

Abstract
Objective
Increasing plasma levels and activity of dipeptidyl peptidase-4 (DPP4 or CD26) are associated with rapid progression of metabolic syndrome to overt type 2 diabetes mellitus (T2DM). While DPP4 inhibitors are increasingly used as anti-hyperglycemic agents, the reason for the increase in plasma DPP4 activity in T2DM patients remains elusive.
Methods
We looked into the source of plasma DPP4 activity in a cohort of 135 treatment naive nonobese (BMI < 30) T2DM patients. A wide array of ex vivo, in vitro, and in silico methods were employed to study enzyme activity, gene expression, subcellular localization, protease identification, surface expression, and protein–protein interactions.
Results
We show that circulating immune cells, particularly CD4+ T cells, served as an important source for the increase in plasma DPP4 activity in T2DM. Moreover, we found kallikrein-related peptidase 5 (KLK5) as the enzyme responsible for cleaving DPP4 from the cell surface by directly interacting with the extracellular loop. Expression and secretion of KLK5 is induced in CD4+ T cells of T2DM patients. In addition, KLK5 shed DPP4 from circulating CD4+ T helper (Th)17 cells and shed it into the plasma of T2DM patients. Similar cleavage and shedding activities were not seen in controls.
Conclusions
Our study provides mechanistic insights into the molecular interaction between KLK5 and DPP4 as well as CD4+ T cell derived KLK5 mediated enzymatic cleavage of DPP4 from cell surface. Thus, our study uncovers a hitherto unknown cellular source and mechanism behind enhanced plasma DPP4 activity in T2DM.
Keywords: DPP4, KLK5, PBMC, T2DM, Th17 cells
Graphical abstract
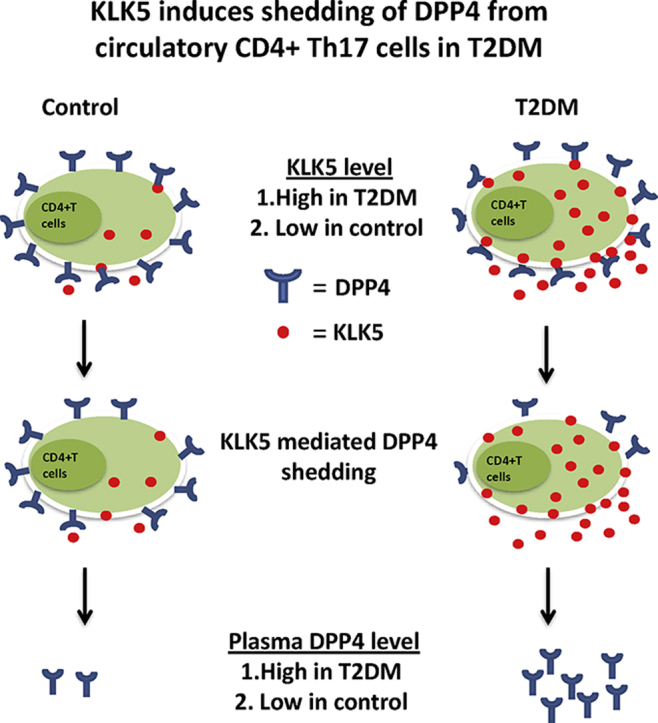
Highlights
-
•
Plasma DPP4 activity is high in T2DM patients.
-
•
PBMC and CD4+ T cells shed increased amount of DPP4 in T2DM patients.
-
•
KLK5 expression is enhanced in PBMC and CD4+ T cells in T2DM patients.
-
•
CD4+ T cell derived KLK5 induces DPP4 shedding in autocrine and paracrine manner.
-
•
Th17 cells are important source of plasma DPP4 in T2DM patients.
1. Introduction
Gut-derived incretin hormones glucagon-like peptide (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) directly activate insulin secretion via binding to their distinct receptors on the pancreatic islet β-cells [1]. Blunted first phase insulin secretion, attributed mainly to GLP-1 deficiency but also partly to GIP resistance, is intimately linked to T2DM [1]. The ubiquitously expressed negative regulator of incretin hormones, DPP4 exists either as a single pass type II transmembrane protein or as a shedded soluble form of serine protease that hydrolyzes proline or alanine from the N-terminus of different polypeptides including incretin hormones [1], [2]. Plasma levels and activity of DPP4 have been found to be associated with progression of metabolic syndrome [3]. Nevertheless, the reason behind this increase in plasma DPP4 activity in T2DM patients and the cellular source and mechanism of DPP4 shedding into plasma remains largely unaddressed.
T2DM, insulin resistance and associated clinical outcomes of metabolic syndrome are causally related to low grade chronic systemic inflammation, or metaflammation, in metabolically active tissues [4]. Recruitment and activation of plethora of immune cells, such as macrophage [5], [6], [7] conventional dendritic cells [5], [6], plasmacytoid dendritic cells [8], T cells [9], and neutrophils [10], in the metabolically active tissues including pancreas, adipose, and liver [7], [8], [11], [12] is well documented in patients as well as in animal models of the disease. Immune cell homing in specific organs leads to obesity associated deregulated metabolic outcome [7] and inflammatory cytokines secreted from immune cell regulate metabolic homeostasis [13] and insulin resistance [14]. Interestingly, DPP4 is expressed in a subset of circulating immune cells, especially T cells, where both the membrane bound as well as soluble DPP4 have been described as co-stimulatory molecules for T cell activation often via interactions with adenosine deaminase [15]. Moreover, it was shown that the DPP4 level in T cells was increased in T2DM patients and linked with long term glycemic control [16]. Of note here, DPP4 expression is maximum in IL-17 producing CD4+ T cells (Th17 cells), which are one of the major participants of inflammation in obesity associated T2DM [17].
In the present study, we tested the hypothesis that circulating immune cells, specifically the Th17 cells, may be the major source of plasma DPP4 abundance and activity in T2DM patients and explored the possible mechanisms that leads to release of soluble DPP4 in plasma of these patients.
2. Materials and methods
2.1. Patient recruitment
A total of 213 individuals were recruited for baseline experiment from Institute of Postgraduate Medical Education & Research (IPGME&R) following the American Diabetes Association (ADA) criteria [18]. Among them, 135 were treatment-naïve type 2 diabetes patients, and 78 were healthy, age-matched controls. All subjects gave informed consent, and research was approved by the ethical committees of IPGME&R hospital and CSIR-IICB. After overnight fasting, blood samples were drawn in EDTA tubes to measure glucose and glycated hemoglobin (HbA1c), total cholesterol (TC), total triglyceride (TG), high density lipoprotein (HDL), low density lipoprotein (LDL), insulin, leptin, GIP, GLP-1, DPP4 concentrations, and plasma DPP4 activity. Blood samples were also drawn 30 min after a 75 g oral glucose load. Plasma samples were obtained after centrifugation and stored at −80 °C until analysis. Homeostatic model assessment (HOMA) was utilized for quantification of insulin resistance (HOMA-IR) and β cell function (HOMA-β). Anthropometric measurements such as weight, height, waist and hip circumference, neck girth, and skin fold thickness at four different sites (abdomen, suprailiac, triceps, and thigh) was measured using Herpenden Skin fold caliper (Baty International Ltd). Skin fold measurements were taken for total body fat percentage calculation. Clinical and biochemical parameters of 213 subjects are shown in Table 1. Additionally, in a subset of randomly select cases (41 treatment naïve T2DM and 48 control subjects), an additional 5 ml of blood samples were taken for peripheral blood mononuclear cells (PBMC) isolation and for other downstream experiments.
