

Abstract
Antivitamins B12 have a growing biomedical impact as robust B12-dummies. Here, the potential antivitamin B12 2,4-difluorophenyl-ethynyl-cobalamin (F2PhEtyCbl) was prepared and its 3D-structure was studied in solution and in the crystal. Chemically inert F2PhEtyCbl resisted thermolysis of its (Co-C)-bond at 100°C, was stable to bright day light and also remained intact during prolonged storage in aqueous solution at room temperature. It bound to the human B12-processing enzyme CblC with high affinity (KD = 130 nM) in the presence of the co-substrate glutathione (GSH). F2PhEtyCbl withstood tailoring by CblC, and it also stabilized the ternary complex with GSH. The crystal structure of this abortive assembly afforded first insights into the binding interactions of an antivitamin B12 with CblC, as well as into the organization of GSH and a base-off cobalamin in the active site of this enzyme.
Keywords: antivitamin, glutathione, enzyme inhibitor, protein crystallography, vitamin B12
Graphical abstract
Antivitamin in Action. Antivitamins B12 are B12-dummies derived from the vitamin. A new, chemically robust antivitamin B12 was used for biochemical analysis of inhibition of CblC, the key B12-processing enzyme of humans. The crystal structure of the abortive enzyme complex provided first detailed insights into CblC loaded by a cobalamin and its co-substrate glutathione.

Vitamin B12 (cyanocobalamin, CNCbl) and other cobalamins (Cbls) are indispensable for the mammalian metabolism,[1] on account of the essential cofactor activities of coenzyme B12 (AdoCbl) and methylcobalamin (MeCbl).[2] In order to provide mammalian cells with AdoCbl and MeCbl, natural Cbls are first tailored to cob(II)alamin (CblII) by the Cbl-processing chaperone CblC (also named MMACHC, for methylmalonic aciduria type C and homocystinuria).[3] CblC binds Cbls base-off and removes their axial Coβ-ligand,[3c, 4] furnishing CblII, the relevant biosynthetic precursor of AdoCbl and MeCbl.[3c] Cbls transported into cells, but not de-ligated there by CblC, remain metabolically inert and have been classified as antivitamins B12.[5] Such inert vitamin B12 derivatives induce symptoms of B12-deficiency in laboratory animals,[6] as does a dysfunctional CblC in humans.[3b]
In order to clarify still puzzling roles of Cbls in humans,[1,7] B12-deficiency is studied in laboratory animals. Application of antivitamins B12[5, 8] is a rather humane means in such studies[6, 9] Indeed, the antivitamin B12 4-ethylphenylcobalamin (1, EtPhCbl) induced functional B12 deficiency in mice.[6, 10] Unfortunately 1 is susceptible to light-induced cleavage of its (Co-C)-bond, ‘uncaging’ its locked-in B12 activity.[10] Herein, we report the preparation of 2-(2,4-difluorophenyl)-ethynylcobalamin (3, F2PhEtyCbl, Figure 1), an analogue of the earlier studied light-stable 2-phenylethynyl-cobalamin (2, PhEtyCbl), made more hydrolysis-resistant by two fluorines.[11,12] We also investigated the interactions of 2 and 3 with the human B12-processing enzyme CblC, and describe the crystal structure of the ternary complex of CblC with glutathione (GSH) and 3, allowing for unprecedented insights into a holo-CblC complex, made abortive by an antivitamin B12.
Figure 1.

Left: General structural formula of important Cbls: vitamin B12 (L = CN, cyanocob(III)alamin, CNCbl), coenzyme B12 (L = 5′-adenosyl, 5′-deoxyadenosyl-cob(III)alamin, AdoCbl), methylcob(III)alamin (L = methyl, MeCbl), cob(II)alamin (L = e−, CblII), 4-ethylphenylcob(III)alamin (1, L = 4-ethylphenyl, EtPhCbl) and aquocob(III)alamin (L = H2O+, H2OCbl); Right: Structural formula of 2-phenylethynylcobalamin (2, PhEtyCbl, X = H) and of 2-(2,4-difluorophenyl)-ethynylcobalamin (3, F2PhEtyCbl, X = F).
