

Abstract
Objectives
Glucagon-like peptide-1 (GLP-1) is secreted from enteroendocrine cells and exerts a broad number of metabolic actions through activation of a single GLP-1 receptor (GLP-1R). The cardiovascular actions of GLP-1 have garnered increasing attention as GLP-1R agonists are used to treat human subjects with diabetes and obesity that may be at increased risk for development of heart disease. Here we studied mechanisms linking GLP-1R activation to control of heart rate (HR) in mice.
Methods
The actions of GLP-1R agonists were examined on the control of HR in wild type mice (WT) and in mice with cardiomyocyte-selective disruption of the GLP-1R (Glp1rCM−/−). Complimentary studies examined the effects of GLP-1R agonists in mice co-administered propranolol or atropine. The direct effects of GLP-1R agonism on HR and ventricular developed pressure were examined in isolated perfused mouse hearts ex vivo, and atrial depolarization was quantified in mouse hearts following direct application of liraglutide to perfused atrial preparations ex vivo.
Results
Doses of liraglutide and lixisenatide that were equipotent for acute glucose control rapidly increased HR in WT and Glp1rCM−/− mice in vivo. The actions of liraglutide to increase HR were more sustained relative to lixisenatide, and diminished in Glp1rCM−/− mice. The acute chronotropic actions of GLP-1R agonists were attenuated by propranolol but not atropine. Neither native GLP-1 nor lixisenatide increased HR or developed pressure in perfused hearts ex vivo. Moreover, liraglutide had no direct effect on sinoatrial node firing rate in mouse atrial preparations ex vivo. Despite co-localization of HCN4 and GLP-1R in primate hearts, HCN4-directed Cre expression did not attenuate levels of Glp1r mRNA transcripts, but did reduce atrial Gcgr expression in the mouse heart.
Conclusions
GLP-1R agonists increase HR through multiple mechanisms, including regulation of autonomic nervous system function, and activation of the atrial GLP-1R. Surprisingly, the isolated atrial GLP-1R does not transduce a direct chronotropic effect following exposure to GLP-1R agonists in the intact heart, or isolated atrium, ex vivo. Hence, cardiac GLP-1R circuits controlling HR require neural inputs and do not function in a heart-autonomous manner.
Keywords: GLP-1, Diabetes, Cardiac, Cardiovascular disease, Heart rate, Autonomic nervous system
Graphical abstract
Highlights
-
•
GLP-1 controls heart rate (HR) through the autonomic nervous system and the cardiac GLP-1R in mice.
-
•
The acute induction of HR by GLP-1R agonists is sensitive to propranolol.
-
•
GLP-1R agonists do not directly increase HR in isolated perfused mouse hearts ex vivo.
-
•
The GLP-1R agonist liraglutide does not directly enhance sinoatrial activity ex vivo.
-
•
GLP-1 does not increase heart rate in a heart autonomous manner.
1. Introduction
Glucagon-like peptide-1 (GLP-1) is a gut hormone synthesized predominantly in enteroendocrine L cells of the small and large intestine. Although GLP-1 levels are low in fasting or interprandial states, GLP-1 secretion and circulating levels of GLP-1 rise rapidly following meal ingestion. Original concepts of GLP-1 action described its role as an incretin hormone that augmented glucose-dependent insulin secretion, via a direct effect on islet β-cells. Subsequent studies extended the islet actions of GLP-1 to encompass stimulation of somatostatin and inhibition of glucagon secretion, mechanisms converging on the reduction of meal-related glycemic excursions. These pancreatic actions of GLP-1 are mediated by a distinct GLP-1 receptor (GLP-1R), expressed on islet β- and δ-cells.
GLP-1 also exerts pleiotropic actions beyond the islet, consistent with the broad extra-pancreatic expression of the GLP-1 receptor. Notably, GLP-1 inhibits gastric emptying through neural pathways and reduces food intake via activation of a hypothalamic and brainstem network of central nervous system GLP-1 receptors [1]. GLP-1 also promotes natriuresis, reduces blood pressure, and inhibits chylomicron secretion, mechanisms mediated through the canonical GLP-1 receptor [2]. The actions of GLP-1 to control blood glucose and body weight are conserved in humans thereby supporting development of multiple GLP-1 receptor agonists for the treatment of type 2 diabetes (T2D).
Although control of blood glucose remains a primary goal in the treatment of T2D, substantial effort is simultaneously directed toward reducing the development of diabetes-associated microvascular and macrovascular complications. Notably, reduction of blood pressure, control of dyslipidemia, and judicious use of anti-platelet agents represent important complimentary goals for management of T2D [3]. The introduction of mandatory cardiovascular safety studies by regulatory authorities has further heightened the interest in whether glucose-lowering agents exert independent actions that modify cardiovascular risk in subjects with nascent or established cardiovascular disease. In this regard, the SGLT-2 inhibitor empagliflozin was recently shown to reduce rates of cardiovascular death in a large cardiovascular outcome study [4], further elevating the importance of understanding the non-glycemic actions of drugs used to treat T2D.
One of the first non-glycemic actions described for native GLP-1 and degradation-resistant GLP-1R agonists was a rapid increase in heart rate (HR) [5], [6]. The increase in HR is mediated through the canonical GLP-1 receptor [6], [7], independent of changes in blood glucose, and associated with activation of autonomic sympathetic preganglionic brainstem neurons in normoglycemic rats [7], [8]. GLP-1 may also modify HR through attenuation of parasympathetic tone [9], however the relative contribution of sympathetic vs. parasympathetic tone to GLP-1R-dependent control of HR remains uncertain [10], [11].
The identification of GLP-1R expression in the atria of mice [12], [13] and in the sinoatrial node of monkey [14] raised the additional possibility that GLP-1 may also modulate HR via a direct effect through the cardiac GLP-1R. Further support for this hypothesis derives from observations that mice with cardiac-selective reduction of GLP-1R expression (Glp1rCM−/−) exhibit a reduction in basal HR assessed over 24 h [15]. Hence, the available data suggest that GLP-1 may control HR indirectly, through modulation of autonomic nervous system activity, as well as directly, through control of pacemaker activity via the atrial GLP-1R.
