

Abstract
Background
Selective kinase and immune checkpoint inhibitors, and their combinations, have significantly improved the survival of patients with advanced metastatic melanoma. Not all patients will respond to treatment however, and some patients will present with significant toxicities. Hence, the identification of biomarkers is critical for the selection and management of patients receiving treatment. Biomarker discovery often involves proteomic techniques that simultaneously profile multiple proteins but few studies have compared these platforms.
Methods
In this study, we used the multiplex bead-based Eve Technologies Discovery assay and the aptamer-based SomaLogic SOMAscan assay to identify circulating proteins predictive of response to immunotherapy in melanoma patients treated with combination immune checkpoint inhibitors. Expression of four plasma proteins were further validated using the bead-based Millipore Milliplex assay.
Results
Both the Discovery and the SOMAscan assays detected circulating plasma proteins in immunotherapy-treated melanoma patients. However, these widely used assays showed limited correlation in relative protein quantification, due to differences in specificity and the dynamic range of protein detection. Protein data derived from the Discovery and Milliplex bead-based assays were highly correlated.
Conclusions
Our study highlights significant limitations imposed by inconsistent sensitivity and specificity due to differences in the detection antibodies or aptamers of these widespread biomarker discovery approaches. Our findings emphasize the need to improve these technologies for the accurate identification of biomarkers.
Electronic supplementary material
The online version of this article (10.1186/s40364-017-0112-9) contains supplementary material, which is available to authorized users.
Keywords: Cytokines, Multiplexing, Luminex, Aptamers, SOMAscan, Biomarkers, Liquid biopsies, Immune checkpoint inhibitors, Melanoma
Background
The identification and validation of biomarkers for monitoring disease progression, and predicting response to therapy and patient outcome is a rapidly growing field in cancer research. Cancer patients are currently being treated with new generations and combinations of targeted drugs [1, 2] and immunotherapy [3, 4] but, the activity of these drugs is hampered by variable response rates and the development of treatment resistance [5–7]. For example, approximately 60% of patients with advanced melanoma respond to the combination of the cytotoxic T lymphocyte antigen-4 (CTLA-4) inhibitor, ipilimumab, with an inhibitor of the programmed death-1 (PD-1) receptor (pembrolizumab or nivolumab). However, the benefit of this drug combination comes with significant toxicity and 40% of patients will show no response to therapy [8, 9]. As a result, the identification and validation of reliable, sensitive and accurate predictive biomarkers is necessary for the improved selection and management of cancer patients.
Circulating biomarkers, identified in liquid biopsies such as serum and plasma, can provide an accurate and minimally invasive means for monitoring disease progression, tumor heterogeneity and treatment response. Analysis of certain circulating biomarkers has already yielded important prognostic and diagnostic information, such as prostate-specific antigen (PSA) in prostate cancer [10] and carcinoembryonic antigen (CEA) in colorectal cancer [11], while others have assisted assessment of treatment response and resistance, including circulating tumor DNA (ctDNA) [12, 13] and cancer antigen CA125 [14].
Biomarker discovery using proteomic analysis of liquid biopsies usually involves bead-based multiplex Luminex assays, aptamer-based assays or unbiased mass spectrometry. A literature search using the terms “cancer biomarker” and “liquid biopsy” (August 1, 2017) for publications reporting on liquid biomarker discovery in cancer found 62 publications applying the bead-based system, 36 publications using aptamer-based assays and 252 publications based on mass spectrometry. Although significant attention has focused on cancer biomarker discovery, few studies have compared commonly used protein detection and quantitation platforms.
In this study, we compared a bead-based multiplex assay (Eve Technologies 65-plex Human Cytokine/Chemokine Discovery assay) and an aptamer-based technology (SomaLogic SOMAscan assay) for biomarker discovery in 47 plasma samples derived from 24 melanoma patients treated with combination pembrolizumab and ipilimumab therapy. We show that these widely used assays have limited correlation in relative protein quantification, and this was largely due to differences in specificity and the dynamic range of protein detection. Further, we found that discrepancies in protein quantification and detection were more apparent when two different detection reagents (i.e. antibodies vs. aptamers) were used. Our findings highlight significant limitations in two common approaches for biomarker discovery, and underscore the need for robust method validation and independent assay assessment for blood-based biomarker discovery.
Methods
Patients, treatment and clinical assessment
This study included 24 metastatic melanoma patients treated with immunotherapy at Westmead Hospital and Melanoma Institute Australia between July 2014 and December 2015. Patients were treated with pembrolizumab in combination with ipilimumab, administered according to the schedule in the MK3475–029 clinical trial (NCT02089685). Informed consent was obtained from all patients under approved human research ethics committee protocols from the Royal Prince Alfred Hospital.