Table 1.
Subject characteristics and biochemical parameters.
| Subjects | Control | T2DM | p-value control vs T2DM |
|---|---|---|---|
| N (M/F) | 78 (50/28) | 135 (91/56) | |
| Age (years) | 44.6 ± 9.63 | 47.2 ± 10.9 | 0.14 |
| BMI (kg/m2) | 23.43 ± 2.3 | 23.24 ± 2.34 | 0.621 |
| Waist circumference (cm) | 87.75 ± 9.52 | 90.24 ± 9.27 | 0.268 |
| Hip circumference (cm) | 93.23 ± 9.02 | 95.15 ± 7.8 | 0.315 |
| Neck girth (cm) | 33.5 ± 3.17 | 34.05 ± 3.61 | 0.508 |
| %FAT(skin fold) | 32.98 ± 6.8 | 31.55 ± 8.47 | 0.396 |
| Fasting blood glucose (mg/dl) | 95.22 ± 8.25 | 165.15 ± 65.23 | <0.001 |
| Post-prandial blood glucose (mg/dl) | 131.78 ± 22.82 | 311.35 ± 106.07 | <0.001 |
| HbA1c (%) | 5.35 ± 0.37 | 8.26 ± 2.54 | <0.001 |
| Triglyceride (mg/dl) | 132.72 ± 72.77 | 156.93 ± 86.7 | 0.017 |
| Total cholesterol (mg/dl) | 183.24 ± 36.69 | 195.14 ± 50 | 0.121 |
| HDL cholesterol (mg/dl) | 46.62 ± 11.59 | 43.04 ± 12.11 | 0.027 |
| LDL cholesterol (mg/dl) | 108.81 ± 29.26 | 117.32 ± 35.61 | 0.085 |
| VLDL cholesterol (mg/dl) | 29.24 ± 27.35 | 31.76 ± 19.03 | 0.05 |
| GGT | 25.85 ± 22.02 | 33.9 ± 26.30 | 0.019 |
| AST | 23.92 ± 17.92 | 29.33 ± 36.40 | 0.752 |
| ALT | 21.09 ± 24.08 | 25.22 ± 38.42 | 0.619 |
| Fasting insulin (μIU/ml) | 7.81 ± 5.71 | 9.33 ± 11.45 | 0.314 |
| HOMA-IR | 1.84 ± 1.37 | 3.61 ± 4.34 | 0.004 |
| HOMA-BETA | 94.37 ± 76.58 | 0.45 ± 0.51 | <0.001 |
| Leptin (μg/ml) | 9.2 ± 12 | 5.015 ± 4.886 | 0.133 |
| GIP fasting (pg/ml) | 122.56 ± 77.58 | 97.5 ± 38.88 | 0.12 |
| 30 min GIP (pg/ml) | 347 ± 98 | 371.72 ± 140 | 0.492 |
| GLP-1 fasting (pg/ml) | 5.35 ± 1.5 | 6.28 ± 2.58 | 0.112 |
| 30 min GLP-1 (pg/ml) | 13.77 ± 9.64 | 11.3 ± 8.6 | 0.421 |
| Plasma DPP4 activity (nmol/min/ml) | 16 ± 18.29 | 24.4 ± 21.31 | <0.001 |
| Plasma DPP4 levels (ng/ml) | 39.30 ± 12.28 | 48.54 ± 21.13 | <0.05 |
2.2. Peripheral blood mononuclear cell (PBMC) culture
PBMCs were isolated using HiSep™ LSM 1077 (Himedia Laboratories). Briefly, EDTA blood was diluted 1:1 with sterile PBS and layered on HiSep™ LSM 1077. Samples were centrifuged for 30 min at 500g without applying a brake. Comparable viability of PBMCs in control and T2DM patients was flow cytometrically determined by AnnexinV apoptosis kit (eBiosciences) (Supplementary Figure 1). The PBMC interface was carefully removed and washed twice with PBS. CD4+ T cells were purified from PBMC by standard MACS protocol (MiltenyiBiotec). Finally, PBMC and CD4+ T cells were cultured in anti-CD3 (eBioscience) coated plates treated with 2 μg/ml anti-CD28 antibodies (BD Pharmingen) in 10% FBS containing RPMI media. For DPP4 and KLK5 secretion analyses, culture supernatants were assayed for DPP4 and KLK5 levels by ELISA (R&D Systems). To identify the specific proteolytic enzyme involved in DPP4 shedding, 10 μM MMP inhibitor (GM 6001; Sigma) and 100 μg/ml KLK inhibitor (aprotinin; Sigma) were used for 24 h and 16 h respectively.
2.3. DPP4 enzyme assay
DPP4 activity in plasma and in PBMC lysates was assayed as described before [19]. Briefly, DPP4 activity was determined as the rate of 7-amino-4-methylcoumarin (AMC) cleavage per minutes per ml from the synthetic substrate H-glycyl-prolyl-AMC (Sigma). AMC fluorescence (excitation/emission – 380/460 nm) was measured in a plate reader (Synergy H1 multi-mode microplate reader, Biotek).
2.4. Protease profiling
After 48 h culture of PBMC, supernatant from 8 different individuals were pooled in a single tube and protease profile was determined using a protease array kit (R&D Systems).
2.5. Cells culture and transfection
Human DPP4 clone (Open Biosystems) and KLK5 clone (MyBioSource) were subcloned with a C-terminal HA-tag into IRES vector and pcDNA™3.1/myc-His(−) B vector respectively. The MMP2 clone was from Origene. HepG2 cells stably expressing HA-DPP4 were generated by transfection followed by puromycin selection (2 μg/ml, InvivoGen). Cells were washed with PBS and incubated with 150 nM human recombinant KLK5 (R&D Systems) for 4 h at 37 °C. DPP4 cleavage was analyzed in culture supernatants by ELISA (R&D Systems). In transient co-transfection experiments, 2 μg of KLK5 plasmid and 200 ng of GFP plasmid were used. A constitutive GFP-expressing vector served as a transfection control. Membrane bound DPP4 expression was detected at 40 h post-transfection.
