

Abstract
C. neoformans is an encapsulated fungal pathogen with defined asexual and sexual life cycles. Due to the availability of genetic and molecular tools for its manipulation, it has become a model organism for studies of fungal pathogens, even though it lacks a reliable system for maintaining DNA fragments as extrachromosomal plasmids. To compensate for this deficiency, we identified a genomic gene-free intergenic region where heterologous DNA could be inserted by homologous recombination without adverse effects on the phenotype of the recipient strain. Since such a site in the C. neoformans genome at a different location has been named previously as “safe haven”, we named this locus second safe haven site (SH2). Insertion of DNA into this site in the genome of the KN99 congenic strain pair caused minimal change in the growth of the engineered strain under a variety of in vitro and in vivo conditions. We exploited this ‘safe’ locus to create a genetically stable highly fluorescent strain expressing mCherry protein (KN99mCH); this strain closely resembled its wild-type parent (KN99α) in growth under a variety of in vitro stress conditions and in the expression of virulence traits. The efficiency of phagocytosis and the proliferation of KN99mCH inside human monocyte-derived macrophages were comparable to those of KN99α, and the engineered strain showed the expected organ dissemination after inoculation, although there was a slight reduction in virulence. The mCherry fluorescence allowed us to measure specific association of cryptococci with leukocytes in the lungs and mediastinal lymph nodes of infected animals and, for the first-time, to assess their live/dead status in vivo. These results highlight the utility of KN99mCH for elucidation of host-pathogen interactions in vivo. Finally, we generated drug-resistant KN99 strains of both mating types that are marked at the SH2 locus with a specific drug resistant gene cassette; these strains will facilitate the generation of mutant strains by mating.
Keywords: Cryptococcus mCherry strain, Second safe haven site, host pathogen interaction, phagocytosis, KN99mCH, gene complementation
1. Introduction
C. neoformans is an encapsulated basidiomycete yeast with a world-wide distribution that has adapted to infect a wide array of organisms, from amoeba and nematodes to mammals, where it withstands a variety of stressful host conditions (Kozubowski and Heitman, 2012; Kronstad et al., 2011; Lin and Heitman, 2006). It has evolved an arsenal of virulence-related traits that has made it a successful fungal pathogen, especially in immunocompromised hosts where C. neoformans meningoencephalitis causes hundreds of thousands of deaths annually (Alspaugh, 2015; J. Park et al., 2014; Litvintseva et al., 2006; Park et al., 2009). The availability of high quality, extensively annotated whole genome sequence; robust animal models; and methods for stable targeted gene deletion has made this species a leading model organism for the study of fungal pathogenesis (Banks et al., 2005; Janbon et al., 2014). The ability to genetically cross strains of opposite mating types (MATα and MATa) has further aided in the characterization of genes critical for sex and virulence of the organism (Hull and Heitman, 2002).
Targeted gene deletion via biolistic transformation is routinely employed for studies of C. neoformans (Davidson et al., 2002; Liu et al., 2008). Deleting cryptococcal genes is challenging compared to deletion in the model yeast S. cerevisiae, which is easily achieved due to the high frequency of homologous recombination and short regions of homology (as 40 bp) required to mediate it. Due to the limited frequency of homologous recombination in C. neoformans, at least 300 bp of homology is required in the transforming DNA construct to routinely achieve insertion at the desired chromosomal locus (Nelson et al., 2003). Furthermore, non-homologous recombination is a major confounding factor, necessitating detailed confirmation of transformants (Kwon-Chung et al., 1998).
To satisfy the Molecular Koch’s Postulates (Falkow, 1988) the association of a gene deletion with specific mutant phenotype(s) must be confirmed by demonstrating phenotype reversal after reintroduction of the gene. For C. neoformans, this complementation is typically performed either by reintroducing the mutated gene at its original locus, or by integrating it randomly into the deletion strain genome. Both approaches have drawbacks. Inserting the target gene fragment at its original locus requires assembly or cloning of complex and often large DNA constructs; these also typically include selectable markers, which may influence nearby genes (Arras et al., 2015). Random ectopic integration of the target gene fragment is itself mutagenic, potentially interrupting other genes due to the highly compact nature of the C. neoformans genome, and thus confounding interpretation of results (Tenney et al., 2004). These challenges were recently demonstrated with attempts to complement an ade2Δ mutant, where neither of the above mentioned gene complementation approaches restored wild-type virulence despite complementation of other phenotypes (Arras et al., 2015). As an alternative strategy, the authors identified an intergenic genomic “Safe Haven” region that could serve as a site for DNA insertion without altering the recipient strain phenotypes (Arras et al., 2015).
Molecular tools such as the Safe Haven are essential for robust analysis of complex host-pathogen interactions. Upon infection, C. neoformans manipulates the host immune response to create an environment conducive for fungal growth and proliferation. It further evades killing by host effector immune cells, a process that is critical to understanding the progression of disease and the development of effective therapeutic interventions. Most in vitro and in vivo experiments designed to elucidate host-fungal interactions have been performed using fungal cells labelled either with capsule-specific antibodies or non-specific fluorescent molecules (Davis et al., 2015; Srikanta et al., 2011). One limitation of such methods is the potential change in surface charge or topography caused by dye conjugation or antibody binding to the yeast cell surface, which may interfere with host-pathogen interactions. Another is that any progeny cells that grow during the study are not labeled and thus cannot be tracked.
A yeast strain that is genetically marked with a stable reporter molecule would be an excellent tool for host-pathogen interaction studies. To this end, Robin May and colleagues engineered GFP-labeled derivatives of C. neoformans and Cryptococcus deuterogattii strain R265, responsible for the Vancouver outbreak (Datta et al., 2009; Galanis et al., 2010; Hagen et al., 2015). These strains displayed growth phenotypes similar to those of wild-type cells (Voelz et al., 2010), but the genomic site at which the GFP expression construct was integrated was not determined.
Our goal in this work was to develop and validate a C. neoformans strain that expressed robust fluorescence from a ‘safe’ site in the genome while retaining the already characterized Safe Haven site to facilitate additional genetic manipulations. To do this, we identified and characterized a second “safe haven” site, the SH2. We then integrated a histone H3 promoter-driven mCherry expression construct at SH2 to create a stable fluorescent transgenic KN99mCH strain. KN99mCH displayed comparable phenotypes to that of its parent under various in vitro growth conditions and in animal infections. We demonstrated the utility of this strain for host-pathogen interaction studies both in vitro, using assays with human monocyte derived macrophages, and in vivo to assess their live/dead status and their association with leukocytes in infected mice from multiple tissues for up to 21 days post infection. Finally, we generated MATa and MATa strains bearing antibiotic resistance markers at the SH2 site that will serve as valuable tools for future experiments, augmenting the molecular toolbox for this important pathogen.
