ultimately the release of the NF-κB transcription factors Dorsal (Dl) and Dorsal-related immunity factor (Dif) that translocate into the nucleus where they activate target gene transcription (Lemaitre and Hoffmann, 2007). When parasitized by the wasp Leptopilina boulardi, mutant larvae for either Toll (Tl), tube or pll fail to encapsulate wasp eggs (Sorrentino et al., 2004). To determine whether this failure is linked to impaired lymph gland response to parasitism, we analyzed lymph gland disruption in mutants of the Toll/NF-κB pathway. 30 hr post-wasp egg laying, 93% of wild-type (wt) larvae lymph glands are disrupted, that is, lymph gland cells are released into circulation (Figure 1A,C). In contrast, in pll2/pll7 mutants, 98% of the lymph glands remain intact (Figure 1B,C). Likewise, more than half of lymph glands are intact in parasitized Dif1 mutant, while the others start disrupting, that is, only some cells at the periphery of the anterior lobes disperse (Figure 1D,F). At a later time point, 48 hr post-parasitism, Dif and pll mutant lymph glands have disrupted (Figure 1H,I), similar to previously reported for Tl and tube mutants (Sorrentino et al., 2004). dl1 lymph glands, however, disrupt as in control larvae (Figure 1E,G). Corroborating these findings, lymph gland dispersal is delayed 30 hr post-parasitism in Dif1/Df(2L)J4 larvae - Df(2L)J4 removes both Dl and Dif - compared to +/Df(2L)J4 controls and dl1/Df(2L)J4 larvae (Figure 1—figure supplement 1). Delayed lymph gland disruption is also observed in Dif1 but not dl1 isogenic mutant larvae, further ruling out a genetic background effect (Figure 1—figure supplement 1). Collectively, these data show that Toll/NF-κB signaling is required for the proper timing of lymph gland dispersal in response to wasp infection and that Dif is the only NF-κB factor involved.
Figure 1. Dif-dependent Toll/NF-κB signaling is required for correct timing of lymph gland dispersal in response to wasp parasitism.
(A, B, D, E) Differential interference contrast (DIC) microscopy of lymph gland anterior lobes 30 hr post-parasitism. Lymph glands were classified into three groups (see Materials and methods): Disrupted (*) in wt (wild type, (A) or dl1 mutant (E); intact in pll2/pll7 mutant (B); disrupting in Dif1 mutant (D). Representative lymph glands are shown. Ring gland, RG; cardiac tube, CT; anterior lobe, AL; posterior lobe, PL. (C, F, G) Quantification of lymph gland disruption (%). The error bars correspond to SEM, ***p<0.001 and ns (not significant) (Pearson’s Chi-squared test). In this and subsequent similar analyses, numbers of lymph glands analyzed are indicated on histograms. (H, I) DIC microscopy of lymph glands in Dif1 (H) and pll2/pll7 (I) mutants 48 hr post-parasitism. Disrupted (*) and disrupting (arrows) lymph glands are shown. (J–M) Perlecan/Trol (white) immunostaining in control (J, L) and Dif1 anterior lobes (K, M). (N–S) αPS4 (green) immunostaining labels lamellocytes in lymph glands in wt (N, Q), Dif1 (O, R) and pll2/pll7 (P, S) mutants. 20 hr post-parasitism, more than 65% (n = 24) of wt lymph glands displayed lamellocytes, whereas none (n = 24) or less than 15% (n = 18) contained lamellocytes in Dif1 and pll2/pll7 mutants, respectively. 30 hr post-parasitism, Dif1 and pll2/pll7 mutant lymph glands have lamellocytes, whereas most wt lymph glands have disrupted. In this and subsequent figures (unless specified), the scale bar represents 20 µm and nuclei are labeled with TOPRO3 or Draq5.

Figure 1—figure supplement 1. Dif but not Dorsal is required for proper lymph gland disruption in response to wasp parasitism.

Lymph gland dispersal in response to wasp parasitism is associated with the loss of the basement membrane surrounding the organ, allowing the release of lamellocytes into circulation (Sorrentino et al., 2002). In contrast to control larvae (Figure 1J,L), immunostaining of parasitized Dif1 mutant lymph glands for an extracellular matrix (ECM) protein, the heparan sulfate proteoglycan Perlecan/Trol (Terribly Reduced Optic Lobes) (Dragojlovic-Munther and Martinez-Agosto, 2013; Grigorian et al., 2013) indicates the presence of a continuous ECM layer surrounding the anterior lobes 20 hr post parasitism (Figure 1K,M). Thus, the delayed lymph gland dispersal observed in Dif1 mutant is associated with basement membrane persistence. We then analyzed lamellocyte differentiation using α-PS4 immunostaining (Krzemień et al., 2007). 20 hr post-parasitism, lamellocytes are detected in control larvae (Figure 1N), whereas none are found in Dif1 and pll2/pll7 mutants (Figure 1O,P). 30 hr post-parasitism, while most control lymph glands have already released lamellocytes into circulation (Figure 1Q), few lamellocytes are detected in mutant lymph glands (Figure 1R–S). Altogether, these data show that Toll/NF-κB signaling, while not required for lamellocyte differentiation per se, is essential for the timely differentiation of lamellocytes in the lymph gland, breakdown of its basement membrane, and lamellocyte release into the hemolymph.
Lymph gland lamellocytes are required for successful wasp egg encapsulation
Upon parasitism, lamellocytes mediate wasp egg encapsulation, which is critical for host survival since it prevents hatching of wasp larvae (Crozatier et al., 2004). Wasp egg hatching, which reliably reflects the success of the immune response (Mortimer et al., 2013; Schmid et al., 2014; Vanha-Aho et al., 2015), is impaired in either Dif1 (Figure 2A–C, Figure 2—figure supplement 1) or pll2/pll21 and Tlrv1/Tlr mutants (Figure 2—figure supplement 1) (Sorrentino et al., 2004) but not in dl1 mutants (Figure 2—figure supplement 1), indicating that Dif-mediated activation of Toll/NF-κB pathway is required for wasp egg encapsulation. These data, together with the delay in lymph gland disruption observed in Dif and pll mutants (Figure 1), strongly suggest that lymph gland-derived lamellocytes are essential for successful wasp egg encapsulation. To strengthen this conclusion, we measured the percentage of circulating lamellocytes just prior, and immediately following lymph gland disruption. Before disruption, which occurs around 20 hr post-parasitism in control larvae and 30 hr post parasitism in Dif1 mutants, less than 10% of circulating hemocytes are lamellocytes (Figure 2D,F,G,I). This indicates that a few lamellocytes differentiate from sessile/circulating hemocytes prior to lymph gland dispersion. Following lymph gland disruption, however, more than 50% of circulating hemocytes are lamellocytes in both control and Dif1 mutant larvae (Figure 2E,F,H,I). We thus conclude that lymph gland disruption causes a rapid and major increase in the number of circulating lamellocytes and that this is delayed in Toll/NF-κB mutants. A delay in both lymph gland disruption (data not shown) and in the increase numbers of circulating lamellocytes, is observed in parasitized wild-type larvae raised at 18°C (Figure 2—figure supplement 2), indicating that the lymph gland response is temperature-dependent.
Figure 2. Dif, lymph gland lamellocyte differentiation and lymph gland disruption are required for wasp egg neutralization.
(A, B) Brightfield images of control (col>, A) and Dif1 mutant larvae 48 hr post parasitism (B). The arrow indicates a melanotic capsule. (C) Quantification (%) of wasp egg hatching in control (col>) and Dif1 mutant. In this and subsequent similar analyses, numbers of (infected) larvae analyzed are indicated on histograms. (D–I) Phalloidin (red) staining of circulating hemocytes in control (hml>, D, E) and Dif1 mutant (G, H) just prior (D, G) and following (E, H) lymph gland disruption. Arrows indicate discoidal-shaped lamellocytes that are larger than plasmatocytes. (F, I) Quantification (%) of circulating plasmatocytes and lamellocytes in control (F) and Dif1 mutant (I). Blue, TOPRO3. (J) Quantification of lymph gland disruption (%) in control larvae (dome-MESO>) and when latran is down-regulated in lymph gland progenitors (dome-MESO > lat RNAi). (K) Quantification of wasp egg hatching in control larvae (dome-MESO>) or (antp>) and when latran is down-regulated in lymph gland progenitors (dome-MESO > lat RNAi) or in PSC cells (antp > lat RNAi). Error bars correspond to SEM, ***p<0.001 and ns (not significant) (Pearson’s Chi-squared test).