To elucidate the relative importance of and inter-dependence of these pathways, we have now studied the regulation of HR in control and Glp1rCM−/− mice treated with lixisenatide or liraglutide, GLP-1R peptide agonists with distinct pharmacokinetic profiles. Notably, lixisenatide is a short-acting exendin-4-derived GLP-1R agonist, whereas the human GLP-1 analog liraglutide, exhibits a more protracted duration of action, via acylation and non-covalent association with albumin [16]. The importance of the autonomic nervous system was examined in mice co-administered propranolol or atropine. Complimentary experiments examined HR responses to GLP-1R agonists in isolated perfused mouse hearts under aerobic and ischemia/reperfusion conditions, and in atrial preparations ex vivo. Our findings reveal temporally distinct contributions from both the sympathetic nervous system and cardiac GLP-1Rs in the HR response to GLP-1R agonism in mice in vivo. In contrast, the isolated perfused mouse heart or dissected atrial preparations did not exhibit a direct chronotropic response to GLP-1R agonists ex vivo. Hence, the atrial GLP-1R mediates a HR response to GLP-1R agonism in the context of the intact mouse heart in vivo, but is not sufficient for transduction of a heart-autonomous chronotropic response ex vivo.
2. Materials and methods
2.1. Mice
All mice used in these studies were adult males housed under pathogen-free conditions in microisolator cages and maintained on a 12-h light/dark cycle with free access to standard rodent diet (#2018, 18% kcal from fat; Harlan Teklad) and water, unless otherwise noted. All experiments were carried out in accordance with protocols approved by the Animal Care Committee at Mt. Sinai Hospital and the Toronto Centre for Phenogenomics and were consistent with ARRIVE guidelines. Glp1rCM−/− mice and Cre WT controls were generated as described [15]. Hcn4-Cre mice were provided by Dr. Andreas Ludwig. The generation of these mice and tamoxifen induction of Cre expression were described previously [17]. Glp1rSAN−/− and GcgrSAN−/− mice and controls (wild type, Cre, floxed) were produced by breeding Hcn4-Cre hemizygous mice with floxed Glp1r mice [18]or floxed Gcgr mice [19], respectively. Wild type C57BL/6J mice (used for experiments in Figure 3, Figure 4, Figure 6) were purchased from The Jackson Laboratory (Bar Harbor, ME).
Figure 3.

Autonomic nervous system-dependent effects of lixisenatide on heart rate. Heart rate (A and B; upper panels) and average heart rates (A and B; lower panels) over a 2–3 h interval in conscious, freely moving wild-type mice following a single ip injection of vehicle (Veh) or lixisenatide (Lixi; 10 μg/kg), in the presence or absence of propranolol (Prop; 5 mg/kg ip; sympathetic nervous system inhibitor) or atropine (Atr; 2 mg/kg ip; parasympathetic nervous system inhibitor). In (A), propranolol or vehicle (solid blue arrow; at time = 15 min) was administered 20 min prior to vehicle or lixisenatide (dotted blue arrow; at time = 35 min). In (B), atropine was administered at the same time as vehicle or lixisenatide (red arrow; at time = 15 min). For (A) and (B), data in panels on the left are from the first 3 h (0–180 min) of heart rate recordings and data in panels on the right are heart rate recordings from 3 to 6 h (180–360 min) after treatment administration. The average heart rate data for (A) and (B) lower left panels was calculated from 60 to 180 min or 40–180 min, respectively, when heart rates were stabilized following ip injections. Values are mean ± SE; n = 4–8 mice/group. Error bars have purposely been omitted from the heart rate data. *p < 0.05; **p < 0.01; ***p < 0.001 (One-way ANOVA). Body weights (g) are 31.0, 29.5, 30.8, and 30.0 for mice in (A) and 34.7, 36.3, 34.5, 34.7, 34.5, 32.4, 35.7, and 32.0 for mice in (B).
Figure 4.

Autonomic nervous system-dependent effects of liraglutide on heart rate. Heart rate (A and B; upper panels) and average heart rate (A and B; lower panels) over a 2–3 h interval in conscious, freely moving wild-type mice following a single ip injection of vehicle (Veh) or liraglutide (Lira; 30 μg/kg), in the presence or absence of propranolol (Prop; 5 mg/kg ip; sympathetic nervous system inhibitor) or atropine (Atr; 2 mg/kg ip; parasympathetic nervous system inhibitor). In (A) and (B), propranolol or atropine was administered at the same time as vehicle or liraglutide (red arrow; at time = 15 min). For (A) and (B), data in panels on the left are from the first 3 h (0–180 min) of heart rate recordings and data in panels on the right are from 3 to 6 h (180–360 min) after treatment administration. The average heart rate data for (A) and (B) lower left panels was calculated from 60 to 180 min or 40–180 min, respectively, when heart rates were stabilized following ip injections. Values are mean ± SE; n = 5–8 mice/group. Error bars have been purposely omitted from the heart rate data. *p < 0.05; **p < 0.01; ***p < 0.001 (One-way ANOVA). Body weights (g) are 33.0, 32.0, 30.0, 34.0, and 30.0 for mice in (A) and 34.5, 36.6, 34.0, 35.2, 35.0, 32.2, 35.6, and 32.2 for mice in (B).
Figure 6.

Lixisenatide has no effect on heart rate or LVDP in isolated mouse hearts ex vivo. (A) Heart rate and left ventricular developed pressure (LVDP) in aerobic perfused ex vivo mouse hearts exposed to vehicle (Veh) or increasing doses of lixisenatide (Lixi). (B) Analysis of heart rate (upper panels) and LVDP (lower panels), at baseline and during reperfusion, following a 30 min period of no-flow global ischemia in ex vivo isolated mouse hearts that were exposed to vehicle (control), 0.5 nM GLP-1 7-36 (GLP-1), 5 nM lixisenatide (Lixi), or ischemia preconditioning (IPC). Heart rate recordings during the ischemic period (upper left panel) are very high due to quivering of the heart and have been omitted from the graph. The panels on the right depict AUC data for heart rate or LVDP at baseline and during the reperfusion period. In (A), isoproterenol (isoprot) was used as a positive control at the end of each perfusion. Values are mean ± SE; n = 5–11 hearts/group. ***p < 0.001 for IPC vs all other treatments (One-way ANOVA of AUC LVDP data).