F2PhEtyCbl (3) was prepared in 58% yield via reduction of aquocob(III)alamin (H2OCbl) with triethylammonium formate in the presence of 2-(2,4-difluorophenyl)-ethynyl iodide, a method developed for the synthesis of PhEtyCbl (2).[11a] The reaction probably follows a radical process initiated by in-situ generated CblII, as proposed for the synthesis 2.[11a] The UV/Vis-spectrum of 3 (Figure 2) is similar to the one of PhEtyCbl (2),[11] but differs significantly from the spectra of the organometallic B12-cofactors AdoCbl and MeCbl.[13] In neutral aqueous solution, F2PhEtyCbl (3) exists in the base-on form, and strong acid is needed in order to de-coordinate the 5,6-dimethylbenzimidazole (DMB) base by protonation to 3-H+. Formation of base-off 3-H+ is accompanied by characteristic hypsochromic shifts of the UV/Vis-absorption maxima (Figure 2). A pKa-value of 0.75 was determined for 3-H+ (see Supporting Information (SI), Figure S11), corresponding to an equilibrium [base-on]/[base-off] of ca. 105 for 3, indicative of strong stabilization of its base-on form.
Figure 2.
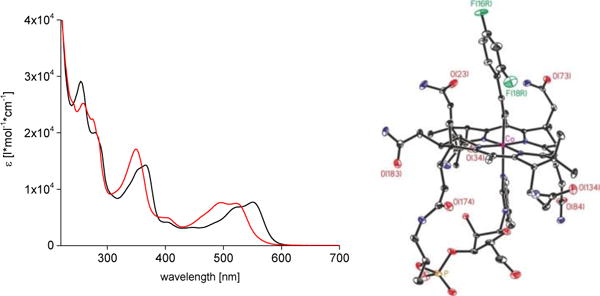
Left: UV/Vis-spectra of an aqueous solution of F2PhEtyCbl (3, c = 55 μM) in H2O (black trace) and immediately after acidification with HClO4 (to 1M) to give base-off H-F2PhEtyCbl+ (3-H+, red trace). Right: Model of the crystal structure of 3 with N, O, P Co and F atoms coloured blue, red, orange, pink, and green, respectively (see SI for details and a stereo-model of 3).
The molecular formula and the basic structure of F2PhEtyCbl (3) were delineated from a MALDI-TOF mass spectrum, an IR-spectrum (signal of the (C-C) triple bond at 2130.5 cm−1, see SI, Figure S2) and detailed 1H-NMR- and 13C-NMR-studies of a solution of 3 in CD3OD (SI, Figure S3). Orthorhombic crystals of 3 (space group P212121) grew from aqueous acetone. The crystal structure of 3 showed a base-on Cbl with an ‘upper’ 2,4-difluorophenylethynyl ligand (Figure 2 and SI, Figures S5 and S6 and Table S1). Geometry and bond distances of the inner coordination sphere of the cobalt center of 3 and of PhEtyCbl (2)[11] are similar. As in 2[11] and in CNCbl,[14,15] the axial bonds of 3 are short (Co-C1L: 1.877(7) Å, Co-N3N: 2.071(5) Å). Likewise, the ethynyl group of 3 exhibits a short (1.206(9) Å) and nearly linear C1L-C2L triple-bond, with bond angles Co-C(1L)-C(2L) of 170.5 (5)° and C(1L)-C(2L)-C(L3) of 176.1(7)°. The plane of the 2,4-difluorophenyl group is nearly parallel to the DMB-base, twisted by only 23.1(3)°.