Figure 2—figure supplement 1. Toll/NF-κB signaling is required for wasp egg encapsulation.

Figure 2—figure supplement 2. At 18°C, lymph gland disruption and the presence of lamellocytes in circulation are delayed.

To further investigate the contribution of lymph gland-derived lamellocytes to the success of wasp egg encapsulation, we specifically impaired lymph gland progenitor differentiation and analyzed the consequence on wasp egg encapsulation. We had previously established that the dominant-negative JAK/STAT receptor latran (lat) (Eye transformer [Kallio et al., 2010]), down-regulates JAK/STAT signaling in lymph gland progenitors following wasp attack, allowing massive differentiation of lymph gland lamellocytes (Makki et al., 2010). Accordingly, we found that knocking-down lat specifically in lymph gland progenitors using double-stranded RNA-mediated interference (RNAi) (dome-MESO > lat RNAi) impairs lymph gland dispersal post-parasitism (Figure 2J). Moreover, 90% of wasp eggs hatched when lat was inactivated in progenitor cells (dome-MESO > lat RNAi) compared to 10% in controls (dome-MESO>), whereas no defect was observed when lat was inactivated in PSC cells (antp > lat RNAi) (Figure 2K). Altogether, these data link lymph gland disruption and lamellocyte release into circulation to successful wasp egg encapsulation.
Fighting wasp parasitism requires Toll/NF-κB activation in PSC cells
Having established that Dif plays an essential role in the lymph gland response to wasp infection, we analyzed whether and in which lymph gland cells Dif activity is required. Immunostaining of wt lymph glands shows that Dif is expressed in PSC cells (Figure 3A–A’’) (Gueguen et al., 2013). 6 hr post-parasitism, it is also expressed in hematopoietic progenitors (Figure 3B–B’’). NF-κB transcriptional activity can be monitored by the expression of a reporter transgene, D4-LacZ, which consists of four tandemly repeated NF-κB-binding sites upstream of lacZ (Flores-Saaib et al., 2001). In agreement with published data, D4–lacZ is expressed in PSC cells in uninfected wt larvae and upregulated in these cells 6 hr post parasitism, in addition to being expressed de novo in lymph gland progenitors (Figure 3C–C’’, D–D’’, E) (Gueguen et al., 2013). In pll2/pll7 mutants, D4-LacZ expression is still observed in the PSC under normal conditions (Figure 3F–F’’), whereas it is not expressed in the PSC in Dif1 mutants (Figure 3I–I’’). This indicates a Toll-independent, basal activity of Dif. 6 hr post-parasitism, no upregulation of D4-LacZ occurs in PSC cells in either pll2/pll7 or Dif1 mutants (Figure 3G–G’’, J–J’’, quantification in Figure 3H). These data show that Toll/NF-κB signaling is activated in the PSC upon parasitism and that this activation requires Dif. Further validating this conclusion, we found that D4-LacZ is not upregulated 6 hr post-parasitism when Dif is specifically down-regulated in PSC cells by driving Dif dsRNA expression with the PSC driver pcol-Gal4 (col > Dif RNAi) (Figure 3K,L; Figure 3—figure supplement 1A,B). 6 hr post-parasitism, unlike in pll2/pll7 mutants (Figure 3G), low levels of D4-LacZ expression persist in Dif1 mutant lymph gland progenitors (compare Figure 3J and D), possibly reflecting Dorsal expression in these cells (Figure 3—figure supplement 1C,D) (Gueguen et al., 2013). Altogether, these data establish that Toll/NF-κB signaling is induced both in the PSC and in lymph gland progenitors in response to parasitism and that Dif is the major NF-κB transactivator involved.
Figure 3. Dif-dependent Toll/NF-kB activation in PSC cells.
(A, B) Dif (red) immunostaining of col > mCD8 GFP (col > GFP, green) lymph glands without (A) and 6 hr post-parasitism (B). (A’, A’’, B’, B’’) enlarged views of the PSC. (C, D, F, G, I, J, K, L) LacZ (red) and Col (green) immunostaining of D4–LacZ of wt (C, D), pll2/pll7 (F, G), Dif1 (I, J), and col > Dif RNAi (K, L) lymph glands without (C, F, I, K), and 6 hr post-parasitism (D, G, J, L). (C’, C'’, D’, D'’, F’, F'’, G’, G”, I’, I”, J’ and J”) enlarged views of the PSC. Arrows (K, L) indicate Col-positive PSC cells that do not express D4–LacZ. (E, H) Quantifications of D4-LacZ mean intensity in PSC cells. Error bars represent SDs, *p<0.1 and ns (not significant) (Mann-Whitney nonparametric test).

Figure 3—figure supplement 1. Dif, Dorsal and Drosomycin-GFP expression in lymph gland cells.

In order to distinguish between possible roles of Toll/NF-κB signaling in the PSC and progenitors following parasitism, we knocked down different components of the pathway, such as Dif (Figure 3—figure supplement 1A,B), pll and Myd88, by expressing RNAi in either PSC cells (col>) or in lymph gland progenitors using domeless-Gal4 (dome>). Toll/NF-κB down-regulation in MZ progenitors (dome > Myd88 RNAi; dome > cact > Dif RNAi) neither significantly reduced lymph gland dispersal (Figure 4—figure supplement 1C,D) nor lymph gland lamellocyte differentiation 20 hr post-parasitism (Figure 4—figure supplement 2A,B). By contrast, downregulation of Toll/NF-κB signaling in PSC cells (col > Dif RNAi; col > Myd88 RNAi; col > pll RNAi) delayed lymph gland dispersal post-parasitism (Figure 4A). Similar results were obtained when antp-Gal4 (antp>), another PSC driver, was used (Figure 4—figure supplement 1A,B). Next, we analyzed lymph gland lamellocyte differentiation 20 hr post-parasitism. Whereas lamellocytes were detected in control lymph glands, few, if any were found when Toll/NF-κB signaling was impaired in PSC cells (Figure 4B,C), corroborating our previous interpretation that lymph gland disruption is dependent on lamellocyte differentiation (Figure 1). Finally, we determined whether Toll/NF-κB signaling in PSC cells is required for successful wasp egg encapsulation. Whereas only 20% of wasp eggs hatched in control larvae (col>), 70% and 80% hatched when Myd88 or Dif were down-regulated in PSC cells (col > Myd88 RNAi; col > Dif RNAi), respectively (Figure 4D). Altogether, these data show that Toll/NF-κB signaling is required in PSC cells and not in lymph gland progenitors for the correct timing of lymph gland lamellocyte differentiation, which leads to lymph gland dispersal and thus a successful cellular immune response against wasp parasitism.
Figure 4. Toll/NF-kB activation in PSC cells controls lymph gland dispersal, lamellocyte differentiation and wasp egg encapsulation.
(A) Quantification (%) of lymph gland disruption post parasitism in col> (control), col > Dif RNAi, col > Myd88 RNAi and col > pll RNAi lymph glands. (B–C) Representative confocal images of lamellocyte differentiation 20 hr post-parasitism (β Integrin, red) in control lymph glands (col > GFP) (B) and when Toll/NF-kB is downregulated in PSC cells (col > GFP > Myd88 RNAi) (C). At least 60% (n = 44) of lymph glands displayed numerous lamellocytes in the control, whereas less than 15% (n = 41) are observed in col > Myd88 RNAi lymph glands. Note early signs of lymph gland disruption in (B, arrow). (D) Quantification (%) of wasp egg hatching 48 hr post-parasitism in col> (control), col > Myd88 RNAi and col > Dif RNAi lymph glands. Error bars correspond to SEM, ***p<0.001 (Pearson’s Chi-squared test).