2.2. Oral glucose tolerance test
Mice were fasted for 6 h (7am–1pm) and then given a single ip injection of vehicle (PBS) or lixisenatide immediately prior to receiving oral glucose (1.5 g/kg body weight) by gavage. Liraglutide was administered as a single ip injection 2 h prior to oral glucose challenge. Blood glucose levels were measured in tail vein blood samples at time 0 and for up to 120 min following oral glucose administration using a hand-held glucose meter (Contour, Bayer Inc., Mississauga, ON).
2.3. Measurement of heart rate via telemetry
Heart rate was measured in conscious, freely moving mice using an implanted radiotelemetry device (PA-C10, Data Sciences International, St. Paul, MN). All mice were allowed a minimum period of 1 week to recover from device implantation surgery prior to initiation of data collection. Mice were given ip injections of vehicle, lixisenatide (10 μg/kg; Sanofi), liraglutide (30 μg/kg; Novo Nordisk), propranolol hydrochloride (5 mg/kg; Sigma Aldrich, Oakville, ON), or atropine methyl nitrate (2 mg/kg; Sigma Aldrich, Oakville, ON). Mice that received liraglutide were not given another injection until 48 h later, to ensure drug wash out. All injections were administered at approximately 4pm each day.
2.4. Nucleic acid isolation and analysis of mRNA expression
Genomic DNA was isolated from mouse tissue using a DNeasy Blood and Tissue Kit (Qiagen, Toronto, ON). PCR amplification of genomic DNA sequences flanking exons 6 and 7 of the Glp1r were generated using the primer pair 5′-GGA TCC GAA CTG AGG TCC TC-3′ and 5′-GGG GTG ATA TTT GGC CAT ATG AG-3.’
Total RNA was extracted from tissues using Tri Reagent (Molecular Research Center Inc., Cincinnati, OH). cDNA was synthesized from DNase I-treated (Thermo-Fisher Scientific, Markham, ON) total RNA using random hexamers and Superscript III (Thermo-Fisher Scientific, Markham, ON). Real-time PCR was carried out using a QuantStudio 5 System and TaqMan Gene Expression Assays (Thermo-Fisher Scientific, Markham, ON) for Glp1r (Exon 5-6; Mm00445292_m1) or Gcgr (Exon 7-8; Mm00433550_g1). Relative mRNA levels were quantified using the 2−ΔCt method and cyclophilin (Ppia) mRNA expression for normalization. Cre cDNA was amplified by PCR using the primer pair 5′-CCC GCG CTG GAG TTT CAA TA-3′ and 5′-CTT CGC CCA GTT GAT CAT GTG-3.′ Glp1r Exons 5-8 cDNA was amplified by PCR using the primer pair 5′-CCT GAG GAA CAG CTC CTG TC-3′ and 5′-ACA ATC CCC CAT GGG ATA AC-3.′ Gcgr Exons 5-13 cDNA was amplified by PCR using the primer pair 5′-AGC ACC GCC TAG TGT TCA AG-3′ and 5′-AAA CAG TAG AGA ACA GCC ACC A-3.′
2.5. Ex vivo determination of heart rate and left ventricular performance
Hearts were removed from mice, cannulated through the aorta, and perfused at constant pressure using a Langendorff apparatus as described [20]. Continuous pressure traces were recorded from a balloon placed in the left ventricle, and all data analysis is based on the 5 min average for a given parameter. For aerobic perfusion experiments, heart rate and left ventricular developed pressure (LVDP) were assessed in response to increasing doses of lixisenatide (0.5–50 nM; Sanofi) or vehicle. Isoproterenol (100 nM; Sigma Aldrich, Oakville, ON) was used as a positive control at the end of each perfusion. For ischemia/reperfusion injury experiments, heart rate and LVDP were determined at baseline (30–40 min aerobic perfusion; analysis is shown for the final 10 min of baseline data prior to ischemia), during 30 min of no-flow global ischemia, and during 40 min of reperfusion. GLP-1 (0.5 nM) or lixisenatide (5 nM) was delivered via a syringe pump into a warmed bubble trap situated directly above the perfused heart. Drug dose calculations were based on an approximation of 3 ml/min total flow through the heart. Ischemia preconditioning was performed before the 30 min ischemic period by 4 cycles of 5 min ischemia, 5 min reperfusion.
2.6. Analysis of sinoatrial node firing rate in isolated atrial preparations and heart rate in anaesthetized mice
Atrial preparations were from 12- to 16-wk-old C57BL/6 mice. Heparinized mice were sacrificed and live atria dissected in Tyrode's solution (35 °C) consisting of (in mmol/L) 140 NaCl, 5.4 KCl, 1.2 KH2PO4, 1.0 MgCl2, 1.8 CaCl2, 5.55 glucose, and 5 HEPES, with pH adjusted to 7.4 with NaOH. Atria were pinned to the bottom of a Sylgard-coated 30-mm petri dish in “butterfly” fashion so that the crista terminalis could be identified and used as a landmark for sinoatrial node position. Atria were incubated in 1 μM di-4-ANEPPS (Molecular Probes, Eugene, OR), a voltage sensitive dye, for 10 min followed by superfusion with 37 °C Krebs solution of the following composition: 118.0 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.0 mM CaCl2, 25.0 mM NaHCO3, 11.1 mM glucose, at a constant flow rate of ∼10 ml/min. The superfusate was continuously bubbled with 95% O2–5% CO2 in a 37 °C water bath, resulting in pH 7.4. Drugs to be tested for their effect on sinoatrial node (SAN) firing rate were added directly to the warmed oxygenated superfusate and allowed to flow directly onto the pinned atrial tissue. Each treatment was applied for 3 min and heart rate values were determined from an area of interest within the sinoatrial node. The integrity of the SAN, and its responsiveness to chronotropic agents was routinely tested at the end of an experiment by the addition of 100 nM isoproterenol. 100% of the atria tested showed a ∼200 b.p.m. increase in their SAN firing rates in response to isoproterenol. The imaging system consisted of illuminating light provided by a 120-W quartz mercury lamp light source (X-Cite exacte, EXFO Life Sciences, Mississauga, ON) through a band-pass filter and off a dichroic mirror (Olympus). A shutter (EXFO Life Sciences, Mississauga, ON) was used to minimize tissue exposure to excess light (only open during image acquisition). The fluorescent light emitted from the preparation passes through the dichroic mirror and is further long-pass (>590 nm) filtered using a Schott glass filter (Melles Griot, Ottawa, ON) before reaching the camera. A charge-coupled device camera (model Cascade 128+, Photometrics, Tucson, AZ) was used in binning mode (64 × 64 pixels, 16-bit resolution, 1000 frames/s). In the present application, it was equipped with a 25-mm focal length lens (Computar, Commack, NY) and a 5-mm spacer, resulting in a 10 × 10 mm field of view (167 × 167 μm/pixel). Image acquisition software ImagePro Plus 5.1.2.59 (Media Cybernetics, Bethesda, MD) and the camera were connected via an image acquisition board (model PCI- 1422, National Instruments, Austin, TX) to a personal computer. All data were processed offline using ImagePro Plus 5.1.2.59 software.