When a solution of F2PhEtyCbl (3) in DMSO was heated to 100°C for 24 h UV/Vis-spectra and HPLC-analyses (SI, Figure S7) indicated complete retention of the (Co-C)-bond. As reported earlier for ethynyl-Cbl[14] and for 2,[11–12] solutions of 3 were stable to sunlight (SI, Figure S8). In aqueous solution, 3 underwent slow pH-dependent hydrolysis to H2OCbl. At pH 2, 3 hydrolyzed with pseudo-first-order kinetics and with a half-life of about 20 h at room temperature. A dependence with roughly first-order upon the H3O+-concentration from pH 0 to pH 4 (SI, Figures S9 and S10) allowed an estimate of the hydrolysis-based half-life of 3 at pH 7 of about 107 h. Hence, 3 should be considered effectively stable under physiological conditions.
Binding of F2PhEtyCbl (3) (or of PhEtyCbl, 2) to CblC was accompanied by blue shifts of the UV/Vis-absorptions, as observed with MeCbl[16] and EtPhCbl.[10] Conversion from base-on 3 (or 2) to corresponding base-off forms was slow, as monitored by UV/Vis-spectroscopic changes accompanying binding (Figure 3 and SI, Figure S12). Addition of the co-substrate glutathione (GSH) caused further hypsochromic shifts of the maxima. Hence, binding of 3 (or of 2) to CblC in the presence of GSH was accompanied by a complete switch to the base-off forms, suggesting a structuring role of GSH-binding in holo-CblC (Figure 3 and SI, Figure S13). Accommodation of the large organometallic moiety of 3 in CblC was consistent with expectations based on the crystal structures of CblC with bound MeCbl.[17] Isothermal calorimetry (ITC) revealed a high binding affinity of the alkynyl-Cbl 3 to human CblC (KD = 129(13) nM) in the presence of glutathione (GSH), about 4 times less than for 2 (KD = 30(5.6) nM) (Figure 3 and SI, Figure S15). Incubation of 17–20 μM solutions of 2 or 3 and 10 mM of GSH together with 50 μM CblC at 20 °C for 12 h did not lead to detectable degradation of either alkynyl-Cbl (SI, Figure S14). Hence, in contrast to MeCbl, which is rapidly de-methylated by CblC and GSH (kobs = 0.6 min−1),[10] 2 and 3 resisted action by this universal ‘B12-tinkerer’ (kobs < 3*10−7 sec−1).
Figure 3.
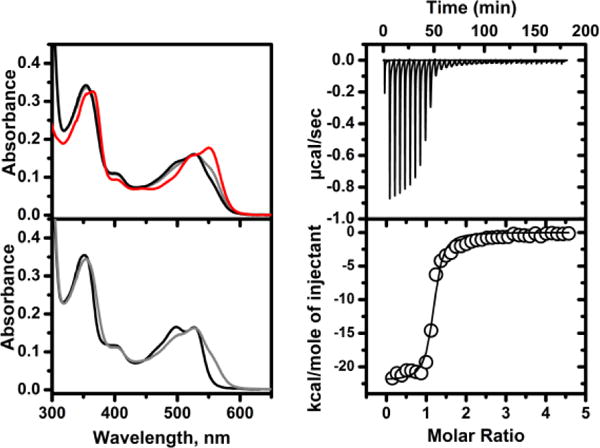
Biochemical studies with CblC, F2PhEtyCbl (3) and GSH. Left top: Time-dependent UV/Vis-spectra associated with binding of 3 to CblC: The original spectrum of 3 is shown in red, the final spectrum after 60 min in black. Left bottom: Spectral changes upon subsequent binding of GSH - initial and final spectra are shown in grey and black, respectively. Right: Isothermal calorimetry (ITC) data for a representative titration of CblC with 3 in the presence of GSH (10 μM CblC, 200 μM 3, 1 mM GSH). The binding isotherm (lower panel) was fitted to a one-binding site model (see SI for details).