Figure 4—figure supplement 1. Toll/NF-kB activation in PSC cells but not in lymph gland progenitors is required for proper lymph gland disruption in response to wasp parasitism.

Figure 4—figure supplement 2. Toll/NF-kB signaling is not required in lymph gland progenitors for lamellocyte differentiation.

Spätzle and SPE are required for Toll/NF-kB activation
Spätzle (Spz), the ligand of the Toll/NF-κB pathway, is synthesized and secreted as an inactive cytokine, and its activation depends on cleavage by the Spätzle Processing Enzyme (SPE) (Jang et al., 2006). Previous studies established that both Spz and SPE are essential for wasp egg encapsulation (Paddibhatla et al., 2010; Sorrentino et al., 2004). Here, we further show that spz null mutant (spzrm7) larvae exhibit a strong lymph gland dispersal delay (Figure 5A). Furthermore, decreasing SPE expression in PSC cells (col > SPE RNAi) severely delays lymph gland dispersal (Figure 5B), whereas knocking down its expression in progenitors (dome > SPE RNAi) has no significant effect (Figure 5C). This suggests that SPE expression in PSC cells is required for Toll/NF-κB activation in the lymph gland. To confirm this, we analyzed D4-lacZ expression in col > SPE RNAi larvae. 6 hr post-parasitism, in contrast to control larvae, D4-LacZ expression is not upregulated either in the PSC or in progenitors (Figure 5D–F). Altogether, these data indicate that, in response to parasitism, SPE expression in PSC cells is essential for the production of active Spz and for Toll/NF-κB activation in all lymph gland cells.
Figure 5. Toll/NF-kB activation depends on SPE expression in PSC cells.
(A–C) Quantification (%) of lymph gland disruption post parasitism in spzrm7 larvae (A) and when SPE is down-regulated in either PSC cells (col > SPE RNAi) (B) or lymph gland progenitors (dome > SPE RNAi) (C). Error bars correspond to SEM, ***p<0.001 and ns (not significant) (Pearson’s Chi-squared test). (D, E) LacZ (red) and Col immunostaining (green) of D4–LacZ expressing lymph glands 6 hr post-parasitism in wt (col>) (D) and when SPE is down-regulated in the PSC (white arrow) (col > SPE RNAi) (E). (F) Quantifications of D4-lacZ mean intensity in PSC cells 6 hr post-parasitism. Error bars represent SDs. **p<0.01 (Mann-Whitney nonparametric test).

Figure 5—figure supplement 1. Psh is required for wasp egg neutralization and timely lymph gland disruption.

SPE itself is activated by proteolytic cleavage (Jang et al., 2006). Two parallel extracellular cascades, namely a pattern recognition receptor-dependent pathway involving the protease ModSP (Modular Serine Protease), and a Psh (Persephone)-dependent pathway are involved in SPE processing upon bacterial and fungal infections (reviewed in [Valanne et al., 2011]). To address whether ModSP and/or Psh are required in the fight against wasp parasitism, we looked at wasp egg hatching in mutants (Figure 5—figure supplement 1A). No significant difference, compared to the control, was observed in modSP1 mutant, whereas survival of wasp larvae was significantly enhanced in psh1 and psh1; modSP1 mutant larvae, indicating that Psh is required for successful wasp egg encapsulation. We further observed that lymph gland disruption is delayed in psh1 mutant larvae (Figure 5—figure supplement 1B), demonstrating that Psh is required for timely lymph gland dispersal. These results strongly suggest that Psh-dependent Spz cleavage activates the Toll/NF-κB pathway in lymph gland cells following parasitism.
Toll/NF-κB activation in PSC cells upon parasitism requires high ROS levels
ROS levels are increased in PSC cells in response to wasp parasitism (Sinenko et al., 2011). We therefore investigated possible cross-regulation between ROS and Toll/NF-κB signaling. To monitor ROS levels, we looked at the expression of the gstD-lacZ transgene, where lacZ is placed under the control of the promoter of Glutathione S-transferase D1, which encodes an anti-oxidant enzyme that is expressed in response to oxidative stress (Sykiotis and Bohmann, 2008). In the absence of parasitism, low, scattered expression of gstD-lacZ was observed in the lymph gland (Figure 6A), consistent with a previous report (Owusu-Ansah and Banerjee, 2009). 6 hr post-parasitism, gstD-lacZ expression is increased in both the PSC (Figure 6B,C) (Sinenko et al., 2011) and the progenitors (Figure 6B,D), indicating that parasitism induces a global oxidative stress in the lymph gland. Interestingly, depleting ROS specifically in PSC cells by expressing an intracellular anti-oxidant catalase (col > catalase) strongly decreased gstD-lacZ expression in both the PSC and progenitors (compare Figure 6E and B, quantified in G, H). In contrast, expressing the extracellular catalase IRC (Immune-Related Catalase) (Ha et al., 2005) in PSC cells (col > IRC), did not prevent gstD-lacZ expression in the lymph gland (Figure 6—figure supplement 1D). Likewise, PSC downregulation of the membrane NADPH oxydase enzymes Duox (dual oxydase) (col > Duox RNAi) or Nox (col > Nox RNAi) (Bae et al., 2010), able to generate extracellular ROS, did not prevent gstD-lacZ upregulation in PSC cells (Figure 6—figure supplement 1B,C). Altogether, these results indicate that following parasitism, ROS are intrinsically produced by PSC cells, which in turn leads to oxidative stress in lymph gland progenitors. Switching off the Toll/NF-κB pathway in PSC cells (col > cact > Dif RNAi) has no effect on gstD-lacZ expression 6 hr post-parasitism (compare Figure 6F and B, quantified in G and H), indicating that the increase of ROS levels does not require Toll/NF-κB activity. In contrast, scavenging ROS in PSC cells both suppresses D4-lacZ expression in progenitors and its upregulation in the PSC (Figure 6I–K), leading us to conclude that Toll/NF-κB activation in PSC cells is a response to increased ROS levels in these cells. To confirm the epistatic relationship between ROS and Toll/NF-κB in PSC cells, we simultaneously reduced ROS levels and activated Toll/NF-κB, through the expression of a constitutively active form of the Toll receptor (Toll10B) in PSC cells (col > catalase > Toll10B). A partial rescue of lymph gland dispersal was observed (Figure 6L). These data indicate that, in response to wasp egg laying, ROS level increase in PSC cells leads to Toll/NF-κB activation and lymph gland dispersal.
Figure 6. Wasp-mediated oxidative stress promotes Toll/NF-kB activation in the PSC and lymph gland disruption.
(A, B, E, F) LacZ (red) and Col (green) immunostaining in gstD-lacZ expressing lymph glands in wt context (col>; A, B), when Catalase is expressed in the PSC (col > catalase) (E) and when Toll/NF-kB is inactivated in PSC cells (col > cact > Dif RNAi)(F). (C, D, G, H) Quantifications of gstD-lacZ mean intensity in PSC and progenitor (MZ) cells. Error bars represent SDs. **p<0.01, ****p<0.0001 and ns (not significant) (Mann-Whitney nonparametric test). (I, J) LacZ (red) and Col (green) immunostaining in D4-LacZ expressing lymph glands in wt (col>) (I) and when an intracellular catalase is expressed in the PSC (col > catalase) (J). (K) Quantifications of D4-lacZ mean intensity in PSC cells. **p<0.01 and ns (not significant) (Mann-Whitney nonparametric test). (L–M) Quantifications of lymph gland disruption post parasitism. (N,O) Quantification (%) of wasp egg hatching. Error bars correspond to SEM, **p<0.01, ***p<0.001 (Pearson’s Chi-squared test).

Figure 6—figure supplement 1. Duox- and Nox-independent generation of ROS in PSC cells 6 hr post-parasitism.

Figure 6—figure supplement 2. ROS are required both in PSC cells and lymph gland progenitors for lamellocyte differentiation.

Figure 6—figure supplement 3. Scavenging ROS in PSC cells or silencing the EGFR pathway in MZ progenitors delays lymph gland disruption upon parasitism.