2. Materials and Methods
2.1. Fungal strains and media
KN99α was the wild-type strain in this study. C. neoformans was routinely grown in yeast peptone dextrose (YPD; 1% yeast extract, 2% Bacto peptone and 2% dextrose) at 30 °C with shaking at 300 rpm. Solid medium contained 2% Bacto agar. Selective medium contained 100 μg/mL nurseothricin (NAT) (Werner BioAgents, Germany), 200 U/mL hygromycin (HYG) (Calbiochem, USA), or 200 μg/mL Geneticin (Gibco Life technologies, USA). RPMI …
2.2. Designing an mCherry-expressing construct to target SH2
We constructed a plasmid pH3mCHSH2 as shown in Fig. 1A in which a mCherry expression construct is flanked by sequences homologous to the C. neoformans SH2 region to mediate targeted integration. To make this we used overlap PCR with Phusion high fidelity PCR system (New England Biolab, USA) to fuse three DNA fragments: 500 bp upstream of the ATG start codon of C. neoformans histone H3 gene (CnH3; CNAG_06745) amplified from KN99α genomic DNA using primers H3PromF and H3PromR (Supplementary Table S1), the mCherry gene amplified from plasmid pLK25 (kindly provided by Dr Peter Williamson) using primers mCherryF and mCherryR (Table S1) and a 410 bp of genomic DNA downstream of the predicted stop codon of CNAG_06125 (EF1-term; translation elongation factor-1 alpha) amplified from KN99α genomic DNA using the primer pair CnEF1TermF and CnEF1TermR (Table S1). The resulting DNA segment (H3mCH) was cloned into pGEM-Easy (Promega, USA) at the NotI site by In-Fusion HD Cloning (Takara Bio, USA) to generate plasmid pH3mCH. A 960_bp genomic region corresponding to 5′ end of the SH2 region was amplified from KN99α genomic DNA using primer pair 5primeSH2F and 5primeSH2R (Table S1) and cloned at the SacII site of the pH3mCH to generate the plasmid pH3mCH1. A G418 resistance cassette flanked by loxP sites was amplified from plasmid JL517 (Baker and Lodge, 2012) using primers G418F and G418R (Table S1) and fused to an 860_bp genomic DNA fragment corresponding to the 3′ end of the SH2 region amplified from KN99α genomic DNA employing 3primeSH2F and 3primeSH2R (Table S1) by overlap PCR to generate fragment G418SH2. Finally, the G418SH2 DNA fragment was cloned at the SpeI site of pH3mCH1 by In-Fusion HD cloning. All the PCR reactions were performed using Phusion® High-Fidelity PCR Master Mix following manufacturer’s instructions (New England Biolab, USA). The sequence of the final construct pH3mCHSH2 was confirmed by DNA sequencing. This plasmid was digested with ApaI and SalI to release a 6.191 kb fragment that included the histone promoter-driven mCherry sequence and the G418 drug resistance marker flanked by sequences homologous to the SH2 region for biolistic transformation. This plasmid has been deposited in Addgene (plasmid id 101053).
Fig. 1. Targeted integration of an mCherry expression construct at the SH2 locus.

A: Schematic illustration of the plasmid pH3mCHSH2 in which fragment expressing mCherry protein from C. neoformans histone 3 promoter (CnH3) is fused to the G418 resistance marker cassette. B: Stable expression of mCherry protein from the SH2 locus on chromosome 11 resulted in a brightly fluorescent transgenic C. neoformans (KN99mCH). Cells were grown in YPD for 24 hr, collected, washed with PBS and visualized under Olympus BX61 microscope and photographs were taken at 1000X magnification. Left panel is bright field image and the right panel is a fluorescence image.
2.3. Integration of foreign DNA at the SH2 locus
DNA fragments for the dominant drug selection markers were amplified using primer pair M13F and M13R (Table S1) from appropriate drug selection marker containing plasmids as templates as previously described (Hua et al., 2000; McDade and Cox, 2001). These drug selection marker fragments were fused to the 5 prime and 3 prime SH2 fragments by overlap PCR (Davidson et al., 2002). Cells to be transformed biolistically with either the mCherry fragment described above or drug resistance modules were grown for 36 h in YPD medium, concentrated and plated onto non-selective YPD agar for transformation as in Toffaletti et al (Toffaletti et al., 1993). Drug resistant primary transformants were serially passaged in YPD medium four times (each 24 hr) before streaking for single colonies on appropriate antibiotic-containing plates. Candidate transformants were screened for integration of the drug resistant cassette at the SH2 locus using a three primer PCR screen as in Lam et al (Lam et al., 2013) with the primers listed in Supplementary Table 1. A positive PCR screen at the 5-prime and the 3-prime junction of the SH2 locus confirms that the transformation module has integrated at the desired SH2 locus in the genome (Fig. S2). A positive long PCR screen confirms that a single DNA fragment has been integrated at the desired genomic locus (Fig. S2). A representative PCR screen results obtained during the generation of strains JLCN 923 (KN99mCH) and JLCN830 (KN99G418) are shown in Fig. S2. To verify that the construct has not integrated elsewhere, genomic DNA from positive isolates prepared as in Lam et al (Lam et al., 2013) was subjected to DNA blot analysis as in Baker and Lodge (Baker and Lodge, 2012) except using DIG DNA labeling and detection per the manufacturer’s instructions (Roche Diagnostics, USA). Blots were probed with a marker specific DIG labelled probe which should yield a single band for a positive isolate confirming a single insertion event in the genome as shown in Fig. S2 (Hua et al., 2000). When mCherry-expressing construct was transformed, we obtained several positive transgenic strains exhibiting different intensities of fluorescence (Fig. S5) and one of the isolate exhibiting strongest fluorescence of all (KN99mCH; JLCN 923; Fig. 1B) was selected for further characterization.
2.4. RNA isolation and real time PCR
C. neoformans strains were grown in either YPD or yeast nitrogen base (YNB), pH 4 at 30 °C or 37 °C to mid-log phase (OD650=3.0–4.0). A 5-mL aliquot was then harvested (10 min; 3,500 g; 4 °C) and the pellet rapidly frozen in liquid nitrogen and stored at −80 °C for use in RNA isolation using the RNeasy Plant Mini kit (QIAGEN GmbH, Germany), with final RNA subjected to on-column RNase free DNase (QIAGEN) digestion. 0.5 μg of total RNA was used for the synthesis of cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, USA) in a 20 μL reaction using random primer mix. 1 μL of cDNA was used for real-time PCR using SsoFast EvaGreen Supermix (Bio-Rad). A BioRad CFX96 thermal cycler was programmed with the following two-step PCR cycles: 95 °C for 30 secs; 95 °C for 5 sec, 60 °C for 10 sec, with a plate read repeated in the second step for a total of 40 cycles. A melting curve was performed at the end of the reaction to confirm the generation of a single product. Standard curves were performed for each primer set and efficiencies calculated. The data were normalized to C. neoformans 18S rRNA expression.
2.5. Cell wall, temperature, oxidative and nitrosative stress sensitivity growth assays
Cultures grown to mid log phase in YPD were diluted to OD650=1.0 and 5 μL portions of the 10-fold serially diluted cell suspensions were spotted on plates containing appropriate stressors and incubated for 5 days at 30 °C. For cell wall stress YPD agar plates were supplemented with sodium dodecyl sulfate (SDS, 0.03%), calco uor white (CFW, Fluorescent Brightener 28, F-3543; Sigma, USA; 1.5 mg/mL), sodium chloride (1 M) or Congo red (C-6767; Sigma;5 mg/mL). For oxidative and nitrosative stresses, 1 mM hydrogen peroxide (H2O2) and 0.75 mM of sodium nitrite (NaNO2) were used, respectively, in solid YNB, pH 4.0, made fresh on the day of the experiment. To test for temperature sensitivity, strains were plated on YPD medium and incubated at either at 25 °C or 30 °C or 37 °C, or 39 °C.