The stable ternary complex of the alkynyl-Cbl 3, GSH and CblC crystallized via the vapor diffusion method. Crystals grew in the P6122 space group with one monomer in the asymmetric unit (SI, Table S2). The structure was solved to 2.5 Å resolution (Figure 4 and SI, Figure S16) by molecular replacement, using a previous structure of CblC as a search model (PDB accession number 3SC0),[17] but omitting the MeCbl cofactor, loops and areas with high B-factors. Clear electron density could be assigned to the Cbl and GSH ligands, which were then modeled and subsequently refined. The antivitamin 3 was bound in a base-off form. In this structure (with 3 and GSH bound to CblC) and in the one with MeCbl bound to CblC[17] position and interactions of the DMB-base were not distinguishable. Likewise, the a- and g-acetamide side chains at rings A and D, as well as the b-propionamide, are tightly H-bonded to the protein part and are found in identical positions in both crystal structures. Among the distinct differences, the d- and e-propionamide and the c-acetamide side chains at rings B and C adopt unique arrangements in the structure of CblC with F2PhEtyCbl and GSH bound (Figures 4 and 5). However, they are not as close to each other and to the Co atom (with Co…N45, Co…N52, N45…N52 distances found at 4.2, 5.1 and 4.3 Å respectively), as in MeCbl bound to CblC, thus allowing better access to the lower axial position. In MeCbl bound to CblC the d- and e-propionamide side chains approach each other and the Co-center even closer (with Co…N45, Co…N52, N45…N52 distances found at 4.1, 4.0 and 3.0 Å respectively). Such striking ‘tuck-in’ structures appear to be a hallmark of Cbls bound to CblC,[17,18] crucial for activation of the bound corrin for ‘tailoring’ by dealkylation of reduction.[3c,4] Remarkably, in the CblC ternary complex with GSH, the H-bonding network between the corrin ring of 3 and its surrounding protein milieu is largely similar to that of the binary complexes of CblC with MeCbl[17] or with AdoCbl.[18]
Figure 4.
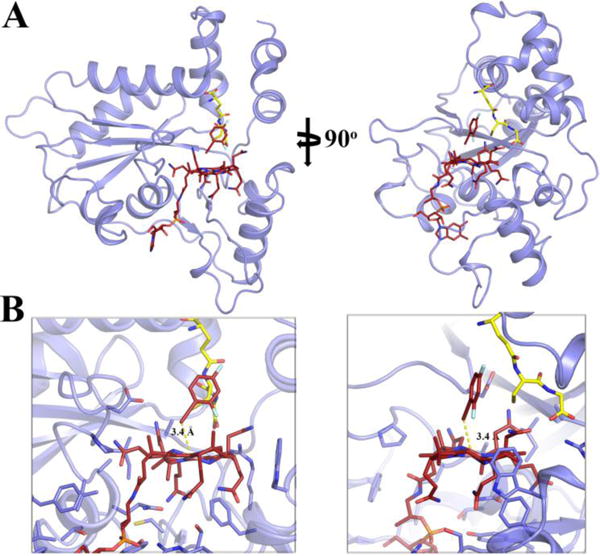
A Two views of structure of GSH and F2PhEtyCbl (3) bound to CblC (light blue) shown as a cartoon. GSH (yellow) and 3 (dark red) ligands are shown as sticks. B Two corresponding close-up views of this ternary complex.
Figure 5.
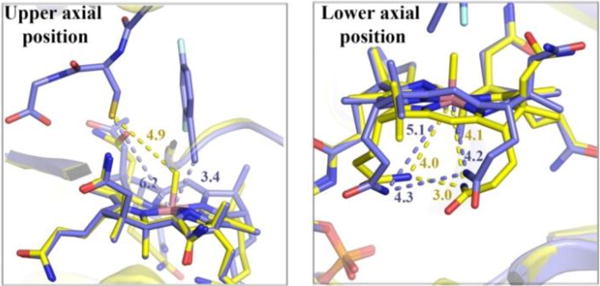
Structural comparison of CblC with MeCbl bound (yellow), and the ternary complex of CblC with GSH and F2PhEtyCbl bound (blue). On the left panel, a close-up of the environment of the upper axial Co position in the vicinity of the GSH ligand is presented. On the right panel a close-up of the environment of the lower axial Co position is shown. Blue and yellow dotted lines represent selected distances of the GSH and F2PhEtyCbl bound and MeCbl bound CblC structures, respectively. Note the arrangement of the ‘tucked-in’ d- and e-propionamide side chains.