Investigator-determined objective response was assessed radiologically with computed tomography scans 12 weeks after start of treatment. Patients were divided into responders (Response Evaluation Criteria In Solid Tumor; RECIST CR and PR) and non-responders (RECIST SD and PD) based on RECIST 1.1 guidelines [15] (Fig. 1).
Fig. 1.
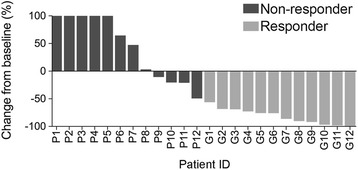
RECIST response of melanoma patients. Melanoma patients were divided into responding (n = 12) or non-responding (n = 12) groups based on RECIST 1.1 (response evaluation criteria in solid tumor) guidelines following the Week 12 computed tomography scan. Patients in the responding group showed RECIST CR (complete response) or PR (partial response) while those in the non-responding group had RECIST SD (stable disease) or PD (progressive disease). The percentage of change in target lesions from baseline to Week 12 is shown
Patient characteristics, clinicopathologic and demographic information including mutation status, lactate dehydrogenase (LDH) levels, disease distribution and American Joint Committee on Cancer (AJCC) M stage (7th edition) were collected (Table 1).
Table 1.
Patient characteristics and treatment outcomes
| Responders | ||||
| Patient ID | RECIST at Week 12 | Baseline LDH (U/L) | PFS (months)a | OS (months)a |
| G1 | PR | 167 | Response ongoing | Alive |
| G2 | PR | 215 | Response ongoing | Alive |
| G3 | PR | 159 | Response ongoing | Alive |
| G4 | PR | 217 | Response ongoing | Alive |
| G5 | PR | 185 | Response ongoing | Alive |
| G6 | PR | 206 | Response ongoing | Alive |
| G7 | PR | 268 | Response ongoing | Alive |
| G8 | PR | 167 | Response ongoing | Alive |
| G9 | PR | 227 | Response ongoing | Alive |
| G10 | PR | 187 | Response ongoing | Alive |
| G11 | PR | 140 | Response ongoing | Alive |
| G12 | CR | 150 | Response ongoing | Alive |
| Non-responders | ||||
| Patient ID | RECIST at Week 12 | Baseline LDH (U/L) | PFS (months)a | OS (months)a |
| P1 | PD | 303 | 2.7 | 5.8 |
| P2 | PD | 568 | 2.7 | 5.1 |
| P3 | PD | 329 | 2.7 | 3.1 |
| P4 | PD | 300 | 1.1 | Alive |
| P5 | PD | 271 | 0.5 | Alive |
| P6 | PD | 267 | 2.7 | Alive |
| P7 | PD | 169 | 2.8 | Alive |
| P8 | SD | 226 | 4 | Alive |
| P9 | SD | 269 | 4.1 | Alive |
| P10 | SD | 176 | 2.7 | 21.7 |
| P11 | SD | 195 | Response ongoing | Alive |
| P12 | SD | 402 | 5.5 | 8.3 |
Abbreviations: PR partial response, SD stable disease, PD progressive disease, CR complete response, LDH lactate dehydrogenase, PFS progression free survival, OS overall survival, U/L units per liter
aData cut off on 30th April, 2017. 11 out of 24 patients had progressed at time of analysis, all of whom were in the non-responders group. Median follow-up for these patients is 24.3 months. Normal range of LDH is 120–250 U/L
Plasma samples
Peripheral blood samples (~10 ml) were collected from patients in EDTA vacutainer tubes (BD Vacutainer Blood Collection Tubes) before treatment (baseline biopsy) and early during therapy (EDT; collected between 1 to 6 weeks after treatment initiation). All patients had an available EDT sample and 23 patients also had a matched baseline biopsy. Blood samples were centrifuged at 1500 rpm (800 x g) for 15 min at room temperature to separate plasma. Approximately 3–4 ml plasma were collected and centrifuged again at 4100 rpm (1600 x g) for 10 min at room temperature. Clarified plasma samples (1 ml aliquots) were stored at −80 °C, and the plasma volume required for the Discovery (155 μl), SOMAscan (130 μl) and Milliplex (50 μl) assays were obtained from the same plasma aliquot.