Since 6 hr post parasitism gstD-lacZ is expressed in all lymph gland cells, we examined in which cells modifying ROS levels could affect the immune response. Decreasing ROS levels either in the PSC (col > catalase) or in progenitors (dome > catalase) delay lymph gland lamellocyte differentiation (Figure 6—figure supplement 2), postpone lymph gland rupture (Figure 6L,M; Figure 6—figure supplement 3A–C) and enhance wasp egg hatching (Figure 6N,O). Thus, increased ROS levels in both PSC cells and progenitors following parasitism are essential for the mounting of an efficient lymph gland response and for wasp egg encapsulation.
Toll/NF-κB and EGFR/Erk pathways act in parallel to trigger lymph gland disruption
The incomplete rescue of lymph gland dispersal when simultaneously scavenging ROS and activating Toll/NF-κB signaling in PSC cells, suggested that additional signaling pathway(s) act downstream of ROS. One obvious candidate is EGFR/Erk which is required for wasp egg encapsulation (Sinenko et al., 2011). According to the authors, increased ROS levels in PSC cells, in response to wasp parasitism, lead to the secretion of Spitz (sSpi) into the hemolymph, which activates EGFR/Erk signaling in circulating hemocytes and triggers their differentiation into lamellocytes. However, a direct role of EGFR activation in lymph gland progenitors was not ruled out. We therefore looked at phosphorylated ERK (p-ERK), an EGFR activation readout (Sinenko et al., 2011). Whereas p-ERK was not, or barely, detected in the lymph gland under normal conditions (Figure 7A–A’’), it was detected in lymph gland progenitors 6 hr post parasitism, indicating EGFR activation (Figure 7B–B’’). Furthermore, decreasing Spitz expression in the PSC (antp >spi RNAi) or down-regulating the EGFR pathway in lymph gland progenitors, by expressing a dominant-negative form of the EGFR receptor (dome > EgfrDN), delays lymph gland dispersal post-parasitism (Figure 7C,D; Figure 6—figure supplement 3D–F). Collectively, these data indicate that, in response to parasitism, the EGFR pathway is activated in lymph gland progenitors, leading to lymph gland dispersal.
Figure 7. Epistatic relationships between EGFR/Erk and Toll/NF-κB signaling.