Distinct electron density was observed in the vicinity above the Co atom where a β-axial ligand would be expected (SI, Fig S17). However, we could not discern an ethynyl carbon directly bound to the Co-center from the clear electron density associated with the F2PhEty group. Based on a calculated unbiased composite omit map, the 2,4-difluorophenyl ring was observed in a different position compared to the free form of F2PhEtyCbl (3) and farther away from the cobalt atom (Figure 4). The planes of the phenyl-rings in free 3 and its CblC-bound form were roughly perpendicular with respect to each other. Moreover, the axis of the aromatic no longer pointed to a Co dz2-orbital. Hence, the 2,4-difluorophenyl-ethynyl group, as it best fits the experimental electron density, is too far away from the Co-center to be coordinated to it. Therefore, we posit that the intense synchrotron radiation during the data collection has led to cleavage of the Co-C bond, possibly by a reductive path,[18–20] furnishing (base-off) CblII and a weakly bound F2PhEty anion. This model gives a distance of 3.4 Å between the CoII-center of Cbl and C1L of the F2PhEty fragment (see Figure 4).
Clear electron density correlating with GSH allowed for a first characterization of its protein environment in CblC and its location with respect to the bound Cbl (Figure 6A and SI, Fig S18). An extended network of protein - GSH interactions anchored this ligand to its binding pocket. Specifically, a combination of 1) multiple H-bond interactions with all three peptide moieties (Glu, Cys, Gly) of GSH, and 2) hydrophobic stacking interactions between the glycine moiety and protein residues (F223, Y224, and A159). Most of the GSH ligand is enclosed by the protein and the bound Cbl, with 70 % of the total GSH surface area buried by the protein and 20% buried by Cbl. In fact, the weakly bound F2PhEty group packs tightly against GSH, and SGSH is near the aromatic ring of the F2PhEty fragment, with distances varying from 4.2 to 5.1 Å. However, the neighboring CH2-group of GSH is found at distances ranging from 3.9 to 4.0 Å from the F2PhEty ring, indicative of C-H-π contacts. In contrast, when modeling-in the structure of intact 3, steric clashes with the GSH ligand occur (see SI, Figures S19 & S20).
Figure 6.

A. GSH-binding environment in the ternary complex with CblC and F2PhEtyCbl (3). GSH and 3 are shown in yellow and dark red sticks, respectively. Protein residues comprising the GSH binding pocket are shown in blue sticks, H-bonds are represented with yellow dotted lines. B. Structural superposition of Apo (pink), MeCbl-bound (yellow) and GSH and F2PhEtyCbl bound CblC (blue). MeCbl (blue), GSH (yellow) and F2PhEtyCbl (yellow) ligands are displayed as sticks. The three major structural differences are highlighted with arrows.
Previous reports based on available structures, sequence conservation and biochemical analysis,[17–18, 21] suggested and identified R230, R161 and R206 as putative residues important for GSH binding. Indeed, our structure determination confirmed the crucial role of these three arginines and identified R161 as anchor of GSH in the CblC active site through interactions of its guanidinium group and the carboxyl group of the cysteine moiety of GSH. However, Y215, D77 and D80 were now additionally shown to H-bond to GSH, pointing to an induced fit by the protein that was not previously predicted. The S-center (SGSH) of GSH, key to the Cbl-dealkylation activity of CblC, is situated in the vicinity of the bound Cbl with a Co to SGSH atom distance found at 6.3 Å. This corresponds to a calculated distance of 4.9 Å between the methyl carbon in a superimposed CblC-bound MeCbl and SGSH in our structure. This position of SGSH vs. the bound corrin does not fulfill the geometric constraints of the proposed SN2-type mechanism of de-alkylation by GSH. Strikingly, the SGSH group also lacks any close contacts that would facilitate its deprotonation and increase its nucleophilicity.