2.6. Capsule analysis
Strains were initially grown in YPD for 24 hr. Cells were collected and washed two times with sterile PBS, pH 7.4. Cells were then suspended in RPMI medium with 10% FBS (Fetal bovine serum) serum at a concentration of 500 yeast cells/μL and incubated at 37°C in the presence of 5% CO2. After 4 days of incubation 8 μL of cell suspension was mixed with 2 μL of 1:4 India ink: H2O solution and photographed on an Olympus BX61 microscope at a magni cation of X60.
2.7. Analysis of melanin production
Cells of each strain were taken from solid YPD medium and resuspended at 5 x 107 cells/mL in 2 mL glucose-free asparagine medium (1 g/L L-asparagine, 0.5 g/L MgSO4.7H2O, 3 g/L KH2PO4 and 1 mg/L thiamine) plus 1 mM L-3,4-dihydroxyphenylalanine (L-DOPA). Cells were shaken at 30 °C for 24 hr, collected and photographed.
2.8. Cellular chitosan measurement
Cellular chitosan was measured as previously described (Banks et al., 2005) except that the alkali extracted cell wall material was digested with chitinase from Trichoderma viride (C 8241, Sigma-Aldrich, USA). Each sample of alkali extracted material was resuspended in 0.2 mL of McIlvaine’s buffer (0.2 M Na2HPO4, 0.1 M citric acid (pH 6.0) and digested with 20 uL of chitinase from T. viride (5 mg/mL in water) for 24 h at 30 °C.
2.9. Assays of macrophage uptake and intracellular fungal proliferation
Uptake of KN99α and KN99mCH by human THP-1 (human monocytic cell line) cells was assayed as previously described (Srikanta et al., 2011). Briefly, fungi were grown in YPD for 24 hr, serum-opsonized, washed in PBS, diluted to 106 cells/mL, and 150 μL/well were added to THP-1 cells in a 96-well plate for an MOI of 10. Plates were incubated for 1 hr at 37 °C in 5% CO2 and then fixed with 4% formaldehyde, stained with DAPI and CellMask Deep Red plasma membrane stain (both from SIGMA, USA), and imaged with a Cytation 3 Cell Imaging Multi Mode Reader (BioTek, USA). Analysis of captured images was performed with the IN Cell Developer Toolbox (GE Healthcare, USA).
To assay intracellular fungal survival, cells were similarly grown and opsonized before addition to THP-1 macrophages at an MOI of 0.1 in 24-well plates. For this survival experiment we wanted yeast cells to be limiting in the hope that majority of the yeast cells added to the well will be taken up by macrophages. Therefore, we used a MOI of 0.1. After 1 hr of incubation as above, the wells were washed twice with PBS to remove non-associated cryptococcal cells and incubation was continued. Host cells in the wells were washed two times with pre-warmed PBS before lysing with H2O at 0, 12, 24, 36, and 48 hr post incubation and the lysate was diluted and plated on YPD agar to determine colony forming units (CFU).
2.10. Animal infection and fungal burden assays
Pulmonary infections and virulence assays were performed as described previously (Baker et al., 2011). For murine infections, C. neoformans strains were grown in YPD for 36 hr at 300 rpm, washed twice with endotoxin-free PBS, and adjusted to 2 x106/mL in PBS. 5–6-week-old female CBA/J mice (Jackson Laboratories, USA) were weighed and intranasally infected with 50 μL of the cell suspension. The viability of the inoculated cells was confirmed after plating the serially diluted inoculum onto YPD plates and incubating them at 30°C for 72 hr for enumerating CFU. Mice were anesthetized with an intraperitoneal injection (200 μL) of ketamine (8 mg/mL)/dexdomitor (0.05mg/mL) mixture which was reversed by an intraperitoneal injection of (200 μL) of antisedan (0.25mg/mL). Mice were monitored daily and those showing any signs of morbidity (75% of pre-inoculation weight loss, extension of the cerebral portion of the cranium) were sacrificed by CO2 asphyxiation and cervical spine dislocation. This infection protocol was reviewed and approved by the Washington University School of Medicine Animal Care and Use Committee (IACUC).
2.11. Preparation of mediastinal lymph node (MDLN) and lung leukocyte single-cell suspensions
MDLNs were dissected from control and mice infected as above at 21 days post-infection (DPI) and added to homogenization C tubes containing 3 mL of tissue dissociation buffer (TDB; PBS, pH 7.2, 0.5% bovine serum albumin, and 2 mM EDTA; Miltenyi Biotec, USA). This was followed by homogenization of MDLN tissue in the gentleMACS Dissociator using program m_spleen_01 (Miltenyi Biotec). After homogenization, samples were gently centrifuged and resuspended in 5 mL of TDB and passed through a 30_μm_pore-size Pre-Separation Filter; the flow-through was the MDLN single-cell suspension. For lung leukocyte preparation, lungs were removed and added to homogenization C tubes containing proprietary catalysts for mechanical and enzymatic digestion (Miltenyi Biotec). This was followed by homogenization of lung tissue in the gentleMACS Dissociator based upon the manufacturer’s instructions (Miltenyi Biotec). Homogenized tissue was then passed through the 70-μm-pore-size MACS SmartStrainer (Miltenyi Biotec) and centrifuged at 300 g for 10 min to pellet cells. Erythrocytes were removed using ACK (ammonium-chloride- potassium) lysing buffer (Gibco, USA). Briefly, 1 mL of ACK buffer was used to suspend pelleted cells. After 10 min, the ACK lysing buffer was diluted 1:10 with complete RPMI medium. Cells were then washed with TDB and counted in a hemocytometer, using trypan blue staining to exclude dead cells.
2.12. Confocal fluorescence microscopy
Single-cell suspensions (3 x 104 cells) from MDLN were cytospun onto poly-L-Lysine coated slides and air-dried overnight. Slides were then fixed with 3% para- formaldehyde (PFA), blocked with 1% bovine serum albumin (BSA), and labeled with anti-CD45 (FITC mouse anti-CD45, clone 30-F11; BioLegend, USA) following the manufactrer’s protocol. After multiple rinses with PBS, anti-photobleaching/fading SlowFade Gold with DAPI (Molecular Probes, USA) was added to the slide to mount the coverslip. Images were collected using a 63x oil objective lens on a ZEISS LSM 510 META confocal laser scanning microscope.
2.13. Flow cytometry
Lung single-cell suspensions were labeled with the fixable viability stain Zombie Aqua (BioLegend), mouse PerCP-anti-CD45 (BioLegend, clone 30-F11), and CFW. Briefly, 2.5×105 cells/well in a microtiter plate were washed in in PBS, resuspended in PBS containing a 1/200 dilution of Zombie aqua, and then incubated at 25 °C for 20 min in the dark. Cells were washed twice in TDB (300 g, 10 min, 4 °C) and resuspended in PerCP-anti-CD45 (2μg/mL). Cells were then incubated on ice for 30 min in the dark, washed once each with TDB and PBS, stained in PBS containing CFW (2.5 μg/mL), and incubated at 25 °C in the dark for 15 min. Cells were next washed twice in TDB, fixed at 4 °C overnight in 1% paraformaldehyde (PFA), washed and resuspended in TDB, and added to a 96-well microtiter plate for high-throughput flow cytometry sampling using a BD LSRFortessa X-20 (BD Biosciences, USA). BD FACSDiva software was used to process samples on the flow cytometer, and FlowJo (FlowJo, USA) was used for data analysis of the acquired samples to facilitate the identification of leukocyte populations. Thirty thousand cell events were recorded, and dead cells and debris were excluded based upon lower forward scatter (FSC) and side scatter (SSC) signals (i.e., smaller size and granularity). The absolute number of each population was determined by multiplying the frequency of the population by the number of cells in the whole lung single-cell suspensions.