(A, B) p-ERK (red) immunostaining in col > GFP (PSC, green) larvae. (A’, A”, B’, B”), enlarged views of PSC cells. (C, D) Quantifications of lymph gland disruption post parasitism. (E, F) p-ERK (red) immunostaining in wt (E) and Dif1 (F) mutant lymph glands. (G) Quantifications (%) of lymph gland disruption post parasitism. Error bars correspond to SEM, ***p<0.001, ns (not significant) (Pearson’s Chi-squared test).
For proper timing of lymph gland dispersal in response to parasitism, EGFR and Toll/NF-κB pathways are required in progenitors and PSC cells, respectively. This raised the question of whether these two pathways are functionally connected. We therefore tested whether Toll/NF-κB activation in PSC cells controls Spitz expression, by looking at p-ERK expression in progenitors of parasitized Dif1 mutant larvae. No significant difference with controls was observed (Figure 7E,F), indicating that Toll/NF-κB activation is not required for EGFR activation. Second, we reciprocally tested whether expressing sSpi in PSC cells could rescue the lymph gland dispersal delay observed upon Toll/NF-κB inactivation in the PSC. Simultaneous overexpression of sSpi and inactivation of Toll/NF-κB in PSC cells (col > cactus > sSpi) did not rescue the lymph gland dispersal defect (Figure 7G), leading us to conclude that Toll/NF-κB and EGFR pathways act in parallel.
Discussion
Toll/NF-κB signaling is well known in Drosophila for its roles in embryonic dorsal/ventral axis formation and in the humoral innate immune response (Lemaitre and Hoffmann, 2007; Moussian and Roth, 2005). In mammals, it plays important roles in various aspects of innate and adaptive immunity (Espín-Palazón and Traver, 2016). Toll/NF-κB function in the Drosophila cellular response induced by wasp parasitism, although long reported (Qiu et al., 1998; Sorrentino et al., 2004), has only recently started to be investigated in some detail (Gueguen et al., 2013; Schmid et al., 2014). Inactivation of the pathway in two immune tissues, namely the fat body and hemocytes, did not impact on either wasp egg encapsulation or lymph gland lamellocyte differentiation and disruption (Schmid et al., 2014) (data not shown). Thus, when and in which cells Toll/NF-κB signaling is activated and required to fight wasp infection remained unknown. Here, we provide evidence that in response to Leptopilina boulardi parasitism, activation of Toll/NF-κB signaling in PSC cells non cell-autonomously controls the timing of wasp-induced lymph gland dispersal and the release of lymph gland lamellocytes into circulation, both required to prevent wasp egg hatching. Toll/NF-κB activation in PSC cells is dependent on both an increase in ROS levels in these cells, and the expression of SPE which cleaves the ligand Spz. Our data show that EGFR pathway activation in lymph gland progenitors is also required for on time lymph gland dispersal. This activation is mediated by the secretion of Spitz from PSC cells, itself dependent upon ROS signaling. In conclusion, our data identify a signaling cascade that links the PSC response to wasp parasitism to lymph gland lamellocyte differentiation, lymph gland disruption and wasp egg neutralization (Figure 8).
Figure 8. Proposed gene regulatory network that controls lymph gland rupture upon wasp parasitism.
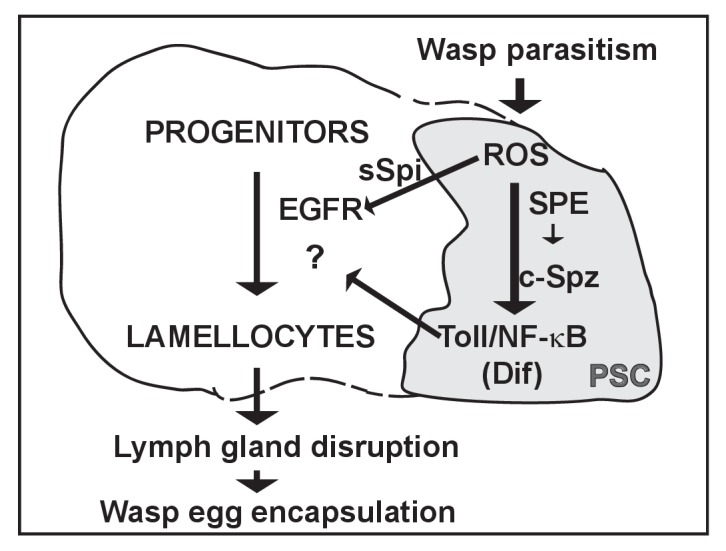
The PSC is drawn in grey. Wasp parasitism increases ROS in PSC cells that activate Toll/NF-κB and Spitz secretion (sSpi). Toll/NF-kB activation in PSC cells requires SPE in the same cells for Spätzle processing (c-Spz). sSpi non cell-autonomously activates the EGFR pathway in lymph gland progenitors. Both EGFR and Toll/NF-κB activation are required for lymph gland lamellocyte differentiation, lymph gland disruption and wasp egg encapsulation.
In the absence of parasitism, hemocyte homeostasis in the lymph gland relies on various signaling pathways active in progenitors and/or differentiating hemocytes, including Hedgehog, Wnt, Pvf and JAK/STAT (for review see Letourneau et al., 2016). Previous work established that downregulation of Col expression or JAK/STAT signaling in progenitors, is a prerequisite for wasp-induced lamellocyte differentiation in the lymph gland (Makki et al., 2010; Oyallon et al., 2016). Our present data show that increasing ROS levels in PSC cells, in response to parasitism, leads to activation of Toll/NF-κB and EGFR signaling in the PSC and hematopoietic lymph gland progenitors, respectively, and that both signaling events are required for timely lamellocyte differentiation. How EGFR signaling, a signal downstream of NF-κB integrates with other signaling pathways active in hematopoietic progenitors, remains to be addressed.
Surviving wasp parasitism is dependent on the ability of the Drosophila larva to reroute basal hematopoiesis and produce, in a timely manner, lamellocytes to neutralize wasp eggs before they hatch in the fly larva. Here, we establish a critical function of Toll/NF-κB signaling in PSC cells in ensuring the proper timing of lamellocyte differentiation and lymph gland disruption in response to parasitism. This temporal control is essential for a successful neutralization of the wasp egg. Drosophila lamellocytes have been reported to differentiate both from sessile/circulating hemocytes of embryonic origin, and from larval lymph gland progenitors (Anderl et al., 2016; Crozatier et al., 2004; Honti et al., 2010; Lanot et al., 2001; Márkus et al., 2009; Sorrentino et al., 2002; Stofanko et al., 2010). This dual origin poses questions as to the relative contributions of these two lamellocyte sources to successful wasp egg encapsulation. We found that, while few lamellocytes are found in circulation before lymph gland disruption, after disruption, their number sharply increases, constituting nearly half of all circulating hemocytes. We further observed that delaying lymph gland disruption impairs the success of wasp egg encapsulation. Altogether, these data indicate that the release of lymph gland lamellocytes is paramount for successful wasp egg neutralisation. One independent study concluded that the sessile compartment is the major source of lamellocytes necessary for fighting parasitism, although melanisation of wasp eggs rather than their hatching was examined (Márkus et al., 2009). In addition, the authors reported a substantial delay of lymph gland disruption (i.e. 48 hr post-wasp egg deposition) in their control condition, compared to 20 hr in our study. The discrepancy between their and our present conclusions could therefore, in part, reflect the use of different conditions for wasp infestation, including the time and/or the temperature since the time scale of lymph gland disruption is highly dependent on temperature (Figure 2—figure supplement 2), or the use of distinct fly genetic backgrounds (Sorrentino et al., 2004). Here, we have investigated the success of wasp egg hatching in different conditions, and our data establish a functional link between timely differentiation of lamellocytes in the lymph gland, their release into circulation and efficient wasp egg neutralization.
In Drosophila, Gram-positive or fungal infections trigger the activation of the Toll/NF-κB pathway, leading to the systemic production of antimicrobial peptides by the fat body, a functional equivalent of the mammalian liver (Lemaitre and Hoffmann, 2007). The antifungal peptide Drosomycin, one main effector of the Toll humoral response, is directly regulated by NF-κB (Ferrandon et al., 1998). We have shown here that wasp parasitism activates Toll/NF-κB in the lymph gland. Drosomycin-GFP expression was, however, not observed in response to wasp parasitism in the lymph gland (Figure 3—figure supplement 1E,F), in agreement with previous observations that antimicrobial peptides are not produced in larvae under these conditions (Nicolas et al., 1996). This suggests that Dif target genes activated in the lymph gland under parasitism are specific to this response. Among these targets, identifying which one(s) code for signal(s) required to trigger efficient lamellocyte differentiation in the lymph gland is of primary interest. Indeed, we found that it directly impacts the timing of lymph gland dispersal. A recent study established that constitutive activation of Toll/NF-κB in PSC cells impairs the permeability barrier of the PSC, possibly facilitating diffusion of signal(s) from the PSC to lymph gland progenitors (Khadilkar et al., 2017). Whether modification of the permeability barrier is also triggered by Toll/NF-κB signaling in PSC cells in response to parasitism to promote lamellocyte differentiation in the lymph gland remains to be investigated.