As a control for the lack of actions of liraglutide in isolated atria, the same stocks of liraglutide were used to assess heart rates in 12–16 wk old anesthetized C57BL/6 mice via implanted Millar 1.4 French Millar blood pressure probe passed through the right common carotid artery into the ascending aorta. Liraglutide or vehicle (5 μl of 0.9% saline) was administered intravenously through a polyethylene catheter (PE10) in the left jugular vein, at a rate of1.5 μl/s and heart rate was recorded for 5 min before addition of next dose. All experiments were performed with experimenters blinded to the drug treatment group until after data analysis was complete.
2.7. Statistical analyses
Results are presented as mean ± SE. Statistical significance was determined by two-tailed Student's t-test or one- or two-way ANOVA with Bonferroni post hoc analysis using GraphPad Prism version 5.02 software (San Diego, CA). A p value < 0.05 was considered statistically significant.
3. Results
3.1. Identification of equipotent doses of lixisenatide and liraglutide for glycemic control
To assess and compare the actions of a short-acting GLP-1R agonist, lixisenatide, and a longer-acting GLP-1R agonist, liraglutide, we first carried out dose-ranging studies to identify doses of these peptides that produced comparable glucose-lowering activity in mice in vivo. Notably, acute administration of 10 μg/kg lixisenatide and 30 μg/kg liraglutide produced comparable glycemic reductions in response to oral glucose (Figure 1A and B) in both control (α-myosin heavy chain promoter-Cre (αMHC-Cre; Cre WT) mice [15] and in mice with cardiomyocyte-selective inactivation of the GLP-1 receptor Glp1rCM−/−). Hence, we selected these two pharmacodynamically equivalent glucoregulatory doses for further analysis of chronotropic activity.
Figure 1.

Oral glucose tolerance in response to different doses of lixisenatide or liraglutide in cardiomyocyte-specific Glp1r knockout mice and controls. Blood glucose levels following oral glucose administration in Cre WT (A) and Glp1rCM−/− (B) mice treated with different doses of lixisenatide (Lixi), liraglutide (Lira), or vehicle (PBS). n = 2–4 mice/group.
3.2. GLP-1R agonist-dependent increases in HR are attenuated in Glp1rCM−/− mice
We next assessed the actions of liraglutide, an extensively studied GLP-1R agonist used in the treatment of diabetes and obesity, on HR in mice. HR was increased for a prolonged period of time, remaining significantly elevated at 16–18 h following a single injection of liraglutide (Figure 2A and Supplementary Figure 1). Furthermore, the actions of liraglutide to increase HR were attenuated in Glp1rCM−/− mice (Figure 2B and Supplementary Figure 1), extending previous findings implicating a role for the cardiomyocyte GLP-1 receptor in the control of basal HR [15]. Consistent with these findings, HR trended higher following lixisenatide administration in WT mice and to a lesser extent in Glp1rCM−/− mice (Supplementary Figure 2).
Figure 2.

Acute administration of liraglutide is associated with prolonged increases in heart rate in mice.Cre WT (A) and Glp1rCM−/− (B) mice were given a single ip injection of vehicle or liraglutide (30 μg/kg), and heart rate was recorded in conscious, freely moving mice for a total of 18 h (upper panels). All injections were administered 15 min after the start of data collection. The data in (A) and (B) are from 1 to 18 h post injection. The bar graphs (lower panels) represent the average heart rate at 3 h intervals following vehicle or liraglutide administration. Values are mean ± SE; n = 3 mice/group. *p < 0.05; **p < 0.01 vs. vehicle (Student's t-test of AUC data for upper panels; two-way ANOVA for lower panels). Body weights (g) are 39.9, 34.1, and 31.6 for Cre WT mice and 38.9, 31.7, and 32.3 for Glp1rCM−/− mice.
3.3. GLP-1R-dependent induction of HR is attenuated by propranolol
As GLP-1R agonists increase activation of autonomic catecholaminergic neurons in the rat central nervous system [7], [8], we next ascertained whether the acute chronotropic effects of lixisenatide or liraglutide were modified by pharmacological blockade using the β-adrenergic antagonist propranolol. Administration of propranolol completely attenuated the acute increase in HR (from 1 to 6 h) observed with lixisenatide (Figure 3A). Although GLP-1R agonists are thought to increase HR in part through inhibition of parasympathetic nervous system activity, the muscarinic receptor antagonist atropine did not eliminate or potentiate the acute lixisenatide-mediated induction of HR over the first 3 h (Figure 3B). In contrast, HR was no longer increased (in the presence of co-administered atropine) from 3 to 6 h after lixisenatide administration (Figure 3B). Propranolol similarly attenuated the acute (0–3 h) induction of HR following liraglutide administration, however a very modest but detectable increase in HR remained evident in mice treated with both liraglutide and propranolol when analyzed from 3 to 6 h after liraglutide injection (Figure 4A). Furthermore, liraglutide continued to increase HR in atropine-treated mice, when analyzed from 1 to 3 h or 3–6 h after liraglutide administration (Figure 4B). Taken together, these findings demonstrate independent, temporally distinct contributions from the sympathetic nervous system and the cardiac GLP-1R in the chronotropic response to GLP-1R agonism in mice.