Proteomic assays
Undiluted baseline and EDT plasma samples from 24 patients were profiled using the 65-plex Discovery assay (Human Cytokine Array/Chemokine Array 65-Plex Panel; Cat no: HD65, Eve Technologies, Alberta, Canada) and the 1310 protein SOMAscan assay (SomaLogic, Inc. CO, USA).
The 65-plex Discovery assay is based on the Luminex technology and utilizes the Millipore assay that comprises of fluorescent color-coded beads pre-coated with capture antibodies targeting 65 specific cytokines. Plasma samples were incubated with the beads before the addition of biotinylated detection antibodies followed by phycoerythrin (PE)-conjugated streptavidin. Bound cytokines were identified and quantitated using the Bio-Rad BioPlex 200 bead analyzer consisting of a dual-laser system which i) activates the fluorescent beads to identify the specific cytokine and, ii) excites the PE conjugate to determine the magnitude of fluorescence, which is in direct proportion to reflect the amount of bound cytokine. This assay utilized 150 μl of plasma per run and each run was performed in duplicate; duplicates did not vary by more than 4%.
The SOMAscan assay uses SOMAmer (Slow Off-rate Modified Aptamer) reagents, which consist of modified short DNA sequences that bind specific protein analytes [16]. Plasma samples were incubated with SOMAmer reagents and protein analytes bound to SOMAmer reagents were then biotinylated before capturing the SOMAmer-protein complexes with streptavidin beads. The SOMAmer-protein complexes were detached, and SOMAmer reagents collected and hybridized to complementary sequences on a microarray chip and quantified by fluorescence, which directly correlates with protein amount in the plasma samples. This assay utilized 130 μl of plasma sample per assay run singly. Five patient plasma samples were run in different batches and data showed high concordance (r = 0.99, p < 0.0001) between batch runs.
Expression of IL-1α, IL-1RA, TNFα and IL-6 in 13 baseline and 15 EDT plasma samples were further validated using the Luminex technology (Milliplex MAP Human Cytokine/Chemokine Panel; Cat no: HCYTOMAG-60 K, Millipore, St. Charles, MO). This bead-based assay is similar to the 65-plex Discovery assay and utilizes fluorescent color-coded beads pre-coated with capture antibodies targeting 4 specific cytokines. Plasma samples were filtered through 0.22 μm spin filters and 25 μl of undiluted plasma was run in duplicates per assay. Duplicates did not vary by more than 5%. Samples were assayed on a robotic liquid handling workstation (epMotion 5075, Eppendorf, Germany) and read with the BioPlex Systems 100 (Bio-Rad) as previously reported [17].
Fluorescence intensity values derived from the Discovery, SOMAscan and Milliplex assays were reported as relative fluorescent units (RFU). Additionally, for the Discovery and Milliplex assays, a protein standard consisting of purified cytokines at known concentrations was included in each batch run; absolute protein concentrations were calculated from the standard curve and reported as pg/ml. Protein standards were not included in the SOMAscan assay and as such, absolute protein concentrations were not determined.
Statistical analysis
Differential protein expression analysis was performed using the limmaGP module in GenePattern [18]. Comparison between two groups was performed using Mann-Whitney test, correlation analysis using Spearman’s correlation coefficient, and patient characteristics compared using a Chi square test in GraphPad Prism (version 7.02). Principal Component Analysis (PCA) was performed using the ClutVis program [19].
Results
Patient response
Twelve patients responded to pembrolizumab in combination with ipilimumab with a complete response (CR, n = 1) or a partial response (PR, n = 11) and twelve patients had no objective response, i.e. stable (SD, n = 5) or progressive disease (PD, n = 7). Clinical characteristics across these two response groups were similar for age, sex and disease volume but were significantly different in the known prognostic factors of baseline LDH and AJCC tumor stage (Tables 1 and 2); earlier AJCC stage and normal LDH are associated with good response to combination immunotherapy.
Table 2.