2.6. Confocal microscopy
HepG2 cells stably expressing HA-DPP4 were grown in a confocal dish and transfected with plasmids. CD4+ T cells were isolated, stimulated with anti-CD3/CD28 antibodies and grown in polylysine coated confocal dishes. Cells were incubated with human recombinant KLK5 (150 nM) in PBS for 4 h at 37 °C and fixed with 4% paraformaldehyde (Sigma) for 20 min, washed with PBS, and blocked in 6% goat serum for 1 h. HA-DPP4 overexpressing HepG2 cells and CD4+ T Cells were immunostained with rabbit anti-HA antibody (1:50; Cell Signaling) and rabbit anti-human DPP4 mAb (1:100; Abcam) in 6% goat serum overnight at 4 °C, followed by incubation with FITC-conjugated goat anti-rabbit and Alexa Fluor 546-conjugated goat anti-rabbit antibody secondary antibody (1:1000; Life Technologies) for 1 h respectively. Nuclei were stained with Hoechst 33342 (Thermofisher Scientific) for 20 min. Cells were imaged with a Nikon A1R confocal imaging system (Tokyo, Japan) and fluorescence intensity in individual cells was analyzed using Image J software.
2.7. Gene expression analysis
Total cellular RNA from PBMC was isolated using TRizol reagent (Life Technologies, USA) and RNA concentration was measured by NanoDrop (Thermo Scientific). cDNA was synthesized from 1 μg RNA using reverse transcription supermix (BioRad). Gene expression was determined by quantitative PCR (LightCycler 96 real time PCR, Roche) using SYBR Green master mix (FastStart Universal SYBR Green Master, Roche) and analyzed by ΔΔCt method and normalized by 18S RNA. The primer sequences are available upon request.
2.8. siRNA electroporation
Isolated CD4+ T cells were cultured for 1 h in complete medium at 37 °C in CO2 incubator. Cells were carefully washed with PBS and resuspended in electroporation buffer (RPMI media, 1× glutamine, 1× sodium pyruvate, no antibiotics or FBS). 100 ng/μl of siKLK5 in 150 μl electroporation buffer containing cells were gently mixed and incubated for 10 min at room temperature. Then, the mixture was transferred into 0.2 cm Biorad gene pulser XL cuvettes and electroporated using 250 V and 950 uF capacitance pulse by Gene Pulser (Bio-Rad). Then, cells were transferred to 200 μl complete RPMI medium in 96 well plates and incubated at 37 °C in CO2 incubator for 3 days. Knockdown was checked after 72 h by qPCR.
2.9. Flow cytometry assay
CD4+ T cells were isolated by standard MACS protocol (Miltenyi Biotec). The enriched CD4+ T cells fraction were cultured overnight in anti-CD3 (eBioscience) coated plates, treated with 2 μg/ml CD28 antibody (BD Pharmingen). Then, 1 μg/ml ionomycine (Sigma) and 10 ng/ml PMA (Sigma) were applied to the media for 2 h before adding brefeldin. Finally, brefeldin was added during the last 4 h of culture. Cells were then stained with following anti-human, fluorophore tagged antibodies: CD8 PerCP (eBioscience) and CD26 PE (eBioscience). To assess phenotype of CD4+ T cells, the cells were permeabilized with saponin and stained with anti-human IFNγ-FITC (BD Bioscience) and IL-17A-EF450 (eBioscience) antibodies. Cells were gated on lymphocytes though forward and side scatter and the doublets were excluded. CD4 T cells (CD8 negative) were checked for the expression of IFNγ (FITC-H) and IL-17 (EF450). IFNγ and IL-17 positive cells were denoted as Th1 and Th17 cells respectively. Then expression of membrane bound CD26 (PE) was checked on both of these populations. Cells were analyzed on BD FACS Aria flow cytometer. Gating strategies are shown in Supplementary Figure 2A–C. For isolation of purified Th1 and Th17 cells, the CD4+ T cells were stained with anti-human CCR5-PE (R&D Systems)/CXCR3-Bv421 (BD Biosciences) and CCR6-APC/CCR4 PerCP (R&D Systems) antibodies respectively. The labeled cells were sorted by MoFlo XDP cell sorter (Beckman Coulter). Gating strategies with exclusion of doublets are shown in Supplementary Figure 2D–G.
2.10. Modeling and molecular dynamic simulation of full length DPP4 protein structure
The protein sequence of DPP4 was subjected to TMPred [20] TMHMM [21] and HMMTop [22] servers and the consensus predicted 22 (region: V7 – L28) residues long region was selected as transmembrane (TM) helix. The sequence (region: M1- T42) encompassing TM helix and flanking loop was subjected to Robetta server [23] for ab-initio modeling and further validated by Errat [24] and Rampage [25] validation programs. The crystal structure of the extracellular domain of DPP4 (PDB ID: 1N1M, region: S39 – P766) and the ab-initio model of the N-terminal part were used to build a three-dimensional (3D) model of full length DPP4 protein. Modeler v9.16 [26] was used to build the models, and top models were filtered based on the Modeler energy parameters, MOLPDF, and DOPE scores. The stereo-chemical properties and the fold compatibility of the final model were validated by Rampage [25] Verify3D [27], and Errat [24] programs. The final DPP4 model was inserted into a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer (dimension: 100 Å × 100 Å × 45 Å), and molecular dynamic (MD) simulation was carried out using Desmond [28] program for 100 ns (nanoseconds) at 300 K temperature. The OPLS_2005 force field parameters [29] were used for the simulation. MD simulation trajectory files were analyzed by various analysis programs of the Desmond package. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) plots suggest that the overall structure does not deviate too much (<1 Å), keeping a lower level of fluctuation for most of the regions of the protein (Supplementary Figure 3A, B). The secondary structural content of the initial structure remains stable throughout the length of the simulation (Supplementary Figure 3C), whereas the overall structure became slightly more compact (Supplementary Figure 3D). However, the energy of the system evolved favorably with the duration of the simulation indicating better stability of the final DPP4 structure (Supplementary Figure 3E).
2.11. Molecular docking and MD simulation of DPP4–KLK5 docked complex
The stable DPP4 structure embedded in POPC bilayer obtained after 100 ns simulation was docked with the crystal structure of KLK5 (PDB ID: 2PSY) using protein–protein docking mode of PatchDock [30] program. Residues 38–40 of DPP4 were considered to be important for binding as the proteolytic cleavage by KLK5 active sites resides at D38-S39-R40 region of DPP4. Hence, region 38–40 of DPP4 and the known catalytic sites of KLK5 were used as seed regions for docking experiment. Approximate interface area and ΔG of binding of the docked complexes were calculated using the PDBePISA server [31]. The docking solutions were screened, and the best DPP4–KLK5 docked complex was identified based on interface area, ΔG of binding, and critical inspection of favorable interactions between the two proteins. The selected docked complex was energetically stable as minimal energy gain (≤1.5%) was obtained even after 100 ns of simulation (Supplementary Figure 1F). The energy gain of the selected complex is relatively lesser and/or comparable to that of known of complexes (Supplementary Figure 1F).