3.4. HCN4-directed Cre expression does not reduce levels of Glp1r mRNA transcripts in the mouse heart
Immunohistochemical analysis of GLP-1 receptor expression in the monkey heart co-localized the sinoatrial node (SAN) GLP-1R with hyperpolarization-activated cyclic nucleotide-gated channel 4 (HCN4), a major ion channel regulating pacemaker function [14]. Accordingly, we hypothesized that mice expressing Cre recombinase under the control of the HCN4 promoter [17] would enable elimination of sinoatrial Glp1r expression in the mouse heart. To this end, we mated Hcn4Cre mice with floxed Glp1rFl/Fl mice to generate Glp1rSAN−/− mice. Unexpectedly, we did not detect reduction of right atrial Glp1r expression (or transcript length) in Glp1rSAN−/− mice (Figure 5A and Supplementary Figures 3 and 4), despite detectable Cre mRNA expression and recombination of Glp1r genomic DNA in the right atrium (Supplementary Figures 3–5). As the structurally related glucagon receptor (Gcgr) has been localized to the mouse SAN [21] and like GLP-1, transduces an acute increase in HR [22], we examined whether Hcn4Cre mice would enable reduction of atrial Gcgr expression. In contrast to findings in Glp1rSAN−/− mice, GcgrSAN−/− mice exhibited a reduction in both the expression and length of Gcgr mRNA transcripts in the right atrium (Figure 5B and Supplementary Figures 6 and 7). Hence, the failure to achieve reduction of atrial Glp1r mRNA transcripts in Glp1rSAN−/− mice (Figure 5A) is consistent with lack of co-expression of HCN4 and GLP-1R in the mouse heart.
Figure 5.

Glp1r mRNA levels are not reduced in floxed Glp1r mice expressing HCN4-Cre.Glp1r (A) and Gcgr (B) mRNA levels were measured in right atria (RA), left atria (LA), ventricle and lung or liver from wild-type control (WT), Glp1rflox/flox (Fl/Fl Glp1r), Gcgrflox/flox (Fl/Fl Gcgr), Hcn4-Cre and sinoatrial node (SAN)-specific Glp1r (Glp1rSAN KO) or Gcgr (GcgrSAN KO) knockout mice. Panels on the right are an expanded view of Glp1r or Gcgr mRNA expression in the right atrium which contains the sinoatrial node. Values are mean ± SE; n = 3–8 mice/group. *p < 0.05 vs. WT (One-way ANOVA).
3.5. GLP-1R agonists do not increase HR in the isolated heart ex vivo
Although endogenous GLP-1 exerts glucoregulatory actions via both neural circuits and activation of the β-cell GLP-1R, GLP-1R agonists robustly stimulate insulin secretion in isolated islets ex vivo. To determine whether the atrial GLP-1R was sufficient for GLP-1R-dependent activation of HR in a heart-autonomous manner, we assessed HR in isolated perfused mouse hearts ex vivo. Neither native GLP-1 nor lixisenatide increased HR under normoxic conditions (Figure 6A); however, HR was robustly increased by the β-adrenergic agonist isoproterenol in the same heart preparations ex vivo (Figure 6A). Similarly, we were unable to detect an effect of GLP-1 or lixisenatide on recovery of left ventricular developed pressure (LVDP) following induction of global no flow ischemia in isolated hearts ex vivo (Figure 6B), whereas ischemic preconditioning robustly increased recovery of LVDP in similar experiments. Moreover, increasing doses of lixisenatide (0.5–50 nM) had no effect on LVDP in aerobic perfused mouse hearts ex vivo (Figure 6A). Hence, although the atrial GLP-1R is required for GLP-1R-dependent control of heart rate in vivo, it is not sufficient for transduction of a GLP-1R-dependent signal to increase HR or ventricular function in denervated perfused hearts ex vivo.
3.6. Liraglutide does not directly increase atrial depolarization and HR ex vivo
To determine whether GLP-1R agonists such as liraglutide directly regulate atrial depolarization, we assessed frequency of the SAN firing rate in isolated mouse atrial preparations ex vivo. Direct application of saline or liraglutide to perfused atria had no measurable effect on SAN firing rates (Figure 7A). In contrast, administration of liraglutide (from the same stock solution) rapidly increased HR (relative to saline vehicle) in intact anesthetized mice (Figure 7B). Hence, GLP-1R agonists increase HR through both the autonomic nervous system and the atrial GLP-1R in vivo, but are unable to increase HR through the atrial GLP-1R in a heart-autonomous manner ex vivo.
Figure 7.

Effects of liraglutide on depolarization frequency of isolated atrial preparations and HR control in anaesthetized mice. (A) Atrial preparations from wild type mice were isolated, loaded with a voltage-sensitive fluorescent dye (di-4-ANEPPS), superfused, and imaged at 1000 frames/sec to determine baseline atrial beating rates (heart rate) as described in the Methods. The indicated doses of liraglutide or saline vehicle were superfused to examine their effects on baseline atrial depolarization frequencies. Data are expressed as the mean % ± SE of the baseline heart rate values for each group (saline baseline; 414 ± 27 bpm, liraglutide baseline; 411 ± 28 bpm). n = 17 atrial preparations/group. At the end of these experiments, atria were subsequently exposed to isoproterenol, which increased sinoatrial node firing rates by more than 200 b.p.m. (B) Heart rate was measured in isoflurane-anaesthetized wild type mice administered the indicated dose of liraglutide or saline. Data are expressed as the mean percentage increase ± SE above basal heart rate prior to drug administration. n = 10 mice/group. For (A) and (B), experimenters were blinded to control and drug group identities until after data processing and analysis.