Summary of patient characteristics
| Characteristics | Responders (n = 12) | Non-responders (n = 12) | P value |
|---|---|---|---|
| Age – no. (%) | |||
| > 65 | 6 (50%) | 2 (17%) | 0.083 |
| ≤ 65 | 6 (50%) | 10 (83%) | |
| Sex – no. (%) | |||
| Male | 9 (75%) | 9 (75%) | >0.999 |
| Female | 3 (25%) | 3 (25%) | |
| LDH – no. (%) | |||
| > 1× ULN | 1 (8%) | 8 (67%) | 0.0032 |
| ≤ 1× ULN | 11 (92%) | 4 (33%) | |
| Disease volume – no. (%) | |||
| SPOD > 1000 mm2 | 7 (58%) | 10 (83%) | 0.178 |
| SPOD ≤ 1000 mm2 | 5 (42%) | 2 (17%) | |
| AJCC tumor stage – no. (%) | |||
| M1a or M1b | 6 (50%) | 1 (8%) | 0.025 |
| M1c | 6 (50%) | 11 (92%) | |
| Mutation – no. (%) | |||
| BRAFV600E/K | 3 (25%) | 5 (42%) | 0.3865 |
| Non-BRAFV600E/K | 9 (75%) | 7 (58%) | |
Abbreviations: LDH lactate dehydrogenase, SPOD sum of product of diameters, AJCC American Joint Committee on Cancer, ULN upper limit of normal. The p value was calculated using the Chi square test for a two by two contingency table
Comparison of assay performance
Baseline and EDT plasma samples from the responding and non-responding patients were analyzed for expression of multiple proteins using the 65-plex Discovery assay (Eve Technologies; 65 proteins detected) and the SOMAscan assay (SomaLogic; 1310 detected proteins).
The 65-plex Discovery assay has a reported dynamic range of 0.64 pg/ml to 10,000 pg/ml, comparable to other Luminex assays, and a minimal limit of detection (sensitivity) ranging from 0.1 pg/ml to 55.8 pg/ml while the inter-assay variability (coefficient of variation; CV) was between 3.5–18.9% for the 65 cytokines included in the panel [20]. Fluorescence intensity values were detected for every protein in all plasma samples in the Discovery assay, and varied from 46.26 RFU to 13,069 RFU, with a median of 184.7 RFU (Fig. 2a). However, despite a dynamic range across five orders of magnitude, absolute protein concentrations could not be calculated for 15 of the 65 cytokines in more than 75% of plasma samples, as the fluorescence values were below the standard curve (Table 3).
Fig. 2.
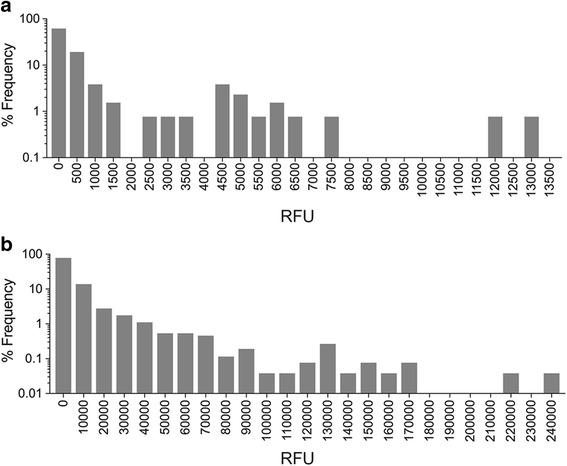
Density distribution of fluorescence intensity values. The distribution, range and frequency of relative fluorescence intensity units (RFU) of proteins detected in all 47 patient samples in the (a). Discovery assay and (b). SOMAscan assay are shown
Table 3.
Detection limits of the Discovery assay
| Cytokine | Samples within standard curvea | Cytokine | Samples within standard curvea | Cytokine | Samples within standard curvea |
|---|---|---|---|---|---|
| BCA-1 | 47/47 (100%) | IL-6 | 43/47 (91%) | MCP-3 | 20/47 (43%) |
| CTACK | 47/47 (100%) | IL-7 | 26/47 (55%) | MCP-4 | 46/47 (98%) |
| EGF | 38/47 (81%) | IL-8 | 39/47 (83%) | MDC | 47/47 (100%) |
| ENA-78 | 47/47 (100%) | IL-9 | 47/47 (100%) | MIP-1α | 34/47 (72%) |
| Eotaxin-1 | 47/47 (100%) | IL-10 | 42/47 (89%) | MIP-1β | 44/47 (94%) |
| Eotaxin-2 | 45/47 (96%) | IL-12P40 | 27/47 (57%) | MIP-1d | 47/47 (100%) |
| Eotaxin-3 | 47/47 (100%) | IL-12P70 | 41/47 (87%) | PDGF-AA | 47/47 (100%) |
| FGF-2 | 47/47 (100%) | IL-13 | 22/47 (47%) | PDGF-BB | 46/47 (98%) |
| Flt-3 Ligand | 20/47 (43%) | IL-15 | 46/47 (98%) | RANTES | 37/47 (79%) |
| Fractalkine | 42/47 (89%) | IL-16 | 47/47 (100%) | sCD40L | 47/47 (100%) |
| G-CSF | 40/47 (85%) | IL-17A | 39/47 (83%) | SCF | 20/47 (43%) |