2.12. Statistics
Statistical analysis was performed using GraphPad Prism 5 software (La Jolla, CA, USA). Descriptive summary of the data has been done by mean and standard deviation. Numerical variables have been compared between groups by independent-samples t-test or Mann–Whitney U test as appropriate. Pearson's correlation coefficient ‘r’ or Spearman's rank correlation coefficient rho (ρ) have been calculated to explore association between variables. P-value less than 0.05 considered statistically significant.
3. Results
3.1. Plasma DPP4 activity is increased in non-obese T2DM patients
To explore the source of plasma DPP4 in T2DM, we recruited 213 treatment naïve subjects (control, n = 78; T2DM, n = 135). The clinical and biochemical characteristics of the cohort are shown in Table 1. Pancreatic β-cell function represented by HOMA-β is impaired while insulin resistance represented by HOMA-IR is increased in T2DM patients. Of note, adiposity among the groups were equivalent as no statistical differences seen in body mass index (BMI), neck girth and plasma leptin levels. Thus, our T2DM cohort represented normoinsulinemic, hyperglycemic, non-obese (BMI < 30) subjects. Fasting plasma DPP4 enzymatic activity was found to be significantly increased in T2DM (p < 0.001) populations from the control group (Figure 1A), in line with previous reports [32]. DPP4 protein levels were also increased; however, we did not find any difference in plasma levels of GLP-1 or GIP either in fasting or 30 min after glucose ingestion (Table 1).
Figure 1.

PBMC is an important source of increased plasma DPP4 activity in T2DM patients. DPP4 activity in plasma and peripheral blood mononuclear cells (PBMC) of treatment naïve type 2 diabetes (T2DM) patients and healthy control subjects. DPP4 expression in PBMC was measured and linear regression analysis was performed with Spearman correlation. (A–C) Comparison of plasma (control n = 78 & T2DM n = 135) PBMC DPP4 activity (control n = 46 & T2DM n = 35) and PBMC DPP4 gene expression (control n = 49 & T2DM n = 41). (D–G) Linear regression analysis of PBMC DPP4 activity with plasma DPP4 activity (D&F), and PBMC DPP4 activity and gene expression (E&G). (H, I) PBMCs from control (n = 22) and treatment naïve T2DM subjects (n = 28) were cultured for 48 h and supernatant (H) as well as cellular DPP4 levels (I) were analyzed by ELISA. Statistical analysis was performed by Mann–Whitney U test and Student's t test with Spearman correlation; *p < 0.05, ***p < 0.001.
3.2. PBMC is an important source for plasma DPP4 activity
To explore the validity of our hypothesis that immune cells circulating in the peripheral blood may be the source of DPP4 abundance in diabetes, we determined DPP4 activity and gene expression in PBMCs isolated from the recruited individuals. Cell viability in the isolated PBMCs in control and T2DM patients was comparable (Supplementary Figure 1). In contrast to increased plasma DPP4 activity, we did not detect any corresponding increase either in PBMC DPP4 activity (Figure 1B) or in gene expression (Figure 1C) in the T2DM population. However, DPP4 enzymatic activity in PBMC homogenate positively correlated with both DPP4 gene expression (r = 0.538, p = 0.03; Figure 1D) and plasma DPP4 activity (r = 0.549, p < 0.001; Figure 1E) in the control population. In T2DM individuals, however, there was no correlation between PBMC DPP4 expression and PBMC DPP4 enzymatic activity (Figure 1G). More interestingly, there was actually a negative correlation between PBMC DPP4 activity and plasma DPP4 activity in T2DM (Figure 1F). These correlation studies indicated that, in the control population, plasma DPP4 activity is contributed by PBMC DPP4 levels and reversal of this correlation pointed to possible sourcing of plasma DPP4 activity from cellular DPP4 in peripheral blood in T2DM. To explore this possibility in vitro, we measured release of DPP4 into the PBMC culture supernatant from control and T2DM patients. As shown in Figure 1H, DPP4 release into the PBMC culture supernatants was significantly enhanced in T2DM patients, compared to controls, thus corroborating the correlation studies. However, we did not find any difference in DPP4 levels in the PBMC homogenate (Figure 1I) suggesting that only a small fraction of total cellular DPP4 is shed from PBMC.
3.3. KLK5 expression and secretion are enhanced from PBMC of T2DM patients
The release of soluble DPP4 protein into PBMC culture supernatants encouraged us to identify the proteases that may be differentially enriched in or secreted from the PBMCs in T2DM patients and which potentially could cleave off DPP4 from the immune cell surface. We pooled PBMC supernatants from eight control subjects and eight T2DM patients to check for the relative abundance of secreted proteases from a panel of 34 proteases. We found that serine protease KLK5 was highly enriched in PBMC culture supernatants of T2DM patients (Figure 2A,B). As expected, the presence of DPP4 in the PBMC supernatants was also increased in T2DM (Figure 2A,B). Gene expression of KLK5 in PBMC was also significantly up regulated in T2DM (Figure 2C). However, we did not find any increase in plasma KLK5 levels in T2DM patients (Figure 2D). Next, to examine whether KLK proteases are responsible for DPP4 shedding we used a broad spectrum KLK inhibitor aprotinin (KLKi). As shown in (Figure 2E), incubation with KLKi markedly reduced DPP4 shedding from both control and T2DM PBMCs while the broad spectrum MMP inhibitor GM 6001 (MMPi) had no such effect (Figure 2F). Thus, unlike adipose and smooth muscle, in which DPP4 is cleaved by specific MMP [33], KLKs serve as the protease for DPP4 shedding from circulatory immune cells.
Figure 2.

KLK5 is the proteolytic enzymes involved in the DPP4 shedding from PBMC. (A, B) PBMC culture supernatants were pooled (n = 8) and a human protease array profile was performed. Spots for KLK5 are marked (A). Data are represented as relative pixel density of significantly altered proteases in T2DM (B). (C) Comparison of PBMC KLK5 gene expression (control n = 17 & T2DM n = 20). (D) Plasma KLK5 levels (control n = 29 & T2DM n = 36). (E, F) PBMC was incubated for 24 h and 16 h with 10 μM MMP inhibitor (MMPi; control n = 6 and T2DM n = 9) and 100 μg/ml KLK inhibitor (KLKi; control n = 6 and T2DM n = 12) respectively. DPP4 release in the culture supernatants were measured by ELISA. Statistical analysis was calculated by Mann–Whitney U test and shown as box plots; p**<0.01, ***p < 0.001.