2.14. Genome sequencing and analysis
Genomic DNA extracted from wild type and KN99mCH cells as in Lam et al (2013) was subjected to RNase treatment and submitted for 101_bp paired-end sequencing at the GTAC (Genome Technology Access Center, Washington University in St. Louis, USA). Each strain was sequenced to approximately 30-fold coverage. Reads were trimmed using Trimmomatic 0.30 (Bolger, Lohse et al. 2014) and mapped to the C. neoformans H99 reference genome (version CNA3, www.broadinstitute.org/annotation/genome/cryptococcus_neoformans/MultiHome.htm, accessed March 2014) using BWA 0.7.5 (Li and Durbin, 2009) and default settings. Duplicate reads were marked using Picard 1.98 (http://broadinstitute.github.io/picard/) and base quality score recalibration and realignment around indels was performed using GATK 3.0 (McKenna et al., 2010). SNPs and indels were identified using GATK 3.0 (DePristo et al., 2011) and structural variation assessed using Breakdancer 1.1.2 (Chen et al., 2009) and Control-FREEC (Boeva et al., 2012). Mappings were visualized and SNP associated amino acid changes were determined using CLC Genomics Workbench (QIAGEN, Germany).
2.15. Mating assay
Strains to be mated were first grown on YPD agar at 30 °C for 2–3 days. Single colonies of each mating type were inoculated into 2 mL YPD, grown overnight, and adjusted to OD650=1.0 before mixing in equal amounts and spotting on V8 agar at 25 °C for 21 days in the dark (Ruff et al., 2009). Mating structures were observed and harvested with a sterile glass pipette. The filaments were re-suspended with vortex mixing in 1 mL PBS, serially diluted, and plated onto selective media for the selection of meiotic F1 progeny.
3. Results
3.1. Identification of an intergenic integration site in the C. neoformans genome for targeted DNA insertions
We searched for long intergenic regions in the C. neoformans var. grubii type strain H99 genome to use for DNA insertion, consulting the initial chromosome-based assembly of the H99 genome made available by the Broad Institute in 2008. We next used BLASTN searches of the NCBI NR database to ensure that the five longest regions contained no open reading frames larger than 20 codons, and chose the longest for further study. This region, which we called SH2, is located between the tandemly transcribed predicted genes CNAG_01561 and CNAG_01562 on chromosome 11 and is 3,278 bp in length (Fig. S1A). We did not find any RNA reads mapping to this part of the chromosome in a previously published expression dataset employing RNA Seq using RNA isolated from wild-type KN99 cells (Fig. S1B and (Donlin et al., 2014). Successful integration at the SH2 locus results in the replacement of a 139 bp chromosomal region with the desired foreign DNA fragment (Fig. S1C).
3.2. Targeted insertion of foreign DNA into the SH2 locus
Drug marker cassettes used routinely for engineering C. neoformans strains include genes that confer resistance to neomycin (G418), nourseothricin (NAT), hygromycin (HYG) and phleomycin (Phleo) (Hua et al., 2000; McDade and Cox, 2001). We used biolistic transformation (see Methods) to insert these cassettes into the SH2 locus of KN99 and generate marked strains in both mating types MATα and MATa (Table 1). We similarly integrated an mCherry expression construct at the SH2 locus of KN99α to generate strain KN99mCH. In all cases, we used PCR screening and subsequent DNA blot analysis (Figs S2 and S3) to identify and confirm isolates with single insertions at the desired SH2 locus and no non-homologous integration events elsewhere in the genome.
Table 1.
Strains created in this study
| Strain number | Marker at the SH2 locus | Mating type |
|---|---|---|
| JLCN 821 | NAT | MATα |
| JLCN 830 | G418 | MATα |
| JLCN 869 | HYG | MATα |
| JLCN 920 | mCherry/G418 | MATα |
| JLCN 922 | mCherry/G418 | MATα |
| JLCN 923 | mCherry/G418 | MATα |
| JLCN 817 | NAT | MATa |
| JLCN 828 | G418 | MATa |
| JLCN 832 | Phleomycin | MATa |
| JLCN 866 | HYG | MATa |
3.3. Integration of foreign DNA at the SH2 locus does not affect the transcription of neighboring genes
The C. neoformans SH2 region is flanked by CNAG_01561, annotated as encoding a protein containing a WD domain, and CNAG_01562, encoding a hypothetical protein. We performed quantitative real time PCR to test the effect of insertion of mCherry expression construct at the SH2 locus on the transcription of flanking genes in strain KN99mCH. We found no statistically significant difference between KN99α and KN99mCH in the expression levels of neighboring genes under any growth conditions tested (Fig. 2). Similar expression levels were observed for CNAG_01563 which is predicted to code for histone deacetylase (Fig. 2). We could not detect RNA corresponding to CNAG_01560 in any strain and therefore did not include it for q-PCR.
Fig. 2. Integration of the mCherry expression construct at the SH2 locus does not affect the transcription of flanking genes.

mRNA abundance of CNAG_01561, CNAG_01562 and CNAG_01563 in KN99α and KN99mCH grown either in rich medium or in nutrient defined YNB medium at room temperature or at host body temperature of 37 oC. Gene expression was normalized to the expression of 18S rRNA. No differences were statistically significant after the analysis of data using two-way analysis of variance (ANOVA) with a Bonferroni post hoc test. Error bars indicate standard deviations.
3.4. Phenotypes and virulence attributes of KN99mCH are equivalent to its parent KN99α
We compared the growth of KN99mCH and KN99α under normal laboratory conditions and in the presence of various stress conditions. We found no differences in growth upon exposure to increased temperature, cell wall stressors (SDS, CFW, NaCl and Congo red) or conditions of oxidative or nitrosative stress (Fig. 3A). We also detected no alteration in virulence related traits such as capsule induction (Fig. 3B). We measured the capsule size and the capsule volume of ~50 cells growing in capsule inducing condition and found no significant difference between KN99α and KN99mCH cells (Fig. 3C). The deposition of melanin was similar in both strains (Fig. 3D). We have previously shown that in addition to its role as a critical component of the cell wall, C. neoformans chitosan (the deacetylated form of chitin) plays a pivotal role in fungal virulence and pathogenicity in the host (Baker et al., 2007; Baker et al., 2011; Upadhya et al., 2016). For this reason, we quantified the amount of cellular chitin and chitosan, and found no significant difference between KN99α and KN99mCH (Fig. 3E). We performed a detailed growth analysis of KN99α, KN99G418 and KN99mCH strains in liquid medium of YPD (nutrient rich medium), YNB (nutrient limited defined medium) at both 30°C and 37°C and in RMPI medium containing 10% FBS with cell growing at 37°C/5% CO2 conditions. KN99G418 served as an additional control which has a G418 antibiotic resistance cassette integrated at the SH2 locus. Genotypically this strain is similar to KN99mCH, except for the lack of the mCherry expressing construct. We did not find any significant difference in the growth rates or doubling time between the strains in all the tested conditions (Fig. 4).