4. Discussion
The development of glucose-lowering agents for the treatment of T2D is now associated with increased scrutiny as to whether and how these drugs independently modulate the risk of developing cardiovascular disease [23]. The non-glycemic actions of GLP-1R agonists encompass reduction of blood pressure and postprandial triglycerides, weight loss, and reduction of inflammation, actions predicted to mitigate the risk of developing cardiovascular complications [2]. Although short-acting drugs such as exenatide and lixisenatide have been associated with modest and transient increases in HR of several beats per minute (bpm) [24], [25], longer-acting agents such as dulaglutide and liraglutide are more potent in vivo and produce greater and more sustained increases in HR up to 10 bpm [26], [27], [28]. As increases in HR may enhance the susceptibility to tachyarrhythmia in at risk patients, understanding the mechanisms through which structurally distinct GLP-1R agonists control HR may have clinical relevance.
Here we show that HR is acutely increased following administration of lixisenatide or liraglutide, in control and Glp1rCM−/− mice. Moreover, the increase in HR is abolished by co-administration of the β−adrenergic antagonist propranolol and not further enhanced by attenuation of cholinergic signaling using the muscarinic receptor antagonist atropine. These findings demonstrate that the cardiac GLP-1R is not essential for the acute chronotropic response to GLP-1R agonists and are consistent with previous studies demonstrating GLP-1R dependent induction of central and peripheral catecholaminergic pathways in rats and mice [7], [8]. Moreover, our findings are in agreement with data demonstrating attenuation of the acute chronotropic response to exendin-4 by propranolol in rats [29]. GLP-1R agonism also potentiated plasma catecholamine responses to exercise in human subjects [30]. Hence the available evidence links acute GLP-1 receptor activation to catecholaminergic activation and induction of HR in vivo.
Although the cardiac GLP-1R was not required for the acute induction of HR by lixisenatide or liraglutide, the sustained induction of HR by liraglutide was attenuated in Glp1rCM−/− mice. It seems likely that once the robust yet transient induction of catecholaminergic signaling wanes, the contribution of intrinsic GLP-1R signaling to the induction of HR becomes evident. Indeed a functional role for the GLP-1R in atrial pacemaker cells was indirectly suggested by Pyke and colleagues who co-localized HCN, and GLP-1R expression within the monkey sinoatrial node [14]. Surprisingly, however, we were unable to attenuate atrial Glp1r expression through generation of Glp1rSAN−/− mice, whereas GcgrSAN−/− mice exhibit a significant reduction of atrial Gcgr expression. These findings indicate that the organization of cellular atrial GLP-1R expression may be species-specific, and the localization of cardiac GLP-1R expression may have evolved to serve a different subset of functions in mice vs. primates.
Several lines of evidence support the existence of the GLP-1R within cardiomyocytes in the mouse atria. First, we have detected expression of full length Glp1r mRNA transcripts in atria [31], and RNA levels of the full length atrial Glp1r were markedly reduced in floxed Glp1r mice, following recombination of the Glp1r allele using Cre under the control of the myosin heavy chain promoter [15]. In addition, we observed robust atrial natriuretic factor secretion in mice [12], [27] but not in humans [27], following administration of GLP-1R agonists. Moreover, GLP-1R agonists rapidly and directly increased cyclic AMP formation and atrial natriuretic factor secretion in isolated mouse atrial cardiomyocyte cultures ex vivo [12]. In contrast to intense GLP-1R expression in the monkey sinoatrial node [14], Richards and colleagues detected only a few scattered random atrial cells expressing a reporter gene under the control of the endogenous Glp1r promoter in transgenic mice [13] and single cell RNASeq has demonstrated low level but detectable expression of the Glp1r mRNA in mouse cardiomyocytes [32]. Nevertheless, the precise cellular identity of the repertoire of GLP-1R+ cells in mouse atria remains incompletely defined and it remains possible that the GLP-1R may also be expressed in non-cardiomyocyte cell types. Taken together, these disparate findings of atrial GLP-1R expression and function highlight the importance of further studies examining whether species-specific patterns of GLP-1R expression and function in the heart might confound interpretation of the cardiovascular actions of GLP-1 in different experimental settings.
Our current findings also indirectly establish the importance of neural and/or vascular inputs essential for coupling atrial GLP-1R activation to an increase in HR. Notably, we were unable to detect an increase in HR using native GLP-1 or lixisenatide in isolated perfused hearts. Furthermore, we failed to demonstrate direct GLP-1R-dependent activation of atrial firing in the mouse atria using preparations of exposed mouse atria ex vivo. In contrast, Glp1rCM−/− mice exhibit reductions in basal HR [15] and attenuated HR responses to liraglutide in vivo. Collectively, these findings imply that although the atrial GLP-1R is essential for HR regulation in mice in vivo, it is unable to activate a signal linked to pacemaker function and HR ex vivo in the absence of intact neurovascular input.
Our study has several limitations. First, we studied the short term effects of GLP-1R activation; however, the relative quantitative importance of the autonomic nervous system vs. the cardiac GLP-1R for the chronic effects of GLP-1R agonists cannot be inferred from our experiments. Second, we used healthy non-obese, normoglycemic mice in our experiments, whereas GLP-1R agonists are used clinically in human subjects with obesity and/or T2D. Third, we did not study the chronotropic actions of GLP-1R agonists in animals with heart disease, or in mouse models with experimental vagotomy or autonomic neuropathy, conditions that may modify the contributions of the cardiac GLP-1R vs the autonomic nervous system to control of HR. Finally, it remains possible that experiments using different concentrations of pharmacological antagonists to achieve parasympathetic or sympathetic nervous system blockade over different time periods, may produce different conclusions.
Nevertheless, our findings clearly extend current concepts defining how GLP-1R agonists acutely increase HR, integrating contributions from both catecholaminergic pathways and the cardiac GLP-1R (Figure 8). Moreover, our genetic data suggest that, unlike data reported for monkeys, atrial GLP-1R expression does not occur in the majority of HCN4+ cardiomyocytes. Finally, the importance of the cardiac GLP-1R for HR control is evident only in the living mouse, and is no longer evident when the atrial GLP-1R is activated in the isolated mouse heart or atrial preparation ex vivo. Collectively, these findings add to our understanding of the cellular sites and pathways linking GLP-1 receptor activation to the control of HR and cardiovascular function.