3.4. KLK5 cleaves DPP4 on the extracellular domain
To examine whether KLK5 could interact and cleave membrane bound DPP4 on the cell surface, we first adopted molecular modeling and docking approaches. The validated model of the full length DPP4 protein embedded within POPC bilayer was stabilized using 100 ns molecular dynamic simulation (Figure 3A). 3D structure of KLK5 (PDB ID: 2PSY) was docked onto the DPP4 model obtained after 100 ns simulation. Most likely mode of interaction was selected based on interface area (1335.5 Å2), ΔG of binding (−8.4 kcal/mol) and critical inspection of favorable interactions (5 probable hydrogen bond and 1 salt bridge interaction). Figure 3B shows the overall binding mode whereas Figure 3C shows the structural proximities (≤5 Å) of critical residues for such interaction leading to proteolytic cleavage of the DPP4 protein. The binding energy of DPP4–KLK5 docking complex is better than or comparable to (Supplementary Figure 4) that of few randomly selected known protein complexes (Supplementary Table 1) having similar sized interface area.
Figure 3.

KLK5 cleaves DPP4 from cell surface through direct interaction. (A) Model structure of the full length DPP4 protein where DPPIV_N domain is marked in purple, Peptidase_S9 domain in green, and linker domain in orange, respectively. The transmembrane (TM) region is marked in blue, embedded in POPC bilayer. Initial (0th ns) and end structure (100th ns) of the 100 ns molecular dynamics simulation. (B) Probable mode of docking of KLK5 (shown in pale blue) with the DPP4. Cleavage site of DPP4 (38DSR40) and the catalytic triad (H57, D103 and S197) of KLK5 are shown in yellow and magenta, respectively. Other associated active sites of KLK5 are shown in chocolate brown. (C) Close up version of the binding region where cleavage site of DPP4 (38DSR40) and the catalytic triad (H57, D103 and S197) of KLK5 are shown in yellow and magenta, respectively. (D) Non-permeabilized HA-DPP4 expressing HepG2 cells were stained with anti-HA antibody (green) and Hoechst (blue) and visualized with fluorescence confocal microscopy. Left panel: Representative fields of control. Right panel: rKLK5 treated cells. Scale bar = 20 μm. (E) Fluorescence intensity for 50 different cells across three fields was measured by Image J software. (F) HepG2 cells expressing HA tagged DPP4 were treated with recombinant KLK5 for 4 h and culture supernatants were analyzed by DPP4 ELISA. (G) Fluorescence confocal microscopy images of the stable HA-DPP4 expressing HepG2 cells co-transfected with KLK5 and GFP plasmids. A constitutive GFP-expressing vector served as a transfection control. Membrane bound DPP4 expression was detected with anti-HA antibody (red) at 40 h post-transfection. Scale bar = 20 μm. (H) Fluorescence intensity was calculated of 40 different GFP positive and negative cells across three fields by Image J software. Data are shown as the mean ± SD. Statistical analysis was performed by Student's t test; *p < 0.05, **p < 0.01, ***p < 0.001.
For the experimental validation of the modeling data in the cellular context, we generated HepG2 cells stably expressing HA-tagged DPP4 (Supplementary Figure 5A–C) and treated them with recombinant KLK5 enzyme. As shown in Figure 3D,E, KLK5 treated cells revealed a ∼3 fold decrease in the surface expression of DPP4, as shown by confocal fluorescence microscopy. Consistently, KLK5 treatment induced ∼5 fold increases in the release of soluble DPP4 in the media (Figure 3F). Further, when KLK5 is overexpressed (Supplementary Figure 5D–F) in these cells, we found a preferential decrease in the surface expression of HA-tagged DPP4 from the KLK5 expressing cells (Figure 3G,H). DPP4 expression was also low in the surrounding untransfected cells, suggesting that KLK5 might cleave membrane bound DPP4 predominantly by paracrine and autocrine fashion. Expression of MMP2, another candidate protease, however, did not induce DPP4 shedding in these cells (Supplementary Figure 5G–I). Taken together, our data suggest that KLK5 cleaves DPP4 from the cell surface by directly interacting with the extracellular loop.
3.5. CD4+ T cell derived KLK5 cleaves DPP4 from cell surface
Th1 and Th17 type CD4+ T cells significantly contribute to the DPP4 levels in PBMC with Th17 cells showing higher DPP4 expression [17]. Regulatory T cells (Treg), on the contrary, do not express DPP4 [34]. As DPP4 shedding from PBMC in T2DM was higher, we wondered if CD4+ T cells from T2DM patients would also share these characteristics. When we stimulated PBMC with anti-CD3 and anti-CD28 antibodies for T cell receptor crosslinking, we found that the release of DPP4 was increased in PBMC culture supernatants in T2DM (Figure 4A) without any change in total cellular protein (Figure 4B). Although DPP4 expression remained similar in the purified CD4+ T cells (Figure 4C), KLK5 expression was significantly enhanced in T2DM (Figure 4D). However, secretion of both DPP4 (Figure 4E) and KLK5 (Figure 4F) from the purified CD4+ T cells was increased in T2DM patients and was significantly correlated (r = 0.68, p ≤ 0.0001; Figure 4G), suggesting that CD4+ T derived KLK5 could be responsible for enhanced DPP4 shedding in T2DM. To further examine whether CD4+ T cell derived KLK5 could cleave DPP4 from these cells, we knocked down KLK5 in the CD4+ T cell from control subjects by electroporating siRNA against KLK5 gene. Knockdown of KLK5 did not alter DPP4 expression (Figure 4H) but significantly reduced secretion of DPP4 from CD4+ T cells (Figure 4I). Moreover, recombinant KLK5 treatment yielded enhanced release of DPP4 in the culture supernatants (Figure 4J) and decreased surface expression of DPP4 in CD4+ T cells from healthy subjects (Figure 4K,L). Taken together, these results thus reveal that CD4+ T cells from T2DM patients shed increased amount of DPP4 through KLK5 dependent catalysis.
Figure 4.

Increased shedding of DPP4 from T cells of T2DM patients. (A, B) PBMC culture supernatants (A) and cell lysates (B) were analyzed for DPP4 by ELISA after 48 h (control n = 9 & T2DM n = 10). (C–D) Relative gene expression in CD4+ T cells for DPP4 and KLK5 in T2DM and control population (control n = 18 & T2DM n = 27). (E–F) Secreted DPP4 as well as KLK5 protein were measured by ELISA (control n = 12 & T2DM n = 27). (G) Linear regression analysis of secreted DPP4 protein with secreted KLK5 protein in overall population. (H–I) siKLK5 was electroporated in CD4+ T cells and gene expression and secreted DPP4 levels were measured after 72 h (n = 7). (J) Stimulated CD4+ T cells were treated with human recombinant KLK5 (150 nM) for 4 h and culture supernatant was analyzed for DPP4 by ELISA (n = 8). (K) Activated CD4+ T cells were cultured in a polylysine coated plate for 16 h, stained with DPP4 mAb (red) and visualized with fluorescence confocal microscopy. Scale bar = 10 μm. (L) Fluorescence intensity was calculated of 100 different cells across four fields by Image J software. Data are shown as the mean ± SD. Statistical analysis was performed by Mann–Whitney U test and Student's t test with Spearman correlation; *p < 0.05, **p < 0.01, ***p < 0.001.