Oxidative stress, in particular reactive oxygen species (ROS), regulates hematopoiesis both in mammals and in Drosophila (Ludin et al., 2014; Owusu-Ansah and Banerjee, 2009; Sinenko et al., 2011). In mammals, there is a complex interplay between ROS and NF-κB signaling during this process. Oxidative stress can either affect IκB kinase complex (IKK) activity in the cytoplasm or NF-κB DNA-binding capacity in the nucleus (Gupta et al., 2014; Siomek, 2012). In the fly lymph gland, increased ROS levels are observed in both the PSC and hematopoietic progenitors in response to parasitism. ROS increase in lymph gland progenitors, which could reflect changes in cell metabolism since progenitors actively divide as an immediate response to parasitism (Krzemien et al., 2010b; Sorrentino et al., 2002), is dependent on ROS production by PSC cells. ROS-induced signaling in both PSC cells and progenitors is required to trigger wasp-induced dispersal of the lymph gland and release of lamellocytes into circulation. This ROS effect is both relayed by Toll/NF-κB activation in PSC cells and EGFR activation in lymph gland progenitors (Sinenko et al., 2011). ROS-dependent Toll/NF-κB activation, following infection by the Wolbachia bacteria, was reported in the mosquito Aedes aegypti, but the mechanism involved was not defined (Pan et al., 2012). How can an oxidative burst in PSC cells lead to Toll/NF-κB activation in the same cells? ROS could control the production by PSC cells of secreted product(s) required for Spätzle activation, like SPE (Figure 5). Alternatively, Spätzle activation could be independent of the oxidative stress in PSC cells, and ROS could regulate intracellular steps of Toll/NF-κB activation, like the DNA-binding properties of NF-κB, as previously reported in mammals (Gloire et al., 2006). What type of regulation is operational remains to be determined.
In mammals, systemic bacterial infection induces an ‘emergency granulopoiesis’ characterized by de novo production of neutrophils in the bone marrow (Zhao and Baltimore, 2015). In this process, the TLR (Toll-like Receptors)/NF-κB pathway is activated via TLR4 in mouse bone marrow endothelial cells, a component of the vascular niche (Boettcher et al., 2014). The cellular immune response to parasitism in Drosophila can be compared to an emergency hematopoiesis. Establishing that Toll/NF-κB activation in the Drosophila hematopoietic niche is essential for this response extends the evolutionary parallels between Drosophila and mouse to the control of stress-induced hematopoiesis. The role of the EGFR pathway in mammalian hematopoiesis is largely unknown (Kerpedjieva et al., 2012). It will be important to determine whether the regulatory network uncovered here in Drosophila, in which ROS dually regulate EGFR and Toll/NF-κB signaling, also operates in vertebrates under emergency hematopoiesis.
Materials and methods
Fly strains
w1118 (wild type, wt), pcol-Gal4, UAS-mCD8-GFP and PG125dome-gal4 (Jung et al., 2005; Krzemień et al., 2007), Antp-gal4 (Mandal et al., 2007), Df(2L)J4 (Meng et al., 1999); gstD-lacZ (Wang et al., 2003), UAS-sSpi (Sudarsan et al., 2002), UAS-EgfrDN (Saxena et al., 2014), D4-lacZ on the third chromosome (Flores-Saaib et al., 2001). A D4-lacZ insertion on the second chromosome, at the integration site attP-51B (II), was generated in this study by re-injecting the original D4/hsp70 construct (Flores-Saaib et al., 2001; Pan and Courey, 1992). UAS-Cact was a gift from S. Govind (The Graduate School and University Center of The City University of New York, New York, USA). The isogenic (iso) strains, w1118, Dif1, pll2, pll21, dl1, Tlrv1, Tlr (Ferreira et al., 2014) were kindly given by Luis Teixeira; modSP1, psh1, double modSP1; psh1 mutant, UAS-Duox RNAi, UAS-Nox RNAi and UAS-IRC by Bruno Lemaitre. Other Drosophila strains were obtained from the Bloomington (BL) stock center: pll2 (BL3111), pll7 (BL3112), Dif1 (BL36559), dl1 (BL3236), spzrm7 (BL55718) (Tzou et al., 2002), UAS-catalase (BL24621), UAS-Toll10B (BL58987)(Schneider et al., 1991), UAS-pll RNAi (BL34733 and BL3577) (Wu et al., 2015), UAS-Dif RNAi (BL30513), or from the Vienna Drosophila RNAi stock center (VDRC): UAS-spi RNAi (3922) (Chen et al., 2016), UAS-Myd88 RNAi (25402) (Schmid et al., 2014), UAS-latran RNAi (19756 and 100881) (Kallio et al., 2010); UAS-SPE RNAi (Jang et al., 2006). For all RNAi experiments, UAS-Dicer2 (BL24650) was introduced and Drosophila development proceeded at 22°C until L1 stage, before shift to 29°C. Controls correspond to Gal4 drivers crossed with w1118.
Generation of dome-MESO-Gal4 transgenic lines
The dome-MESO sequence from pCasHs43DomeMESO-lacZ (Rivas et al., 2008) was sub-cloned into the Gal4 pBPGUw (17575, Addgene). The resulting plasmid was used to generate dome-MESO-Gal4 (dome-MESO>) transgenic flies using attP/attB technology (Bischof et al., 2007). Two independent Drosophila lines were created by integration at the attP-51B (II) and attP-68A4 (III) sites. domeMESO-Gal4 > GFP-positive cells overlapped with domeless-Gal4 > GFP (PG125; [Krzemień et al., 2007]) progenitors in lymph gland anterior lobes at L3 stage. At L2 stage, in addition to its expression in progenitors, a weak and transient expression was also detected in a subset of PSC cells.
Immunostaining
Lymph glands were dissected and processed as previously described (Krzemień et al., 2007). Antibodies used were rabbit anti-αPS4 (1/200) (Krzemień et al., 2007), mouse anti-Col (1/100) (Krzemień et al., 2007), rabbit anti-Dif (1/500, gift from D. Ferrandon), rabbit anti-Trol (1/1000, gift from S. Baumgartner), chicken anti-βgal (1/1000, Abcam), mouse anti-diphosphorylated Erk kinase (pERK) antibody (1/100, Sigma-Aldrich), mouse anti-β Integrin (Developmental Studies Hybridoma Bank, CF.6G11c, 1/100) (Irving et al., 2005). Nuclei were labeled with TOPRO3 (Thermo Fisher Scientific, Waltham, USA) or Draq5 (Thermo Fisher Scientific). Images were captured using a Zeiss 710 or a Leica SP8 confocal microscope. Three independent biological replicates were analyzed, and one representative image of at least 20 lymph glands analyzed is shown.
Wasp parasitism, lymph gland rupture and wasp egg hatching assays
Late second instar or early third instar Drosophila larvae raised at 27°C or 29°C (for RNAi experiments) were subjected to parasitism for 1 hr at 22°C by Leptopilina boulardi (G486 avirulent strain; [Russo et al., 1996]). Larvae were allowed to develop further at 27°C or 29°C before dissection 6, 20, 30, or 48 hr post-parasitism. Quantification of lymph gland dispersal and wasp egg hatching was performed in un-fixed infected larvae. Quantification of lymph gland dispersal was performed 20 hr post-parasitism for the isogenic strains, modSP1, psh1 mutants, double modSP1; psh1 mutant, col> , dome> , domeMESO> and antp> lines or 30 hr post-parasitism for wt, Dif1, pll2/pll7, dl1, spzrm7, +/Df(2L)J4, dl1/Df(2L)J4 and Dif1/Df(2L)J4 mutants. Lymph glands were classified into three groups: disrupted when anterior lobes were absent or rudimentary, disrupting when some cells had escaped the anterior lobes, or intact when the anterior lobe border was regular. The percentage of lymph gland dispersal was calculated by scoring the number of lymph glands in each group, divided by the total number of infected larvae. Wasp egg hatching was analyzed 48 hr post-parasitism and the number of fly larvae containing melanized/unhatched wasp eggs or living hatched wasp larvae, was counted. The % of wasp egg hatching was calculated by scoring the number of living hatched wasp larvae inside the body of the dissected fly larvae, divided by the total number of infected fly larvae. In all experiments, genotypes were analyzed in parallel. Each experiment was repeated independently at least three times, and quantification represents the mean of three independent biological replicates. Graphs and statistical analyses t-test (Mann–Whitney nonparametric test) were performed using GraphPad Prism five software.
Larval bleeding and hemocyte counting
Individual parasitized larvae were bled on microscope coverslips. Hemolymph samples were dried 10 min, fixed for 10 min in 4% paraformaldehyde in 1XPBS, then stained 2 hr with Phalloidin (1/100; Sigma). Samples were mounted in Vectashield (Vector Laboratories) and examined using a Leica SP8 confocal microscope. In parallel, each larva was examined for lymph gland disruption. At least 10 independent bleedings corresponding to intact or disrupted lymph gland larvae were analyzed for control (hml>) and Dif1 mutant, and 4 and 3 samples, respectively, were quantified. The percentage of lamellocytes and plasmatocytes was calculated relative to the total number of circulating hemocytes. Lamellocytes are easily distinguishable from plasmatocytes by their elongated shape and large size.
Quantification of expression intensity
Expression intensities of D4-lacZ and gstD-lacZ reporters in PSC cells were quantified using Fiji software as described previously (Morin-Poulard et al., 2016). Briefly, ROIs (Regions Of Interest) corresponding to PSC cells, labeled by Col, were defined, and the mean intensity of reporter staining by pixel in each ROI was quantified. For quantifying expression intensity in lymph gland progenitors (gstD-lacZ), representative areas of reporter staining were defined as ROIs in each lymph gland primary lobe, and the mean intensity by pixel was quantified using Fiji software. For each experiment, middle slides of a Z stack were analyzed. In all experiments, genotypes were analyzed in parallel. Each experiment was repeated independently at least three times, and one quantification is shown. Graphs and statistical analyses t-test (Mann–Whitney nonparametric test) were performed using GraphPad Prism five software.
Acknowledgements