The protein fold of the here studied abortive ternary CblC complex reveals distinct differences, when compared to the previous structures of apo-CblC (PDB ID 3SBZ) and of MeCbl-bound CblC (MeCbl:CblC; PDB ID 3SC0)[17] (Figure 6). Loop LDE is found in a unique arrangement as result of GSH-binding in the ternary complex. In order to accommodate for its glutamate moiety, the co-substrate GSH also promotes a rearrangement of the N-terminal part of loop LDE, as the well as of the C-terminal region of preceding helix (HD), including residues 77–80. Moreover, the terminal helix (HL) in the four-helical cap region occurs in different conformations in the CblC structures with MeCbl or F2PhEtyCbl (3) bound. However, helix HL appears in the same conformation in both the Apo- and F2PhEtyCbl-bound CblC structures, hinting at the dynamic nature of this helix.
The base-off forms of 2 or 3 bound effectively to CblC in the presence of glutathione (GSH). However, as shown here also, abortive ternary complexes were assembled in this way, incapable of generating CblII. The lack of reactivity of the antivitamin B12 3 was employed to trap the co-substrate GSH in a previously unattainable form of the ternary complex, allowing for a first crystal structure of holo-CblC with GSH and with an antivitamin B12 bound. This study pinpointed the specific protein and Cbl contributions in anchoring GSH, providing a rational structural basis for future work with mutant proteins. For example, two severe pathogenic mutations of CblC (R161G and R161Q) can now be rationalized, based on the key role of R161 in binding GSH. Antivitamins B12, more resistant against X-ray reduction than 3 in the complex with GSH, may help define more precisely the structural basis for the de-alkylation activity of CblC.
As explored here for a B12-processing enzyme, antivitamins B12 producing abortive enzyme complexes may be helpful for solving their crystal structures. Future studies are also required to learn more about the scope of antivitamins B12 like 3. In fact, EtPhCbl (1) not only induced ‘functional B12-deficiency’ in mice,[6] but it also helped in eradicating resistant gram negative human pathogens.[22] Studies of antivitamins B12 and related Cbls as anti-cancerous agents have also raised interest.[5,8,23] Indeed, 3 is a promising, light stable alternative to the antivitamin B12 1,[6,10] and may also be a very versatile biomedical tool. Long-term experiments with animals in close to natural habitats[6] may be feasible with 3 that will specifically help in expanding our knowledge of the physiological effects of B12-deficiency, and in reducing the disturbing gap in understanding its downstream pathophysiological effects in humans.
Experimental Section
See Supporting Information (SI) for materials, instruments, synthetic procedures, spectral characterization of compounds and enzyme complexes, biochemical studies and X-ray crystallography.
X-ray crystallography: The supplementary crystallographic data of F2PhEtyCbl (3) were deposited as CCDC-1530214. These data are provided, free of charge, by the Cambridge Crystallographic Data Centre. Coordinates and structure factors for the structure of the complex F2PhEtyCbl:GSH:CblC have been deposited in the Protein Data Bank as entry 5UOS.
Supplementary Material
Acknowledgments
We are grateful to the Austrian Science Foundation (FWF) for support of the research in Innsbruck (current project FWF P-28892). We also would like to thank the National Institute of General Medical Sciences and National Cancer Institute Collaborative Access Team (GM/CA CAT) at the Advanced Light Source for beam time. This work was supported in part by the American Heart Association (13SDG14560056, to MK) and by the National Institute of Health (NIH, grant DK45776, to RB).
Footnotes
Supporting information for this article is given via a link at the document’s end.
Contributor Information
Dr. Markus Ruetz, Institute of Organic Chemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck (Austria) University of Michigan Medical School, Ann Arbor, MI 48109-0600 (USA).
Aranganathan Shanmuganathan, Department of Biochemistry, Uniformed Services University of the Health Sciences, Bethesda, MD 28104 (USA); Department of Biochemistry, Uniformed Services University of the Health Sciences, Bethesda, MD 28104 (USA).
Dr. Carmen Gherasim, University of Michigan Medical School, Ann Arbor, MI 48109-0600 (USA) Department of Pathology, University of Utah School of Medicine, Salt Lake City, UT. (USA).