3.6. Shedding of DPP4 from Th17 cells is enhanced in T2DM
To investigate any preferential increase in the DPP4 expressing CD4+ T cell subsets, we measured gene expressions of TBX21 and RORC, the master regulator transcription factors driving Th1 and Th17 phenotypes of CD4+ T cells, respectively, and the percentage of Th1 (defined by IFNγ+CD4+ T cells) and Th17 (defined by IL-17+CD4+ T cells) cells in the PBMC by flow cytometry. We did not detect any difference either in gene expression (Supplementary Figure 6A, B) or in the percentage of Th1 or Th17 cells of control and T2DM populations (Supplementary Figure 6C, D). However, KLK5 expression was significantly higher in the purified Th17 cells (defined by CCR6+CCR4+CD4+ T cells) from T2DM patients but not in the Th1 cells (defined by CCR5+CXCR3+CD4+ T cells) (Figure 5A,B). Given that PBMC derived DPP4 contributes to plasma DPP4 activity and that shedding of DPP4 from CD4+ T cells could be one important mechanism in T2DM, we next sought to examine whether T helper cell subsets contribute to plasma and PBMC DPP4 activity. We looked at the correlations of plasma and PBMC DPP4 activity with the percentages of IL-17+CD26+ T cells (Th17 cells) in circulation. Interestingly, we found a significant negative correlation with plasma DPP4 activity and a positive association with PBMC DPP4 activity with fraction of CD26+IL17+ subset within CD4+ T cells (although the later did not reach statistical significance; r = 0.443, p = 0.098) (Figure 5C,D). In contrast, percentages IFNγ+CD26+ T cells (Th1 cells) revealed a significant correlation with PBMC DPP4 activity but had no association with plasma DPP4 activity (Figure 5E,F). Our findings thus suggested that abundance of DPP4 activity in plasma of T2DM patients possibly results from an enhanced shedding of soluble DPP4 proteins from the Th17 cells. When we looked at surface expression of DPP4/CD26 on these T helper cell subsets, we found no difference in the percentage of IFNγ+CD26+ T cells between control and T2DM (Figure 5G,H). In contrast, we found a significant decrease of the DPP4/CD26 surface expression in IL-17+CD26+ T cells from T2DM patients (Figure 5I,J). These results thus confirmed that rather than Th1 cells, Th17 cell-derived DPP4 contributes to plasma DPP4 activity in T2DM.
Figure 5.

Decreased surface expression of DPP4 in Th17 cells of T2DM patients. (A, B) Comparison of relative KLK5 gene expression in sorted Th1 and Th17 cells (control, n = 10 & T2DM, n = 14). (C, D) Correlations of percentage of CD26+IL17A+CD4+ T cells with plasma DPP4 activity (A; n = 26) and PBMC DPP4 activity (B; n = 16). (E, F) Correlations of percentage of CD26+IFNγ+CD4+ T cells with plasma DPP4 activity (C; n = 30) and PBMC DPP4 activity (D; n = 15). (G, H) Representative flow plots for IFNγ and CD26 staining (E) and the summary (F) of all the analyzed samples are shown (control n = 17& T2DM n = 12). (I, J) IL-17A and CD26 staining from healthy control (n = 20) and treatment naïve T2DM (n = 19) were determined by flow cytometry. Statistical analysis was performed by Mann–Whitney U test and Student's t test with Spearman correlation; *p < 0.05, ***p < 0.001.
4. Discussion
DPP4 is ubiquitously expressed, but the sources of the soluble form of plasma DPP4 in T2DM is not precisely known. In rodents, bone marrow derived cells contribute 25–40% of plasma DPP4 [35], [36] while liver contributes significantly to plasma DPP4 pool in human [37]. Although immune cells contribute to the circulating DPP4 pool in mice, DPP4 in endothelial cells is crucial for the inactivation of incretin hormones, suggesting that an increase in circulatory DPP4 per se might not be critical for incretin response and hence glycemic control [36]. Thus, the physiological implications of immune cell derived DPP4 is still elusive and moreover in humans the contribution of circulating immune cells in sourcing plasma DPP4 specifically in the context of T2DM has not yet been investigated. Here we show that increased plasma DPP4 activity in T2DM patients is linked with the enhanced shedding of DPP4 from circulating Th17 cells. Moreover, we identify KLK5 as the putative protease that cleaves DPP4 from the CD4+ T cell surface. We also found that Th17 cells in T2DM patients showed reduced surface expression of DPP4/CD26, and the level of this reduction was positively correlated with plasma DPP4 activity in the same patients. This study thus provides data that define a major cellular source and mechanism behind plasma DPP4 abundance in T2DM.
Consistent with the notion that T2DM patients usually do not exhibit lowered GLP-1 secretion in response to glucose ingestion [38], we did not find any alteration in either basal or glucose induced surge in GLP-1 and GIP levels. In agreement with results from various population groups [32], [39], our data depict that plasma DPP4 activity is significantly increased in the Indian T2DM patients. However, mechanisms for the increase in plasma DPP4 activity in T2DM have not yet been thoroughly addressed. Obesity and associated metaflammation have been postulated for this phenomenon, but, as the present cohort is comprised of treatment naive, non-obese subjects, our data indicate that hyperglycemia alone could be responsible for increased plasma DPP4 activity.
It is becoming evident that an increase in pro-inflammatory Th1 and Th17 subsets along with decrease in anti-inflammatory Treg cells are associated with obesity and T2DM [40]. A predisposition toward Th17 cell response has been shown in obese women [41] and in diet-induced obesity model in mice [42]. T cell inflammation in obesity-associated T2DM patients has been characterized by elevated number of circulatory Th17 cells, increased Th17-derived cytokine production, and activation of Th17 signature genes [41], which is supported by the inflammatory milieu conferred by B cells [43]. Thus, in addition to the detrimental role of Th17 in type 1 diabetes [44], [45], enhanced Th17-associated pro-inflammatory skewing in T2DM also implicates Th17 cells as major players in the metaflammation associated with T2DM. However, we did not find any increase in the circulatory Th17 cell number, either in the freshly isolated PBMC or following CD4+ T cell culture and activation with anti-CD3/CD28 antibodies. These differences could be due to our non-obese, treatment naïve cohort with shorter duration of disease.