We thank S Govind, WJ Lee, A Courey, B Lemaitre, L Teixeira, the Bloomington and Vienna Stock Centers for plasmids and fly stocks; D Ferrandon, I Ando, A Moore and T Trenczek for antibodies; L Bataillé, A Davy, M Meister, C Monod, for critical reading of the manuscript. We are grateful to the Toulouse RIO imaging patform, J Favier, and V Nicolas for fly culture. This work was supported by the CNRS, Ministère de la Recherche (ANR) and Association pour la Recherche sur le Cancer (ARC).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Michèle Crozatier, Email: michele.crozatier-borde@univ-tlse3.fr.
Nathalie Vanzo, Email: nathalie.vanzo@univ-tlse3.fr.
Bruno Lemaître, Ecole Polytechnique Fédérale de Lausanne, Switzerland.
Funding Information
This paper was supported by the following grants:
Fondation ARC pour la Recherche sur le Cancer Graduate Student Fellowship to Isabelle Louradour.
Centre National de la Recherche Scientifique to Alain Vincent, Michèle Crozatier, Nathalie Vanzo.
Agence Nationale de la Recherche Bench grant and post-doc fellowship to Michèle Crozatier.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Visualization, Methodology, Writing—original draft.
Formal analysis, Visualization, Methodology.
Formal analysis, Methodology.
Methodology.
Conceptualization, Formal analysis, Funding acquisition, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Data curation, Formal analysis, Supervision, Funding acquisition, Investigation, Writing—original draft, Project administration, Writing—review and editing.
Conceptualization, Data curation, Formal analysis, Supervision, Investigation, Visualization, Methodology, Writing—original draft, Project administration, Writing—review and editing.
Additional files
References
- Anderl I, Vesala L, Ihalainen TO, Vanha-Aho LM, Andó I, Rämet M, Hultmark D. Transdifferentiation and proliferation in two distinct hemocyte lineages in drosophila melanogaster larvae after wasp infection. PLoS Pathogens. 2016;12:e1005746. doi: 10.1371/journal.ppat.1005746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YS, Choi MK, Lee WJ. Dual oxidase in mucosal immunity and host-microbe homeostasis. Trends in Immunology. 2010;31:278–287. doi: 10.1016/j.it.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Benmimoun B, Polesello C, Haenlin M, Waltzer L. The EBF transcription factor Collier directly promotes Drosophila blood cell progenitor maintenance independently of the niche. PNAS. 2015;112:9052–9057. doi: 10.1073/pnas.1423967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. PNAS. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, Kopf M, Manz MG. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. 2014;124:1393–1403. doi: 10.1182/blood-2014-04-570762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015;126:2443–2451. doi: 10.1182/blood-2015-07-533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xu N, Huang H, Cai T, Xi R. A feedback amplification loop between stem cells and their progeny promotes tissue regeneration and tumorigenesis. eLife. 2016;5:e14330. doi: 10.7554/eLife.14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozatier M, Ubeda JM, Vincent A, Meister M. Cellular immune response to parasitization in Drosophila requires the EBF orthologue collier. PLoS Biology. 2004;2:E196. doi: 10.1371/journal.pbio.0020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragojlovic-Munther M, Martinez-Agosto JA. Extracellular matrix-modulated Heartless signaling in Drosophila blood progenitors regulates their differentiation via a Ras/ETS/FOG pathway and target of rapamycin function. Developmental Biology. 2013;384:313–330. doi: 10.1016/j.ydbio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espín-Palazón R, Traver D. The NF-κB family: Key players during embryonic development and HSC emergence. Experimental Hematology. 2016;44:519–527. doi: 10.1016/j.exphem.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Ferrandon D, Jung AC, Criqui M, Lemaitre B, Uttenweiler-Joseph S, Michaut L, Reichhart J, Hoffmann JA. A drosomycin-GFP reporter transgene reveals a local immune response in Drosophila that is not dependent on the Toll pathway. The EMBO Journal. 1998;17:1217–1227. doi: 10.1093/emboj/17.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ÁG, Naylor H, Esteves SS, Pais IS, Martins NE, Teixeira L. The Toll-dorsal pathway is required for resistance to viral oral infection in Drosophila. PLoS Pathogens. 2014;10:e1004507. doi: 10.1371/journal.ppat.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Saaib RD, Jia S, Courey AJ. Activation and repression by the C-terminal domain of Dorsal. Development. 2001;128:1869–1879. doi: 10.1242/dev.128.10.1869. [DOI] [PubMed] [Google Scholar]
- Gao H, Baldeosingh R, Wu X, Fossett N. The friend of gata transcriptional co-regulator, u-shaped, is a downstream antagonist of dorsal-driven prohemocyte differentiation in drosophila. PLoS One. 2016;11:e0155372. doi: 10.1371/journal.pone.0155372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochemical Pharmacology. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Gold KS, Brückner K. Macrophages and cellular immunity in Drosophila melanogaster. Seminars in Immunology. 2015;27:357–368. doi: 10.1016/j.smim.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian M, Liu T, Banerjee U, Hartenstein V. The proteoglycan Trol controls the architecture of the extracellular matrix and balances proliferation and differentiation of blood progenitors in the Drosophila lymph gland. Developmental Biology. 2013;384:301–312. doi: 10.1016/j.ydbio.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian M, Mandal L, Hartenstein V. Hematopoiesis at the onset of metamorphosis: terminal differentiation and dissociation of the Drosophila lymph gland. Development Genes and Evolution. 2011;221:121–131. doi: 10.1007/s00427-011-0364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen G, Kalamarz ME, Ramroop J, Uribe J, Govind S. Polydnaviral ankyrin proteins aid parasitic wasp survival by coordinate and selective inhibition of hematopoietic and immune NF-kappa B signaling in insect hosts. PLoS Pathogens. 2013;9:e1003580. doi: 10.1371/journal.ppat.1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Patel AK, Shah N, Chaudhary AK, Jha UK, Yadav UC, Gupta PK, Pakuwal U. Oxidative stress and antioxidants in disease and cancer: a review. Asian Pacific Journal of Cancer Prevention. 2014;15:4405–4409. doi: 10.7314/APJCP.2014.15.11.4405. [DOI] [PubMed] [Google Scholar]
- Ha EM, Oh CT, Ryu JH, Bae YS, Kang SW, Jang IH, Brey PT, Lee WJ. An antioxidant system required for host protection against gut infection in Drosophila. Developmental Cell. 2005;8:125–132. doi: 10.1016/j.devcel.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Holz A, Bossinger B, Strasser T, Janning W, Klapper R. The two origins of hemocytes in Drosophila. Development. 2003;130:4955–4962. doi: 10.1242/dev.00702. [DOI] [PubMed] [Google Scholar]
- Honti V, Csordás G, Márkus R, Kurucz E, Jankovics F, Andó I. Cell lineage tracing reveals the plasticity of the hemocyte lineages and of the hematopoietic compartments in Drosophila melanogaster. Molecular Immunology. 2010;47:1997–2004. doi: 10.1016/j.molimm.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Irving P, Ubeda JM, Doucet D, Troxler L, Lagueux M, Zachary D, Hoffmann JA, Hetru C, Meister M. New insights into Drosophila larval haemocyte functions through genome-wide analysis. Cellular Microbiology. 2005;7:335–350. doi: 10.1111/j.1462-5822.2004.00462.x. [DOI] [PubMed] [Google Scholar]
- Jang IH, Chosa N, Kim SH, Nam HJ, Lemaitre B, Ochiai M, Kambris Z, Brun S, Hashimoto C, Ashida M, Brey PT, Lee WJ. A Spätzle-processing enzyme required for toll signaling activation in Drosophila innate immunity. Developmental Cell. 2006;10:45–55. doi: 10.1016/j.devcel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Jung SH, Evans CJ, Uemura C, Banerjee U. The Drosophila lymph gland as a developmental model of hematopoiesis. Development. 2005;132:2521–2533. doi: 10.1242/dev.01837. [DOI] [PubMed] [Google Scholar]
- Kallio J, Myllymäki H, Grönholm J, Armstrong M, Vanha-aho LM, Mäkinen L, Silvennoinen O, Valanne S, Rämet M. Eye transformer is a negative regulator of Drosophila JAK/STAT signaling. The FASEB Journal. 2010;24:4467–4479. doi: 10.1096/fj.10-162784. [DOI] [PubMed] [Google Scholar]
- Kerpedjieva SS, Kim DS, Barbeau DJ, Tamama K. EGFR ligands drive multipotential stromal cells to produce multiple growth factors and cytokines via early growth response-1. Stem Cells and Development. 2012;21:2541–2551. doi: 10.1089/scd.2011.0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadilkar RJ, Vogl W, Goodwin K, Tanentzapf G. Modulation of occluding junctions alters the hematopoietic niche to trigger immune activation. eLife. 2017;6:e28081. doi: 10.7554/eLife.28081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Suda T, Takubo K. How hematopoietic stem/progenitors and their niche sense and respond to infectious stress. Experimental Hematology. 2016;44:92–100. doi: 10.1016/j.exphem.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Krzemien J, Crozatier M, Vincent A. Ontogeny of the Drosophila larval hematopoietic organ, hemocyte homeostasis and the dedicated cellular immune response to parasitism. The International Journal of Developmental Biology. 2010a;54:1117–1125. doi: 10.1387/ijdb.093053jk. [DOI] [PubMed] [Google Scholar]