Fig. 3. KN99mCH grows like KN99α under various in vitro stress conditions and shows similar virulence trait expression.

A: Comparison of the growth of KN99α and KN99mCH strain under various in vitro stress conditions. C. neoformans strains were grown to mid log phase in YPD. Cells were harvested and diluted to OD650 = 1.0. Cell suspension was serially diluted (10 fold) and 4 μL of the original and the diluted cell suspensions were spotted on YPD agar containing appropriate stress reagent. Plates were incubated at 30 oC except where noted. Plates were photographed after 5 days of incubation. Top row, KN99α; bottom row, KN99mCH. B: Visualization of the capsule after India ink staining of the strains which were incubated in capsule inducing conditions. C: Quantitative analysis of the capsule length and the volume between KN99α and KN99mCH. D Comparison of cell associated melanin after incubating the cells in melanin inducing medium. E: Chitin and chitosan content of the indicated strains grown to saturation in YPD. There was no significant difference between KN99α and KN99mCH after Bonferroni multiple comparison test employing two-way ANOVA using Graph Pad Prism 7 software. Mean of three independent biological experiments are plotted with standard deviation.
Fig. 4. Growth rate of KN99mCH is comparable to KN99α under multiple nutrient and growth conditions.

Indicated strains were grown initially in YPD for 24 hr at 300rpm. Cells were collected by centrifugation washed two times with PBS and suspended in the different media at an OD650=0.01–0.05. At different time intervals OD650 of the culture was recorded. Data is the average of three independent biological experiments.
3.5. KN99mCH shows wild type interactions with phagocytes in vitro
We expect the brightly fluorescent KN99mCH to be useful in analysis of host pathogen interactions. We therefore wanted to be sure that fluorophore expression from the SH2 site did not perturb either the engulfment of fungi by host cells or their intracellular proliferation. To address this, we incubated unopsonized or serum-opsonized KN99α and KN99mCH cells with a human macrophage-like cell line (THP-1) and measured the phagocytic index (number of fungi per 100 host cells) using a sensitive image-based assay (Srikanta et al., 2011). We also determined the rate of proliferation of internalized cells within the host phagocytes over a 48-hour period after engulfment. In all respects, results were equivalent for KN99α and KN99mCH (Fig. 5), supporting the utility of KN99mCH to probe host-pathogen interactions in vitro.
Fig. 5. KN99mCH exhibits normal phagocytic index and intracellular proliferation rate.

A: Phagocytic index (number of fungi per 100 host cells) of KN99α and KN99mCH with or without serum-opsonization, after exposure to THP-1 host cells. B: Fold-change in colony forming units of fungi lysed from host cells at the indicated times after an initial 1-hr uptake interval, normalized to the initial time point.
3.6. KN99mCH in a murine infection model
We next analyzed the strains for their virulence in a mouse model of cryptococcal infection. When we infected CBA/J mice with 105 CFU of KN99, KN99G418, or KN99mCH via intranasal inhalation we noted a median survival time for KN99 and KN99G418 of 16 days, while mice infected with KN99mCH showed a slight but statistically significant extended median survival time of 19-days post infection (p<0.05) (Fig. 6). Notably, fungal burden in lung, brain and the spleen at the time of sacrifice was same for all strains (Fig 6B–D), suggesting that KN99mCH disseminates normally. We considered that an altered growth rate in mouse lung might explain the slight difference in virulence between the wild-type and KN99mCH strains. Assessment of fungal organ burden after infecting mice with a 1:1 mixture of KN99α and KN99mCH showed that KN99mCH did grow more slowly in the host context, with reduced counts in lung at 3, 7, and 14 DPI (Fig. 7). We conclude that integration of foreign DNA at the SH2 locus alone does not affect strain virulence, because KN99G418 behaved like wild type in animal infections. Instead we attribute the extended time course of infection with KN99mCH to its slower growth in vivo, likely due to high expression of mCherry or due to genomic changes described below.
Fig. 6. Virulence of KN99mCH.

A: Group of five mice (female CBA/J) were subjected to intranasal inoculation with 105 cells of the indicated strains and survival was followed by monitoring the body weight. Mice that lost 25% of their starting bodyweight were considered ill and were sacrificed. Data shown represent two independent experiments with KN99α and KN99mCH (p<0.05) and one for KN99G418. At the end point of survival experiment, organ homogenates were plated to calculate colony forming units for lung (B), brain (C) and spleen (D). No significant difference in the CFU between either KN99α or KN99mCH or KN99G418 after one-way ANOVA employing Dunnett’s multiple comparison test (GraphPad Prism Ver7).
Fig. 7. Comparative growth fitness of KN99α and KN99mCH in the infected murine lung.
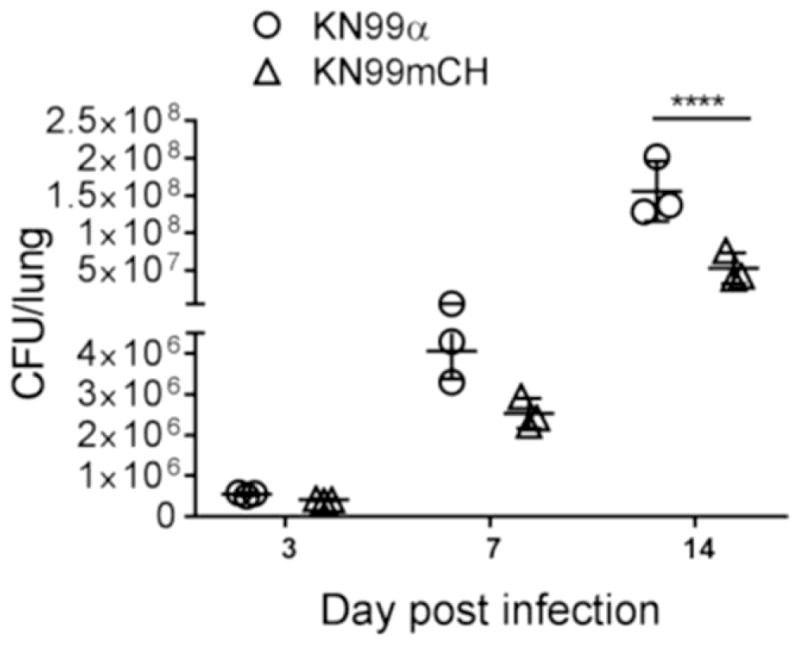
A mixture of KN99 and KN99mCH strains (5x104 CFU each) was inoculated intranasally; Lungs of the infected animals were excised at 3, 7, and 14 DPI. Organ homogenate was subjected to CFU enumeration. Means were compared following Sidak’s multiple comparison test. ****p<0.0001.
3.7 Whole genome sequencing of KN99mCH
Another possible explanation for the slightly slower growth in vivo of KN99mCH compared to wild type is that it acquired additional DNA changes during transformation or laboratory passage. We performed whole-genome sequencing of KN99mCH and KN99α and identified three single nucleotide polymorphisms that distinguished these strains from the reference strain H99: two unique mutations in KN99mCH and a single mutation in KN99α (Table 2). One mutation in KN99mCH introduces a premature stop codon within CNAG_02699 on chromosome 3, truncating the protein at residue 542 of 1,186. This gene is annotated as encoding a hypothetical protein with an atrophin-1 domain, which is associated with several neurodegenerative diseases (pfam03154). It has homology to similarly uncharacterized proteins in other Cryptococcus species, including ~80% identity with C. neoformans var. neoformans and ~70% identity with C. gattii. This conservation suggests the protein has an important functional role and its truncation in KN99mCH could contribute to the altered growth of the strain observed in host lung conditions. The whole genome sequencing data has been deposited at the NCBI database with the accession numbers SAMN07440305, SAMN07440306.