Agnes Karasik, Department of Biochemistry, Uniformed Services University of the Health Sciences, Bethesda, MD 28104 (USA).
Dr. Robert Salchner, Institute of Organic Chemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck (Austria) Watercryst GmbH & Co, Kematen (Austria).
Christoph Kieninger, Institute of Organic Chemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck (Austria).
Dr. Klaus Wurst, Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, A-6020 Innsbruck (Austria)
Prof. Ruma Banerjee, University of Michigan Medical School, Ann Arbor, MI 48109-0600 (USA)
Prof. Markos Koutmos, Department of Biochemistry, Uniformed Services University of the Health Sciences, Bethesda, MD 28104 (USA)
Prof. Bernhard Kräutler, Institute of Organic Chemistry and Center for Molecular Biosciences, University of Innsbruck, A-6020 Innsbruck (Austria).
References
- 1.Carmel R, Green R, Rosenblatt DS, Watkins D. Hematology Am Soc Hematol Educ Program. 2003:62–81. doi: 10.1182/asheducation-2003.1.62. [DOI] [PubMed] [Google Scholar]
- 2.a) Kräutler B, Arigoni D, Golding BT, editors. Vitamin B12 and B12 Proteins. Wiley-VCH; Weinheim: 1998. [Google Scholar]; b) Banerjee R, editor. Chemistry and Biochemistry of B12. John Wiley & Sons; New York, Chichester: 1999. [Google Scholar]; c) Banerjee R, Ragsdale SW. Ann Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]; d) Matthews RG, Koutmos M, Datta S. Curr Opin Struct Biol. 2008;18:658–666. doi: 10.1016/j.sbi.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lerner-Ellis JP, Tirone JC, Pawelek PD, Dore C, Atkinson JL, Watkins D, Morel CF, Fujiwara TM, Moras E, Hosack AR, Dunbar GV, Antonicka H, Forgetta V, Dobson CM, Leclerc D, Gravel RA, Shoubridge EA, Coulton JW, Lepage P, Rommens JM, Morgan K, Rosenblatt DS. Nature Genetics. 2006;38:93–100. doi: 10.1038/ng1683. [DOI] [PubMed] [Google Scholar]; b) Hannibal L, DiBello PM, Yu M, Miller A, Wang SH, Willard B, Rosenblatt DS, Jacobsen DW. Mol Genet Metab. 2011;103:226–239. doi: 10.1016/j.ymgme.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Banerjee R, Gherasim C, Padovani D. Curr Opin Chem Biol. 2009;13:484–491. doi: 10.1016/j.cbpa.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannibal L, Kim J, Brasch NE, Wang SH, Rosenblatt DS, Banerjee R, Jacobsen DW. Mol Genet Metab. 2009;97:260–266. doi: 10.1016/j.ymgme.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kräutler B. Chem Eur J. 2015;21:11280–11287. doi: 10.1002/chem.201502072. [DOI] [PubMed] [Google Scholar]
- 6.Mutti E, Ruetz M, Birn H, Kräutler B, Nexo E. Plos One. 2013;8:e75312. doi: 10.1371/journal.pone.0075312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Scalabrino G. Progress in Neurobiol. 2009;88:203–220. doi: 10.1016/j.pneurobio.2009.04.004. [DOI] [PubMed] [Google Scholar]; b) Quadros EV. British J Haematol. 2009;148:195–204. doi: 10.1111/j.1365-2141.2009.07937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Solomon LR. Blood Rev. 2007;21:113–130. doi: 10.1016/j.blre.2006.05.001. [DOI] [PubMed] [Google Scholar]; d) Moreno-Garcia MA, Rosenblatt DS, Jerome-Majewska LA. Nutrients. 2013;5:3531–3550. doi: 10.3390/nu5093531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zelder F, Sonnay M, Prieto L. ChemBioChem. 2015;16:1264–1278. doi: 10.1002/cbic.201500072. [DOI] [PubMed] [Google Scholar]