- Krzemien J, Oyallon J, Crozatier M, Vincent A. Hematopoietic progenitors and hemocyte lineages in the Drosophila lymph gland. Developmental Biology. 2010b;346:310–319. doi: 10.1016/j.ydbio.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Krzemień J, Dubois L, Makki R, Meister M, Vincent A, Crozatier M. Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature. 2007;446:325–328. doi: 10.1038/nature05650. [DOI] [PubMed] [Google Scholar]
- Lanot R, Zachary D, Holder F, Meister M. Postembryonic hematopoiesis in Drosophila. Developmental Biology. 2001;230:243–257. doi: 10.1006/dbio.2000.0123. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annual Review of Immunology. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- Letourneau M, Lapraz F, Sharma A, Vanzo N, Waltzer L, Crozatier M. Drosophila hematopoiesis under normal conditions and in response to immune stress. FEBS Letters. 2016;590:4034–4051. doi: 10.1002/1873-3468.12327. [DOI] [PubMed] [Google Scholar]
- Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, Lapidot T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxidants & Redox Signaling. 2014;21:1605–1619. doi: 10.1089/ars.2014.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhijani K, Alexander B, Tanaka T, Rulifson E, Brückner K. The peripheral nervous system supports blood cell homing and survival in the Drosophila larva. Development. 2011;138:5379–5391. doi: 10.1242/dev.067322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makki R, Meister M, Pennetier D, Ubeda JM, Braun A, Daburon V, Krzemień J, Bourbon HM, Zhou R, Vincent A, Crozatier M. A short receptor downregulates JAK/STAT signalling to control the Drosophila cellular immune response. PLoS Biology. 2010;8:e1000441. doi: 10.1371/journal.pbio.1000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal L, Martinez-Agosto JA, Evans CJ, Hartenstein V, Banerjee U. A hedgehog- and antennapedia-dependent niche maintains drosophila haematopoietic precursors. Nature. 2007;446:320–324. doi: 10.1038/nature05585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Agosto JA, Mikkola HK, Hartenstein V, Banerjee U. The hematopoietic stem cell and its niche: a comparative view. Genes & Development. 2007;21:3044–3060. doi: 10.1101/gad.1602607. [DOI] [PubMed] [Google Scholar]
- Matova N, Anderson KV. Rel/NF-kappaB double mutants reveal that cellular immunity is central to Drosophila host defense. PNAS. 2006;103:16424–16429. doi: 10.1073/pnas.0605721103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matova N, Anderson KV. Drosophila Rel proteins are central regulators of a robust, multi-organ immune network. Journal of Cell Science. 2010;123:627–633. doi: 10.1242/jcs.060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márkus R, Laurinyecz B, Kurucz E, Honti V, Bajusz I, Sipos B, Somogyi K, Kronhamn J, Hultmark D, Andó I. Sessile hemocytes as a hematopoietic compartment in Drosophila melanogaster. PNAS. 2009;106:4805–4809. doi: 10.1073/pnas.0801766106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Khanuja BS, Ip YT. Toll receptor-mediated Drosophila immune response requires Dif, an NF-kappaB factor. Genes & Development. 1999;13:792–797. doi: 10.1101/gad.13.7.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin-Poulard I, Sharma A, Louradour I, Vanzo N, Vincent A, Crozatier M. Vascular control of the Drosophila haematopoietic microenvironment by Slit/Robo signalling. Nature Communications. 2016;7:11634. doi: 10.1038/ncomms11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer NT, Goecks J, Kacsoh BZ, Mobley JA, Bowersock GJ, Taylor J, Schlenke TA. Parasitoid wasp venom SERCA regulates Drosophila calcium levels and inhibits cellular immunity. PNAS. 2013;110:9427–9432. doi: 10.1073/pnas.1222351110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussian B, Roth S. Dorsoventral axis formation in the Drosophila embryo--shaping and transducing a morphogen gradient. Current Biology. 2005;15:R887–R899. doi: 10.1016/j.cub.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Nicolas E, Nappi AJ, Lemaitre B. Expression of antimicrobial peptide genes after infection by parasitoid wasps in Drosophila. Developmental & Comparative Immunology. 1996;20:175–181. doi: 10.1016/0145-305X(96)00017-1. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyallon J, Vanzo N, Krzemień J, Morin-Poulard I, Vincent A, Crozatier M. Two independent functions of collier/early b cell factor in the control of drosophila blood cell homeostasis. PLoS One. 2016;11:e0148978. doi: 10.1371/journal.pone.0148978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddibhatla I, Lee MJ, Kalamarz ME, Ferrarese R, Govind S. Role for sumoylation in systemic inflammation and immune homeostasis in Drosophila larvae. PLoS Pathogens. 2010;6:e1001234. doi: 10.1371/journal.ppat.1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Courey AJ. The same dorsal binding site mediates both activation and repression in a context-dependent manner. The EMBO Journal. 1992;11:1837–1842. doi: 10.1002/j.1460-2075.1992.tb05235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Zhou G, Wu J, Bian G, Lu P, Raikhel AS, Xi Z. Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. PNAS. 2012;109:E23–E31. doi: 10.1073/pnas.1116932108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Pan PC, Govind S. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development. 1998;125:1909–1920. doi: 10.1242/dev.125.10.1909. [DOI] [PubMed] [Google Scholar]
- Rivas ML, Cobreros L, Zeidler MP, Hombría JC. Plasticity of Drosophila Stat DNA binding shows an evolutionary basis for Stat transcription factor preferences. EMBO Reports. 2008;9:1114–1120. doi: 10.1038/embor.2008.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, Dupas S, Frey F, Carton Y, Brehelin M. Insect immunity: early events in the encapsulation process of parasitoid (Leptopilina boulardi) eggs in resistant and susceptible strains of Drosophila. Parasitology. 1996;112:135–142. doi: 10.1017/S0031182000065173. [DOI] [PubMed] [Google Scholar]
- Saxena A, Denholm B, Bunt S, Bischoff M, VijayRaghavan K, Skaer H. Epidermal growth factor signalling controls myosin II planar polarity to orchestrate convergent extension movements during Drosophila tubulogenesis. PLoS Biology. 2014;12:e1002013. doi: 10.1371/journal.pbio.1002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MR, Anderl I, Vesala L, Vanha-aho LM, Deng XJ, Rämet M, Hultmark D. Control of Drosophila blood cell activation via Toll signaling in the fat body. PLoS One. 2014;9:e102568. doi: 10.1371/journal.pone.0102568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DS, Hudson KL, Lin TY, Anderson KV. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes & Development. 1991;5:797–807. doi: 10.1101/gad.5.5.797. [DOI] [PubMed] [Google Scholar]
- Sinenko SA, Shim J, Banerjee U. Oxidative stress in the haematopoietic niche regulates the cellular immune response in Drosophila. EMBO Reports. 2011;13:83–89. doi: 10.1038/embor.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomek A. NF-κB signaling pathway and free radical impact. Acta Biochimica Polonica. 2012;59:323–331. [PubMed] [Google Scholar]
- Sorrentino RP, Carton Y, Govind S. Cellular immune response to parasite infection in the Drosophila lymph gland is developmentally regulated. Developmental Biology. 2002;243:65–80. doi: 10.1006/dbio.2001.0542. [DOI] [PubMed] [Google Scholar]
- Sorrentino RP, Melk JP, Govind S. Genetic analysis of contributions of dorsal group and JAK-Stat92E pathway genes to larval hemocyte concentration and the egg encapsulation response in Drosophila. Genetics. 2004;166:1343–1356. doi: 10.1534/genetics.166.3.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofanko M, Kwon SY, Badenhorst P. Lineage tracing of lamellocytes demonstrates Drosophila macrophage plasticity. PLoS One. 2010;5:e14051. doi: 10.1371/journal.pone.0014051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsan V, Pasalodos-Sanchez S, Wan S, Gampel A, Skaer H. A genetic hierarchy establishes mitogenic signalling and mitotic competence in the renal tubules of Drosophila. Development. 2002;129:935–944. doi: 10.1242/dev.129.4.935. [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Developmental Cell. 2008;14:76–85. doi: 10.1016/j.devcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzou P, Reichhart JM, Lemaitre B. Constitutive expression of a single antimicrobial peptide can restore wild-type resistance to infection in immunodeficient Drosophila mutants. PNAS. 2002;99:2152–2157. doi: 10.1073/pnas.042411999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valanne S, Wang JH, Rämet M. The Drosophila Toll signaling pathway. The Journal of Immunology. 2011;186:649–656. doi: 10.4049/jimmunol.1002302. [DOI] [PubMed] [Google Scholar]
- Vanha-Aho LM, Anderl I, Vesala L, Hultmark D, Valanne S, Rämet M. Edin Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in Drosophila melanogaster. PLoS Pathogens. 2015;11:e1004895. doi: 10.1371/journal.ppat.1004895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Developmental Cell. 2003;5:811–816. doi: 10.1016/S1534-5807(03)00323-X. [DOI] [PubMed] [Google Scholar]
- Wu C, Chen C, Dai J, Zhang F, Chen Y, Li W, Pastor-Pareja JC, Xue L. Toll pathway modulates TNF-induced JNK-dependent cell death in Drosophila. Open Biology. 2015;5:140171. doi: 10.1098/rsob.140171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, Baltimore D. Regulation of stress-induced hematopoiesis. Current Opinion in Hematology. 2015;22:286–292. doi: 10.1097/MOH.0000000000000149. [DOI] [PMC free article] [PubMed] [Google Scholar]