Table 2.
Unique mutations identified in KN99mCH and KN99α
| Strain | Chr | Position | Reference | Allele | Gene | Mutation |
|---|---|---|---|---|---|---|
| KN99mCH | 3 | 979,413 | C | G | CNAG_02699 | Ser542* |
| KN99mCH | 7 | 1,004,455 | T | A | 5′ UTR of CNAG_05836 | |
| KN99α | 11 | 78,195 | G | A | CNAG_01482 | Gly155Asp |
To further explore the potential possibility of the mutation in CNAG_02699 influencing fungal virulence, we obtained CNAG_02699Δ strain from the fungal genetics stock center (2015 Madhani plates; UCSF_2015_1st_22_Cryptococcus_library). We included this strain for measuring growth in RPMI/5% CO2 and for determining its virulence in murine model. Deletion of CNAG_02699 did not affect growth in RPMI medium (Fig. S4A). However, CNAG_02699Δ did show a slight delay in virulence similar to KN99mCH with a median survival time of 20-day post infection compared to 16-day post infection for KN99α (Fig. S4B).
3.8. Use of KN99mCH for long-term studies of host response to infection
Host immune response to infection, that includes an adaptive response, can show peak activity occurring after 14 days. Therefore, to help elucidate host immune response mechanisms that are induced by KN99mCH infection, we must be able to identify their; i) long-term characteristics (e.g., host cell interactions and live/dead status) in vivo from ii) multiple tissues while using iii) multiple fluorescent measuring platforms. Since commonly-used methods of external fluorescent staining of C. neoformans prevents long-term specific characterization (i.e., >3 days) of host cell-C. neoformans interactions, we explored the use of KN99mCH for their in vivo characterization. To examine the utility of KN99mCH for characterizing the long-term host immune response we made single-cell suspensions of MDLN from infected mice, 21 DPI. These single-cell suspensions were cytospun onto slides and then labeled with fluorochrome-conjugated anti-CD45 and DAPI. Confocal microscopy revealed that KN99mCH were detected in the MDLN and that they could be found colocalized externally with leukocytes (Figure 8A).
Fig. 8. KN99mCH fluorescence and cell staining for analysis of fungal: host interactions.

Mice were intranasally inoculated with either PBS (sham control) or 5 x 104 KN99mCH and single-cell suspensions were prepared at on 1, 7, and 21 days PI from MDLN and lungs. (A) A KN99mCH cell being engulfed by a MDLN leukocyte (CD45-labeled) at day 21 PI. (B) Quantitation of externally or internally co-localized KN99mCH with host leukocytes and analysis strategy. Lung single-cell suspensions were stained with a fluorochrome-conjugated antibody towards CD45 (BioLegend), and were then stained with CFW (Sigma-Aldrich). Samples were then fixed (1% PFA), and then examined by flow cytometry. Following forward vs side scatter exclusion of cellular debris, Fluorescence minus one (FMO) controls were used to objectively set the gates to identify the leukocyte population (CD45+). This population was then further delineated for KN99mCH being internally (mCherry+CFW−) or externally (mCherry+CFW+) colocalized with leukocytes from 21 DPI. Rare cell events not shown. (C) To better understand the differences in colocalization of KN99mCH between those that were internally and externally colocalized with leukocytes, collected cells were compared on 1, 7, and 21 DPI. Statistical analysis was done using a two-tailed unpaired Student’s t test comparing KN99mCH externally colocalized to KN99mCH internally colocalized. Data is presented as the mean +/− standard deviation with an N = 3. *p < 0.05, and ****p < 0.0001.
Next we harvested lungs from mice infected with KN99mCH at 1, 7, and 21 DPI. Single-cell suspensions were made from these lungs, labeled with fluorochrome-conjugated anti-CD45 and stained with calcofluor-white (CFW), a fluorescent dye that binds to chitin, and then examined by flow cytometry. This optimized staining strategy enabled us to not only differentiate C. neoformans and leukocytes from other mammalian cells, but provided the identification of KN99mCH colocalized internally (CD45+mCherry+CFW−) and externally (CD45+mCherry+CFW+) with leukocytes. A representative contour smoothed zebra density plot for the 21 DPI sample is shown in Fig. 8B. We identified at 1 DPI, KN99mCH in the lung resided both within leukocytes (52%) and outside of them (48%) (Fig. 8C). Over time, this balance shifted, with fewer fungi within leukocytes (23%) on days 7 and 21 PI (Fig. 8C). However, during this interval the fungal population also increased, so that the absolute numbers of internalized yeast increased, by 5.7 fold and 34.3 fold on day 7 and day 21, respectively, compared to day 1 levels (Fig. 8C).
Beyond the localization of KN99mCH fungal cells with respect to multiple tissues and association with leukocytes, we also wished to determine their viability in the host. Using BioLegend’s (USA) Zombie fixable viability dye, we determined that under in vitro conditions, using fluorescent counting beads detected by flow cytometry, we could identify and enumerate the live/dead status of KN99mCH that were combined at different ratios with heat-killed KN99mCH (i.e., 0%, 25%, 50%, 75% and 100%; Fig. 9A). The legitimacy of this method was confirmed by taking an aliquot of the cell suspension used for flow cytometry, to detect the live/dead status of KN99mCH, and examine them also for CFUs. This comparison identified no differences between the enumeration of the live/dead status of KN99mCH between flow cytometry and CFUs (Fig. 9B). Based upon this empirical evidence we used the fixable viability dye to detect the live/dead status of KN99mCH in vivo at 3 separate time points from mouse lungs: 1, 7, and 21 DPI (Figs 9C and D). Long-term in vivo data collection from the lung identifies a substantial increase in live KN99mCH over 21 days (766 fold), despite there being an increase in KN99mCH death over time (356 fold; Fig. 9D). Combined, these data indicate that KN99mCH can be detected for multiple conditions in vivo by different fluorescent platform systems in long-term studies.
Fig. 9. Zombie Aqua fixable viability dye can be used to detect the live/dead status of KN99mCH in vitro and in vivo.

(A&B): KN99mCH cells were grown overnight in YPD, washed in 1x PBS, counted by hemocytometer and split into 2 groups. One group was heat-killed (HK) by incubating for 15 minutes at 70 °C. The two groups of cells were combined to create new groups that contained 0%, 25%, 50%, 75%, and 100% HK cells. These new groups were split into two additional groups: Group I) Flow cytometry analysis and Group II) CFUs. Group I was stained with Zombie Aqua live/dead dye and then fixed in 1% PFA for flow cytometry analysis. Also, Molecular Probes CountBright Absolute Counting beads were added prior to flow cytometry analysis for direct counting of KN99mCH cells. (C&D): Lung single cell suspensions from infected mice (with 5x104 of yeast cells) were subjected to flow cytometry at 1, 7 and 21 days PI for the identification and quantification of live and dead KN99mCH cells. Specifically, single-cell suspensions were stained with Zombie Aqua fixable viability dye, fixed (1% PFA), and then examined by flow cytometry. Following forward vs side scatter exclusion of cellular debris, and use of FMO controls from the lung single cell suspension, cells were gated on the CD45− population and then analyzed as shown. The rare cell events are not shown in order to reduce noise, but are calculated as part of quadrant percentages which are noted in the flow diagrams. Live and dead KN99mCH are represented as mCherry+Zombie Aqua− and mCherry+Zombie Aqua+, respectively. Statistical analysis was done using a one-way ANOVA test. Data is presented as the mean +/− standard deviation with an N = 3. *p < 0.05, and **p < 0.01.