- 9.Scalabrino G, Peracchi M. Trends Mol Med. 2006;12:247–254. doi: 10.1016/j.molmed.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Ruetz M, Gherasim C, Fedosov SN, Gruber K, Banerjee R, Kräutler B. Angew Chem Int Ed. 2013;52:2606–2610. doi: 10.1002/anie.201209651. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:2668–2672. [Google Scholar]
- 11.a) Ruetz M, Salchner R, Wurst K, Fedosov S, Kräutler B. Angew Chem Int Ed. 2013;52:11406–11409. doi: 10.1002/anie.201305206. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:11617–11620. [Google Scholar]; b) Chrominski M, Lewalska A, Gryko D. Chem Comm. 2013;49:11406–11408. doi: 10.1039/c3cc47210h. [DOI] [PubMed] [Google Scholar]
- 12.Miller NA, Wiley TE, Spears KG, Ruetz M, Kieninger C, Kräutler B, Sension RJ. J Am Chem Soc. 2016;138:14250–14256. doi: 10.1021/jacs.6b05299. [DOI] [PubMed] [Google Scholar]
- 13.Giannotti C. In: B12. Dolphin D, editor. I. Wiley & Sons; New York: 1982. pp. 393–430. [Google Scholar]
- 14.Pratt JM. Inorganic Chemistry of Vitamin B12. Academic Press; New York: 1972. [Google Scholar]
- 15.Randaccio L, Geremia S, Nardin G, Würges J. Coord Chem Rev. 2006;250:1332–1350. [Google Scholar]
- 16.Kim J, Hannibal L, Gherasim C, Jacobsen DW, Banerjee R. J Biol Chem. 2009;284:33418–33424. doi: 10.1074/jbc.M109.057877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koutmos M, Gherasim C, Smith JL, Banerjee R. J Biol Chem. 2011;286:29780–29787. doi: 10.1074/jbc.M111.261370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Froese DS, Krojer T, Wu XC, Shrestha R, Kiyani W, von Delft F, Gravel RA, Oppermann U, Yue WW. Biochem. 2012;51:5083–5090. doi: 10.1021/bi300150y. [DOI] [PubMed] [Google Scholar]
- 19.a) Gruber K, Reitzer R, Kratky C. Angew Chem Int Ed. 2001;40:3377–3380. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2001;113:3481–3884. [Google Scholar]; b) Randaccio L, Geremia S, Würges J. J Organomet Chem. 2007;692:1198–1215. [Google Scholar]
- 20.Lexa D, Savéant JM. Acc Chem Res. 1983;16:235–243. [Google Scholar]
- 21.a) Gherasim C, Ruetz M, Li Z, Hudolin S, Banerjee R. J Biol Chem. 2015;290:11393–11402. doi: 10.1074/jbc.M115.637132. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lerner-Ellis JP, Anastasio N, Liu JH, Coelho D, Suormala T, Stucki M, Loewy AD, Gurd S, Grundberg E, Morel CF, Watkins D, Baumgartner MR, Pastinen T, Rosenblatt DS, Fowler B. Human Mutation. 2009;30:1072–1081. doi: 10.1002/humu.21001. [DOI] [PubMed] [Google Scholar]
- 22.Guzzo MB, Nguyen HT, Pham TH, Wyszczelska-Rokiel M, Jakubowski H, Wolff KA, Ogwang S, Timpona JL, Gogula S, Jacobs MR, Ruetz M, Kräutler B, Jacobsen DW, Zhang GF, Nguyen L. Plos Pathog. 2016;12:e1005949. doi: 10.1371/journal.ppat.1005949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Zelder F, Alberto R. In: Handbook of Porphyrin Science. Kadish KM, Smith KM, Guilard R, editors. Vol. 25. 2012. pp. 84–132. World Scientific. [Google Scholar]; b) Rossier J, Hauser D, Kottelat E, Rothen-Rutishauser B, Zobi F. Dalton Trans. 2017;46:2159–2164. doi: 10.1039/c6dt04443c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.