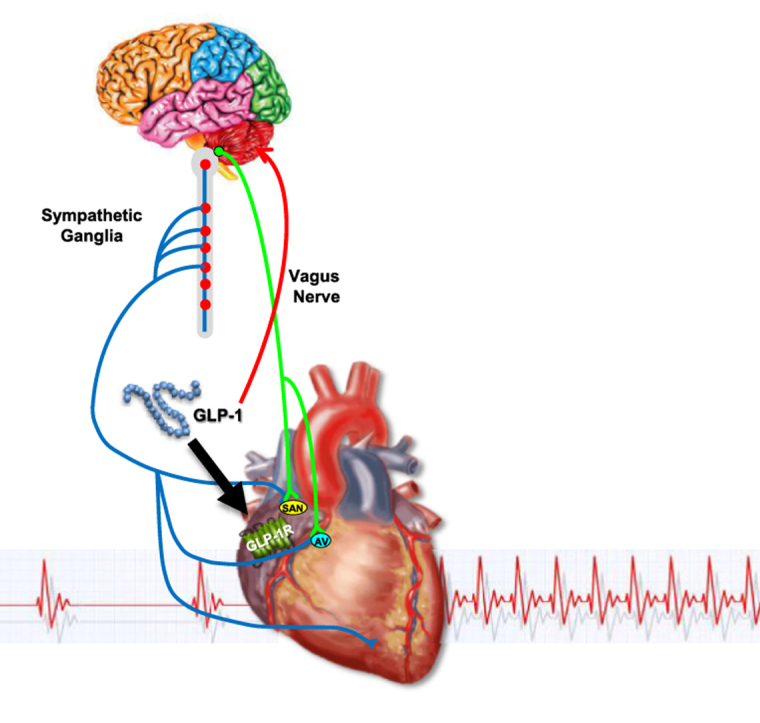
Figure 8.

Direct and indirect mechanisms linking GLP-1R signaling to control of heart rate. GLP-1 engages GLP-1 receptors in the central, peripheral, and autonomic nervous systems to enhance sympathetic nervous system (SNS) activity, and reduce parasympathetic nervous system (PNS) activity. GLP-1 additionally activates atrial GLP-1 receptors which also contribute, with inputs from the SNS and PNS, to control of heart rate.
Acknowledgments
These studies were supported in part by CIHR grant 123391, and investigator-initiated grants from Sanofi Inc. and Novo Nordisk Inc.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2017.08.010.
Conflict of interest
Dr. Drucker has served as an advisor or consultant within the past 12 months to Arisaph Pharmaceuticals Inc., Intarcia Therapeutics, Merck Research Laboratories, Novo Nordisk Inc., and Pfizer Inc. Neither Dr. Drucker nor his family members hold stock directly or indirectly in any of these companies. Dr. Heximer served as an advisor for Novo Nordisk during the course of these studies. Dr. Seeley has received research support from and/or has served as an advisor or consultant within the past 12 months to Ethicon Endo-Surgery/Johnson & Johnson, Orexigen, Novo Nordisk Inc., Daiichi Sankyo, Janssen/Johnson & Johnson, Novartis, Paul Hastings Law Firm, Zafgen, MedImmune, Sanofi, and Kallyope.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Burmeister M.A., Ayala J.E., Smouse H., Landivar-Rocha A., Brown J.D., Drucker D.J. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes. 2017;66(2):372–384. doi: 10.2337/db16-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drucker D.J. The cardiovascular biology of glucagon-like Peptide-1. Cell Metabolism. 2016;24(1):15–30. doi: 10.1016/j.cmet.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Stamler J., Vaccaro O., Neaton J.D., Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993;16(2):434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 4.Zinman B., Wanner C., Lachin J.M., Fitchett D., Bluhmki E., Hantel S. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New England Journal of Medicine. 2015;373(22):2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 5.Barragan J.M., Rodriguez R.E., Blazquez E. Changes in arterial blood pressure and heart rate induced by glucagon-like peptide-1-(7-36 amide) in rats. American Journal of Physiology. 1994;266:E459–E466. doi: 10.1152/ajpendo.1994.266.3.E459. [DOI] [PubMed] [Google Scholar]
- 6.Barragán J.M., Rodríguez R.E., Eng J., Blázquez E. Interactions of exendin-(9–39) with the effects of glucagon-like peptide-1-(7–36) amide and of exendin-4 on arterial blood pressure and heart rate in rats. Regulatory Peptides. 1996;67(1):63–68. doi: 10.1016/s0167-0115(96)00113-9. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto H., Lee C.E., Marcus J.N., Williams T.D., Overton J.M., Lopez M.E. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. Journal of Clinical Investigation. 2002;110(1):43–52. doi: 10.1172/JCI15595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto H., Kishi T., Lee C.E., Choi B.J., Fang H., Hollenberg A.N. Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. Journal of Neuroscience. 2003;23(7):2939–2946. doi: 10.1523/JNEUROSCI.23-07-02939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffioen K.J., Wan R., Okun E., Wang X., Lovett-Barr M.R., Li Y. GLP-1 receptor stimulation depresses heart rate variability and inhibits neurotransmission to cardiac vagal neurons. Cardiovascular Research. 2011;89(1):72–78. doi: 10.1093/cvr/cvq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharucha A.E., Charkoudian N., Andrews C.N., Camilleri M., Sletten D., Zinsmeister A.R. Effects of glucagon-like peptide-1, yohimbine, and nitrergic modulation on sympathetic and parasympathetic activity in humans. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2008;295(3):R874–R880. doi: 10.1152/ajpregu.00153.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumarathurai P., Anholm C., Larsen B.S., Olsen R.H., Madsbad S., Kristiansen O. Effects of liraglutide on heart rate and heart rate variability: a randomized, double-blind, placebo-controlled crossover study. Diabetes Care. 2017;40(1):117–124. doi: 10.2337/dc16-1580. [DOI] [PubMed] [Google Scholar]
- 12.Kim M., Platt M., Shibasaki T., Quaggin S., Backx P.H., Seino S. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nature Medicine. 2013;19(5):567–575. doi: 10.1038/nm.3128. [DOI] [PubMed] [Google Scholar]
- 13.Richards P., Parker H.E., Adriaenssens A.E., Hodgson J.M., Cork S.C., Trapp S. Identification and characterisation of glucagon-like peptide-1 receptor expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224–1233. doi: 10.2337/db13-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pyke C., Heller R.S., Kirk R.K., Orskov C., Reedtz-Runge S., Kaastrup P. GLP-1 receptor localization in monkey and human tissue; Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155(4):1280–1290. doi: 10.1210/en.2013-1934. [DOI] [PubMed] [Google Scholar]