Enhanced expression and secretion of CD4+ T cell-derived KLK5 in T2DM is suggestive of a functional paracrine network that leads to augmented DPP4 cleavage. Moreover, conspicuous presence of KLK5 transcript with a significant positive association between secreted KLK5 with shed DPP4 from CD4+ T cells further indicates that KLK5 can also work through an autocrine system. Enhanced expression of KLK5 in Th17 cells from T2DM patients further demonstrate that circulatory Th17 pool could be an important source of plasma DPP4. KLK5 expression is induced by cytokines in neutrophils [46] and Th17 response is usually associated with neutrophil infiltration [47], [48], suggesting that neutrophils can also be another source of the protease that leads to DPP4 shedding from the Th17 cells. Homing of circulatory Th17 cells in tissues such as intestine, lung, and skin has been well characterized [49], and, interestingly, KLK5 is maximally expressed in skin [50]. Thus, skin being a site of KLK5 and Th17 cell interactions can also be one of the possibilities. However, this warrants further investigations.
Immune cell derived DPP4 contributes significantly to plasma DPP4 pool [37]; thus, DPP4 expressed in immune cells represents a novel target for various inflammatory diseases including T2DM [51]. Indeed, DPP4 inhibitors such as sitagliptin exert an anti-inflammatory effect in T2DM by reducing plasma inflammatory cytokine levels as well as inflammatory gene expression in PBMC [52]. Interestingly, nonobese patients of Asiatic origin respond better to gliptin therapy [53], which at least partially could be implicated to T cell inflammation. Moreover, our data on KLK5 mediated shedding of DPP4 opens up the possibility of utilizing this interaction and catalysis for therapeutic targeting in metaflammation and T2DM.
5. Conclusion
Collectively, our results show that in T2DM patients, circulating CD4+ T cells, specifically cells having the Th17 phenotype, shed cleaved DPP4 protein into plasma due to the enzymatic action of KLK5. Expression and secretion of KLK5 is enhanced in CD4+ T cells of T2DM patients. We also gathered structural insights into the interaction of KLK5 with DPP4. Thus, we uncovered a hitherto unknown link between T cell inflammation and aberrant plasma DPP4 abundance in T2DM.
Author contributions
TN did the patient sampling and performed most of the experiments. KK performed all the in silico experiments. ARG and DS conducted the flow cytometry experiments. AS and DR contributed to Th17 cell experiments and analyzed data. SM performed the clinical evaluation and stratification of recruited patients. SC contributed to data analysis of in silico work. PC and DG contributed to the study concept and design, data analysis, and writing of the manuscript.
Acknowledgments
We thank the volunteer participants in this study. This work has been supported by grants to PC by Department of Biotechnology, West Bengal (578(Sanc)/BT(Estt)/RD16/2015) and Council of Scientific and Industrial Research (CSIR), India (BSC 0206) and to DG by Government of India's Science and Engineering Board's Ramanujan Fellowship (SR/S2/RJN-92/2011). TN received a research fellowship from CSIR (31/2 (956)/2013-EMR-I) and research grant from Research Society for the Study of Diabetes in India (RSSDI/WB/RG-3). KK received a research fellowship from DBT, Govt. of India. We thank Utpalendu Ghosh and Pragnya Chakraborty for help with confocal microscopy and cell biology experiments respectively. The corresponding author takes full responsibility to all the data in the study, including study design and data analysis.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2017.09.004.
Contributor Information
Dipyaman Ganguly, Email: dipyaman@iicb.res.in.
Partha Chakrabarti, Email: pchakrabarti@iicb.res.in, partha.iicb@gmail.com.
Conflict of interest
Authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Drucker D.J. The biology of incretin hormones. Cell Metabolism. 2006;3:153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Drucker D.J., Sherman S.I., Bergenstal R.M., Buse J.B. The safety of incretin-based therapies—review of the scientific evidence. The Journal of Clinical Metabolism & Endocrinology. 2011;96:2027–2031. doi: 10.1210/jc.2011-0599. [DOI] [PubMed] [Google Scholar]
- 3.Yang F., Zheng T., Gao Y., Baskota A., Chen T., Ran X. Increased plasma DPP4 activity is an independent predictor of the onset of metabolic syndrome in Chinese over 4 years: result from the China national diabetes and metabolic disorders study. PloS One. 2014;9:e92222. doi: 10.1371/journal.pone.0092222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruce B.D., Maria I.S., James S.P., Christie M.B., David C., Alvaro V. Low-grade systemic inflammation and the development of type 2 diabetes the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- 5.Chien P.L., Seongah H., Takafumi S., Alan R.T. The macrophage at the crossroads of insulin resistance and atherosclerosis. Circulation Research. 2007;100:1546–1555. doi: 10.1161/CIRCRESAHA.107.152165. [DOI] [PubMed] [Google Scholar]
- 6.Pickup J.C. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 7.Donath M.Y., Shoelson S.E. Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh A.R., Bhattacharya R., Bhattacharya S., Nargis T., Rahaman O., Duttagupta P. Adipose recruitment and activation of plasmacytoid dendritic cells fuel metaflammation. Diabetes. 2016;65:3440–3452. doi: 10.2337/db16-0331. [DOI] [PubMed] [Google Scholar]
- 9.Markus F., Laura H., Daniela C., Afia N., Jamie W., Ali N. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampson M.J., Davies I.R., Brown J.C., Ivory K., Hughes D.A. Monocyte and neutrophil adhesion molecule expression during acute hyperglycemia and after antioxidant treatment in type 2 diabetes and control patients. Arteriosclerosis Thrombosis and Vascular Biology. 2002;22:1187–1193. doi: 10.1161/01.atv.0000021759.08060.63. [DOI] [PubMed] [Google Scholar]
- 11.Kreier F., Kap Y.S., Mettenleiter T.C., van Heijningen C., van der Vliet J., Kalsbeek A. Tracing from fat tissue, liver, and pancreas: a neuroanatomical framework for the role of the brain in type 2 diabetes. Endocrinology. 2006;147:1140–1147. doi: 10.1210/en.2005-0667. [DOI] [PubMed] [Google Scholar]
- 12.Maarten E.T., Mathijs C.B., Petra J.P., Saskia B., Jan Hein T.W., Roger K.S. Pancreatic fat content and β-cell function in men with and without type 2 diabetes. Diabetes Care. 2007;30:2916–2921. doi: 10.2337/dc07-0326. [DOI] [PubMed] [Google Scholar]
- 13.Maya E., Kotas, Ruslan M. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steven E.S., Jongsoon L., Allison B.G. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorrell M.D., Gysbers V., McCaughan G.W. CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scandinavian Journal of Immunology. 2001;54:249–264. doi: 10.1046/j.1365-3083.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 16.Lee S.A., Kim Y.R., Yang E.J., Kwon E.J., Kim S.H., Kang S.H. CD26/DPP4 levels in peripheral blood and t cells in patients with type 2 diabetes mellitus. The Journal of Clinical Endocrinology and Metabolism. 2013;98:2553–2561. doi: 10.1210/jc.2012-4288. [DOI] [PubMed] [Google Scholar]
- 17.Bengsch B., Seigel B., Flecken T., Wolanski J., Blum H.E., Thimme R. Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26) The Journal of Immunology. 2012;188:5438–5447. doi: 10.4049/jimmunol.1103801. [DOI] [PubMed] [Google Scholar]