3.9. Demonstration of the use of KN99mCH and other SH2 “marked” strains for studies employing genetic crosses
One goal of generating drug resistant “marked” strains of C. neoformans was for their potential utility in genetic studies, including for the identification and characterization of genes that are critical for mating, filamentation and sporulation. Therefore, we first tested whether these strains produce hyphae when mixed with opposite mating type and incubated in V8 agar medium. We found that all SH2 marked strains produced comparable amounts of hyphae when mixed with opposite mating type strain and incubated in V8 agar medium (data not shown).
To further evaluate the ability of these strains to produce viable spores, we incubated MATα type strains marked at their SH2 locus with hygromycin (JLCN869) and G418 resistance cassettes (JLCN830 and JLCN923; Table 1) with a NAT resistant mpk1Δ strain generated in KN99 of MATa mating type background (Donlin et al., 2014). We also crossed a strain with the NAT marker at the SH2 locus (JLCN821) with a G418 resistant mkk2Δ generated in KN99 MATa background (Donlin et al., 2014). We randomly chose these deletion strains since they had the compatible drug resistant cassette and were of the appropriate mating type. None of the SH2 marked strains had defect in the levels of filamentation compared to their progenitor KN99α strain (data not shown). We qualitatively determined the ability of the spores of selected mating events to germinate and to produce new mutant strains. Due to the presence of different drug resistant genes at the SH2 locus of either parents, potential F1 progeny can be selected on media containing two antibiotics. We then selected and germinated spores in the presence of two antibiotics and screened them by PCR for mating type and for the presence of specific markers at the SH2 locus together with the expected gene deletion to confirm they were F1 offspring. For crosses that involved KN99mCH, the red color of the progenies indicated presence of that marker. In the case of mpk1Δ the newly generated mCherry expressing mpk1Δ strain exhibited expected cell wall stress phenotypes like its parent mpk1Δ strain (data not shown). Together these results confirm that strains marked at the SH2 locus can be used for studies involving mating and sporulation.
4. Discussion
The SH2 locus characterized in this study is the second ‘safe haven’ site reported in the genome of C. neoformans. A previously characterized gene-free region, 1,544 bp in length and located on chromosome 1 between the convergently arranged genes CNAG_00777and CNAG_00778, was suggested to be a good site for gene complementation (Arras et al., 2015). The SH2 locus, located between two tandemly arranged genes on chromosome 11, is 3,278 bp long. This allows over 1 kb of sequence to be left unperturbed on either side of heterologous DNA insertion, which is sufficient to limit any effects of integration on the transcription of neighboring genes.
Our successful integration of multiple sequences at the SH2 locus shows that this chromosomal region is amenable for foreign DNA insertions. We also showed robust expression of mCherry from this region under a variety of conditions, both in vitro and in vivo. The presence of mCherry protein did not affect virulence trait expression in KN99mCH cells, their growth on normal or stress media, or their engulfment by macrophages or intracellular proliferation in vitro. These characteristics, coupled with the bright fluorescence of these cells, make them extremely useful for in vitro host-pathogen interaction studies, as has already been demonstrated in studies of how C. neoformans crosses the blood-brain barrier (Santiago-Tirado et al., 2017).
KN99mCH differed from the parental KN99α strain in its modestly delayed virulence in an intranasal model of cryptococcal infection and its decreased growth in the context of the mouse lung. These phenotypes may be attributable to the observed point mutation resulting in the premature STOP codon for CNAG_02699 in the genome of JLCN923. Even though truncation of a protein is different from the deletion of the entire gene from the genome, the observation that CNAG_02699Δ showed attenuated virulence suggests that mutation of CNAG_02699 may have played a role in the delayed virulence of KN99mCH (Fig. S4). During the generation of KN99mCH (JLCN 923) we have obtained additional mCherry expressing fluorescent strains (Table 1, strains JLCN 920 and JLCN 922). The fluorescence intensity of these strains is lower than JLCN 923 (Fig. S5A). But they were as virulent as KN99α when tested employing murine model of infection as shown in Fig. S5B with median survival time of 15 and 15.5 days respectively for JLCN 920 and JLCN 922 compared to 16 days for KN99α. Even though fluorescence signal of these strains is lower than JLCN 923, they still possess robust fluorescence signal and full virulence and therefore offer a useful tool for studies of host: pathogen interactions in vivo.
We had hoped to monitor infection with KN99mCH by non-invasive whole body animal imaging. Therefore, we chose JLCN 923 which had by far the strongest fluorescent signal of all the mCherry expressing strains we obtained. Although we did detect fluorescent signal upon direct examination of lungs isolated from the infected animals, we could not detect it from whole animals. It may be that expressing a brighter far-red fluorescent protein such as iRFP (Filonov et al., 2011), mCardinal (Chu et al., 2014) or Katushka2S (Luker et al., 2015) from the SH2 locus would create strains useful for quantitative infection studies employing non-invasive whole animal imaging techniques.
Common methods of in vivo dissemination analysis of C. neoformans to study their pathogenesis within a host involve labeling them with fluorescent dyes such as fluorescein isothiocyanate (FITC) (Shi et al., 2010), Uvitex-2B (Nicola et al., 2012), (Davis et al., 2015) or 5-chloromethylfluorescein diacetate (CMFDA; (Alanio et al., 2015; Nicola et al., 2011). Although these studies have helped characterize important in vivo relationships that C. neoformans has with host cells, labeling C. neoformans with dyes to gain an understanding of their dissemination strategies, is cell-division limited. Specifically, as C. neoformans divides they lose the labels described above, in turn, their fluorescent signal, preventing long-term characterization and understanding of their in vivo pathogenesis. As shown by this work, since mCherry is integrated in the genome of KN99mCH and constitutively expressed, a decrease in the mCherry fluorescent signal does not happen, allowing for its use in long-term in vivo host-pathogen studies. Furthermore, fluorescent protein expression further avoids potential perturbation or alteration of surface properties of the yeast that might alter their host interactions. Understanding that the adaptive immune response, which is critical for protection of the host from infection, does not develop until greater than 3 days after the initial exposure of the host to the pathogen, this response, in vivo, can only be studied using a reporter C. neoformans strain such as KN99mCH (Siegrist, 2008).
Another identified benefit of KN99mCH is the ability to use multiple fluorescence detection platforms combined with other fluorescent stains and fluorochrome-conjugated antibodies to simultaneously characterize various traits associated with its host pathogenesis. This current work identified long-term internal and external colocalization of leukocytes with KN99mCH and, for the first time, the live/dead status of C. neoformans during their infection. Using these methods combined with additional fluorochrome-conjugated antibody markers for specific populations and subpopulations of leukocytes or other mammalian cell populations, will further help delineate the in vivo host response to C. neoformans pathogenesis.
Supplementary Material
Highlights.