- 15.Ussher J.R., Baggio L.L., Campbell J.E., Mulvihill E.E., Kim M., Kabir M.G. Inactivation of the cardiomyocyte Glucagon-Like Peptide-1 Receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Molecular Metabolism. 2014;3(5):507–517. doi: 10.1016/j.molmet.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meier J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nature Reviews Endocrinology. 2012;8(12):728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 17.Hoesl E., Stieber J., Herrmann S., Feil S., Tybl E., Hofmann F. Tamoxifen-inducible gene deletion in the cardiac conduction system. Journal of Molecular and Cellular Cardiology. 2008;45(1):62–69. doi: 10.1016/j.yjmcc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Wilson-Pérez H.E., Chambers A.P., Ryan K.K., Li B., Sandoval D.A., Stoffers D. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like peptide-1 receptor deficiency. Diabetes. 2013;62(7):2380–2385. doi: 10.2337/db12-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longuet C., Robledo A.M., Dean E.D., Dai C., Ali S., McGuinness I. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes. 2013;62(4):1196–1205. doi: 10.2337/db11-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sauve M., Ban K., Momen A., Zhou Y.-Q., Henkelman R.M., Husain M. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes following myocardial infarction in mice. Diabetes. 2010;59(4):1063–1073. doi: 10.2337/db09-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vedantham V., Galang G., Evangelista M., Deo R.C., Srivastava D. RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circulation Research. 2015;116(5):797–803. doi: 10.1161/CIRCRESAHA.116.305913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukharji A., Drucker D.J., Charron M.J., Swoap S.J. Oxyntomodulin increases intrinsic heart rate through the glucagon receptor. Physiology Reproductive. 2013;1(5) doi: 10.1002/phy2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drucker D.J., Goldfine A.B. Cardiovascular safety and diabetes drug development. Lancet. 2011;11(377):977–979. doi: 10.1016/S0140-6736(10)62299-4. [DOI] [PubMed] [Google Scholar]
- 24.Gill A., Hoogwerf B.J., Burger J., Bruce S., Macconell L., Yan P. Effect of exenatide on heart rate and blood pressure in subjects with type 2 diabetes mellitus: a double-blind, placebo-controlled, randomized pilot study. Cardiovascular Diabetology. 2010;96 doi: 10.1186/1475-2840-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier J.J., Rosenstock J., Hincelin-Mery A., Roy-Duval C., Delfolie A., Coester H.V. Contrasting effects of lixisenatide and liraglutide on postprandial glycemic control, gastric emptying, and safety parameters in patients with type 2 diabetes on optimized insulin glargine with or without metformin: a randomized, open-label trial. Diabetes Care. 2015;38(7):1263–1273. doi: 10.2337/dc14-1984. [DOI] [PubMed] [Google Scholar]
- 26.Ferdinand K.C., White W.B., Calhoun D.A., Lonn E.M., Sager P.T., Brunelle R. Effects of the once-weekly glucagon-like peptide-1 receptor agonist dulaglutide on ambulatory blood pressure and heart rate in patients with type 2 diabetes mellitus. Hypertension. 2014;64(4):731–737. doi: 10.1161/HYPERTENSIONAHA.114.03062. [DOI] [PubMed] [Google Scholar]
- 27.Lovshin J.A., Barnie A., DeAlmeida A., Logan A., Zinman B., Drucker D.J. Liraglutide promotes natriuresis but does not increase circulating levels of atrial natriuretic peptide in hypertensive subjects with type 2 diabetes. Diabetes Care. 2015;38(1):132–139. doi: 10.2337/dc14-1958. [DOI] [PubMed] [Google Scholar]
- 28.Kumarathurai P., Anholm C., Fabricius-Bjerre A., Nielsen O.W., Kristiansen O., Madsbad S. Effects of the glucagon-like peptide-1 receptor agonist liraglutide on 24-h ambulatory blood pressure in patients with type 2 diabetes and stable coronary artery disease: a randomized, double-blind, placebo-controlled, crossover study. Journal of Hypertension. 2017;35(5):1070–1078. doi: 10.1097/HJH.0000000000001275. [DOI] [PubMed] [Google Scholar]
- 29.Gardiner S.M., March J.E., Kemp P.A., Bennett T. Autonomic nervous system-dependent and -independent cardiovascular effects of exendin-4 infusion in conscious rats. British Journal of Pharmacology. 2008;154(1):60–71. doi: 10.1038/bjp.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khoo E.Y., Wallis J., Tsintzas K., Macdonald I.A., Mansell P. Effects of exenatide on circulating glucose, insulin, glucagon, cortisol and catecholamines in healthy volunteers during exercise. Diabetologia. 2010;53(1):139–143. doi: 10.1007/s00125-009-1579-1. [DOI] [PubMed] [Google Scholar]
- 31.Ali S., Ussher J.R., Baggio L.L., Kabir M.G., Charron M.J., Ilkayeva O. Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Molecular Metabolism. 2014;4(2):132–143. doi: 10.1016/j.molmet.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quaife-Ryan G.A., Sim C.B., Ziemann M., Kaspi A., Rafehi H., Ramialison M. Multi-cellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.117.028252. AHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.