- 18.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care Supplement. 2013;1:S67–S74. doi: 10.2337/dc13-S067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirino Y., Sei M., Kawazoe K., Minakuchi K., Sato Y. Plasma dipeptidyl peptidase activity correlates with body mass index and the plasma adiponectin concentration in healthy young people. Endocrine Journal. 2012;59:949–953. doi: 10.1507/endocrj.ej12-0158. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann K., Stoffel W. Tmbase – a database of membrane spanning proteins segments. Biological Chemistry Hoppe-Seyler Journal. 1993;374:166. http://www.ch.embnet.org/software/tmbase/TMBASE_doc.html [Google Scholar]
- 21.Krogh A., Larsson B., von Heijne G., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 22.Tusnady G.E., Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 23.Kim D.E., Chivian D., Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Research. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colovos C., Yeates T.O. Verification of protein structures: patterns of non-bonded atomic interactions. Protein Science. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lovell S.C., Davis I.W., Arendall W.B., 3rd, De Bakker P.I., Word J.M., Prisant M.G. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 26.Webb B., Sali A. Current protocols in bioinformatics. John Wiley & Sons, Inc; 2014. Comparative protein structure modelling using modeller.http://onlinelibrary.wiley.com/doi/10.1002/0471250953.bi0506s47/abstract [DOI] [PubMed] [Google Scholar]
- 27.Luthy R., Bowie J.U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 28.Bowers K.J., Chow E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A. 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida. [Google Scholar]
- 29.Kaminski G., Friesner R.A., Tirado-Rives J., Jorgensen W.L. Evaluation and reparameterization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. The Journal of Physical Chemistry B. 2001;105:6474–6487. [Google Scholar]
- 30.Duhovny S.D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Research. 2005;33(Web Server issue):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. Journal of Molecular Biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Mannucci E., Pala L., Ciani S., Bardini G., Pezzatini A., Sposato I. Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia. 2005;48:1168–1172. doi: 10.1007/s00125-005-1749-8. [DOI] [PubMed] [Google Scholar]
- 33.Röhrborn D., Eckel J., Sell H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Letters. 2014;588:3870–3877. doi: 10.1016/j.febslet.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 34.Mandapathil M., Hilldorfer B., Szczepanski M.J., Czystowska M., Szajnik M., Ren J. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. The Journal of Biological Chemistry. 2010;285:7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z., Grigo C., Steinbeck J., Hörsten S.V., Amann K., Daniel C. Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides. 2014;57:109–117. doi: 10.1016/j.peptides.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Mulvihill E.E., Varin E.M., Gladanac B., Campbell J.E., Ussher J.R., Baggio L.L. Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metabolism. 2016;25:152–165. doi: 10.1016/j.cmet.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Itou M., Kawaguchi T., Taniguchi E., Sata M. Dipeptidyl peptidase-4: a key player in chronic liver disease. World Journal of Gastroenterology. 2013;19:2298–2306. doi: 10.3748/wjg.v19.i15.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calanna S., Christensen M., Holst J.J., Laferrère B., Gluud L.L., Vilsbøll T. Secretion of glucagon-like peptide-1 in patients with type 2 diabetes mellitus: systematic review and meta-analyses of clinical studies. Diabetologia. 2013;56:965–972. doi: 10.1007/s00125-013-2841-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng T., Baskota A., Gao Y., Chen T., Tian H., Yang F. Increased plasma DPP4 activities predict new-onset hyperglycemia in Chinese over a four-year period: possible associations with inflammation. Metabolism. 2015;64:498–505. doi: 10.1016/j.metabol.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Bogdan M.J., McDonnell M.E., Shin H., Rehman Q., Hasturk H., Apovian C.M. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. The Journal of Immunology. 2011;186:1162–1172. doi: 10.4049/jimmunol.1002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez F.Z., Auguet T., Aragonès G., Jurado E.G., Berlanga A., Martinez S. Interleukin-17A gene expression in morbidly obese women. International Journal of Molecular Science. 2015;16:17469–17481. doi: 10.3390/ijms160817469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winer S., Paltser G., Chan Y., Tsui H., Engleman E., Winer D. Obesity predisposes to Th17 bias. European Journal of Immunology. 2009;39:2629–2635. doi: 10.1002/eji.200838893. [DOI] [PubMed] [Google Scholar]
- 43.Ip B., Cilfone N., Belkina A.C., Furia J.D., Bogdan M.J., Zhu M. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity. 2016;24:102–112. doi: 10.1002/oby.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emamaullee J.A., Davis J., Merani S., Toso C., Elliott J.F., Thiesen A. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. 2009;58:1302–1311. doi: 10.2337/db08-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferraro A., Socci C., Stabilini A., Valle A., Monti P., Piemonti L. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes. 2011;60:2903–2913. doi: 10.2337/db11-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelletier M., Maggi L., Micheletti A., Lazzeri E., Tamassia N., Costantini C. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood. 2010;115:335–343. doi: 10.1182/blood-2009-04-216085. [DOI] [PubMed] [Google Scholar]
- 47.Lizama A.J., Andrade Y., Colivoro P., Sarmiento J., Matus C.E., Gonzalez C.B. Expression and bioregulation of the kallikrein-related peptidases family in the human neutrophil. Innate Immunity. 2015;6:575–586. doi: 10.1177/1753425914566083. [DOI] [PubMed] [Google Scholar]
- 48.Mantovani A., Cassatella M.A., Costantini C., Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nature Reviews Immonology. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 49.Tesmer L.A., Lundy S.K., Sarkar S., Fox D.A. Th17 cells in human disease. Immunological Reviews. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prassas I., Eissa A., Poda G., Diamandis E.P. Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nature Reviews Drugs Discovery. 2015;14:183–202. doi: 10.1038/nrd4534. [DOI] [PubMed] [Google Scholar]
- 51.Lamers D., Famulla S., Wronkowitz N., Hartwig S., Lehr S., Ouwens D.M. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60:1917–1925. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Makdissi A., Ghanim H., Vora M., Green K., Abuaysheh S., Chaudhuri A. Sitagliptin exerts an antinflammatory action. The Journal of Clinical Endocrinology & Metabolism. 2012;97:3333–3341. doi: 10.1210/jc.2012-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta V., Kalra S. Choosing a gliptin. Indian Journal of Endocrinology and Metabolism. 2011;15:298–308. doi: 10.4103/2230-8210.85583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.