A second safe haven site (SH2) in the C. neoformans genome that is amenable for targeted heterologous DNA insertions.
A strain encoding mCherry at the SH2 site (KN99mCH) is highly fluorescent.
KN99mCH phenotypes are comparable to those of the parent KN99α strain.
KN99mCH is ideal for in vitro and in vivo host-pathogen interaction studies employing fluorescence detection.
C. neoformans marked at the SH2 locus will facilitate studies involving genetic crosses.
Acknowledgments
We thank Mark Shabsovich and Kimberly J. Gerik for technical help with generation of SH2 marked strains. This work is supported by NIH grants R01 AI072195 and R01 AI125045 to JKL, R01 AI102882 to TLD and NHMRC grant 455980 to JAF. ALC is partially supported by a Stephen I. Morse fellowship from the Department of Molecular Microbiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alanio A, et al. Cryptococcus neoformans host adaptation: toward biological evidence of dormancy. MBio. 2015:6. doi: 10.1128/mBio.02580-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alspaugh JA. Virulence mechanisms and Cryptococcus neoformans pathogenesis. Fungal Genet Biol. 2015;78:55–8. doi: 10.1016/j.fgb.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SD, et al. A genomic safe haven for mutant complementation in Cryptococcus neoformans. PLoS One. 2015;10:e0122916. doi: 10.1371/journal.pone.0122916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LG, Lodge JK. Multiple gene deletion in Cryptococcus neoformans using the Cre-lox system. Methods Mol Biol. 2012;845:85–98. doi: 10.1007/978-1-61779-539-8_6. [DOI] [PubMed] [Google Scholar]
- Baker LG, et al. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell. 2007;6:855–67. doi: 10.1128/EC.00399-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LG, et al. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell. 2011;10:1264–8. doi: 10.1128/EC.05138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks IR, et al. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell. 2005;4:1902–12. doi: 10.1128/EC.4.11.1902-1912.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–5. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, et al. Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat Methods. 2014;11:572–8. doi: 10.1038/nmeth.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, et al. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis. 2009;15:1185–91. doi: 10.3201/eid1508.081384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RC, et al. A PCR-based strategy to generate integrative targeting alleles with large regions of homology. Microbiology. 2002;148:2607–15. doi: 10.1099/00221287-148-8-2607. [DOI] [PubMed] [Google Scholar]
- Davis MJ, et al. Cryptococcus neoformans-induced macrophage lysosome damage crucially contributes to fungal virulence. J Immunol. 2015;194:2219–31. doi: 10.4049/jimmunol.1402376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin MJ, et al. Cross talk between the cell wall integrity and cyclic AMP/protein kinase A pathways in Cryptococcus neoformans. MBio. 2014:5. doi: 10.1128/mBio.01573-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–6. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- Filonov GS, et al. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol. 2011;29:757–61. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis E, et al. Epidemiology of Cryptococcus gattii, British Columbia, Canada, 1999–2007. Emerg Infect Dis. 2010;16:251–7. doi: 10.3201/eid1602.090900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen F, et al. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet Biol. 2015;78:16–48. doi: 10.1016/j.fgb.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Hua J, et al. Development of positive selectable markers for the fungal pathogen Cryptococcus neoformans. Clin Diagn Lab Immunol. 2000;7:125–8. doi: 10.1128/cdli.7.1.125-128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, Heitman J. Genetics of Cryptococcus neoformans. Annu Rev Genet. 2002;36:557–615. doi: 10.1146/annurev.genet.36.052402.152652. [DOI] [PubMed] [Google Scholar]
- Park J, et al. Update on the global burden of cryptococcosis. Mycoses. 2014;57:5–32. [Google Scholar]
- Janbon G, et al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet. 2014;10:e1004261. doi: 10.1371/journal.pgen.1004261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozubowski L, Heitman J. Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol Rev. 2012;36:78–94. doi: 10.1111/j.1574-6976.2011.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronstad JW, et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol. 2011;9:193–203. doi: 10.1038/nrmicro2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon-Chung KJ, et al. Fate of transforming DNA in pathogenic fungi. Med Mycol. 1998;36(Suppl 1):38–44. [PubMed] [Google Scholar]
- Lam WC, et al. Role of Cryptococcus neoformans Rho1 GTPases in the PKC1 signaling pathway in response to thermal stress. Eukaryot Cell. 2013;12:118–31. doi: 10.1128/EC.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Heitman J. The biology of the Cryptococcus neoformans species complex. Annu Rev Microbiol. 2006;60:69–105. doi: 10.1146/annurev.micro.60.080805.142102. [DOI] [PubMed] [Google Scholar]
- Litvintseva AP, et al. Multilocus sequence typing reveals three genetic subpopulations of Cryptococcus neoformans var. grubii (serotype A), including a unique population in Botswana. Genetics. 2006;172:2223–38. doi: 10.1534/genetics.105.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu OW, et al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–88. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker KE, et al. Comparative study reveals better far-red fluorescent protein for whole body imaging. Sci Rep. 2015;5:10332. doi: 10.1038/srep10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade HC, Cox GM. A new dominant selectable marker for use in Cryptococcus neoformans. Med Mycol. 2001;39:151–4. doi: 10.1080/mmy.39.1.151.154. [DOI] [PubMed] [Google Scholar]
- McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RT, et al. Sequence length required for homologous recombination in Cryptococcus neoformans. Fungal Genet Biol. 2003;38:1–9. doi: 10.1016/s1087-1845(02)00510-8. [DOI] [PubMed] [Google Scholar]
- Nicola AM, et al. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun. 2012;80:3065–76. doi: 10.1128/IAI.00358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AM, et al. Nonlytic exocytosis of Cryptococcus neoformans from macrophages occurs in vivo and is influenced by phagosomal pH. MBio. 2011:2. doi: 10.1128/mBio.00167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BJ, et al. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS. 2009;23:525–30. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- Ruff JA, et al. Three galactose inducible promoters for use in C. neoformans var. grubii. Fungal Genet Biol. 2009;46:9–16. doi: 10.1016/j.fgb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Tirado FH, et al. Trojan Horse Transit Contributes to Blood-Brain Barrier Crossing of a Eukaryotic Pathogen. MBio. 2017:8. doi: 10.1128/mBio.02183-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, et al. Real-time imaging of trapping and urease-dependent transmigration of Cryptococcus neoformans in mouse brain. J Clin Invest. 2010;120:1683–93. doi: 10.1172/JCI41963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanta D, et al. A sensitive high-throughput assay for evaluating host-pathogen interactions in Cryptococcus neoformans infection. PLoS One. 2011;6:e22773. doi: 10.1371/journal.pone.0022773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney AE, et al. Gene prediction and verification in a compact genome with numerous small introns. Genome Res. 2004;14:2330–5. doi: 10.1101/gr.2816704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffaletti DL, et al. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol. 1993;175:1405–11. doi: 10.1128/jb.175.5.1405-1411.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya R, et al. Induction of Protective Immunity to Cryptococcal Infection in Mice by a Heat-Killed, Chitosan-Deficient Strain of Cryptococcus neoformans. MBio. 2016:7. doi: 10.1128/mBio.00547-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelz K, et al. Automated analysis of cryptococcal macrophage parasitism using GFP-tagged cryptococci. PLoS One. 2010;5:e15968. doi: 10.1371/journal.pone.0015968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.