| GM-CSF | 40/47 (85%) | IL-18 | 43/47 (91%) | SDF-1 | 46/47 (98%) |
| GRO pan | 45/47 (96%) | IL-20 | 45/47 (96%) | TARC | 47/47 (100%) |
| IFNα2 | 42/47 (89%) | IL-21 | 22/47 (47%) | TGF-a | 36/47 (77%) |
| IFNγ | 36/47 (77%) | IL-23 | 45/47 (96%) | TNFα | 47/47 (100%) |
| IL-1α | 46/47 (98%) | IL-28A | 46/47 (98%) | TNFβ | 22/47 (47%) |
| IL-1B | 35/47 (74%) | IL-33 | 29/47 (62%) | TPO | 39/47 (83%) |
| IL-1RA | 40/47 (85%) | IP-10 | 47/47 (100%) | TRAIL | 47/47 (100%) |
| IL-2 | 28/47 (60%) | I-309 | 47/47 (100%) | TSLP | 42/47 (89%) |
| IL-3 | 47/47 (100%) | LIF | 33/47 (70%) | VEGF-A | 32/47 (68%) |
| IL-4 | 19/47 (40%) | MCP-1 | 47/47 (100%) | 6CKINE | 47/47 (100%) |
| IL-5 | 36/47 (77%) | MCP-2 | 47/47 (100%) |
aPercentage of the 47 plasma samples with target protein fluorescence intensity values within the protein standard curve. Absolute cytokine concentration for 15/65 cytokines (shown in bold) could not be calculated in more than 75% of plasma samples because fluorescence intensity values were below the standard curve
Fluorescence intensity values have been shown to be more robust indicators of protein expression compared to absolute concentrations in terms of reproducibility, and for statistical differential analysis [21, 22]. Boxplot graphs showed median fluorescence intensity distributions of the plasma samples were within the range of the standard curve for most of the 65 cytokines in the Discovery assay (Standard 1–7; Additional file 1: Figure S1a and b). However, five of the 65 cytokines (Eotaxin-3, IL-21, IL-3, IL-9 and TSLP) had median fluorescence distributions that were below the standard curve range (Additional file 1: Figure S1b). Detailed analysis of these 5 cytokines showed a symmetrical distribution of RFU (Additional file 1: Figure S2), suggesting that these fluorescence values are unlikely to be background artefacts despite the low readings. As such, fluorescence intensity readings for all 65 cytokines were used in our analyses instead of derived concentrations.
The SOMAscan assay has a larger dynamic range compared to the Discovery assay, detecting protein level from fM to μM across eight orders of magnitude. The average minimal limit of detection is 1.6 pg/ml and the CV varied between 2.9–12.6% for all 1310 protein analytes [23]. Fluorescence data for all 1310 proteins were detected in all plasma samples, and ranged from 45.33 RFU to 238,857 RFU, with a median of 1254 RFU (Fig. 2b). Protein standards of known concentrations were not included for the analytes in the SOMAscan assay, thus absolute protein concentrations could not be calculated and fluorescence data were used instead.
It is important to mention that although fluorescence intensity readings reflect relative protein quantity, they are not directly comparable across different analytes in the SOMAscan assay. For example, a two fold increase in RFU values does not indicate a two fold increase in protein quantity.
Comparison of protein identification and quantification
The Discovery and SOMAscan assays have 49 proteins in common (Fig. 3a) and the fluorescence intensity values for each of these 49 proteins were median collapsed and analyzed for correlation.
Fig. 3.
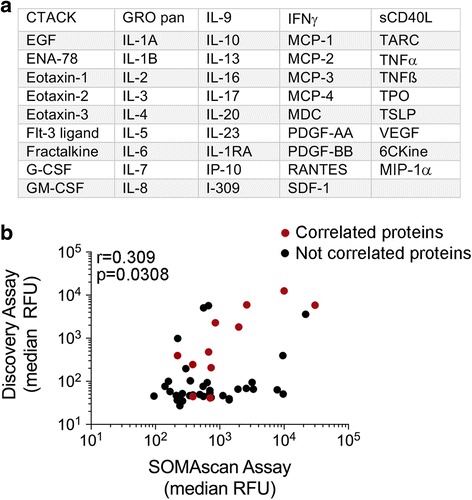
Correlation of common protein targets. a List of the 49 proteins shared by the SOMAscan and Discovery Assays. b The median relative fluorescence units (RFU) of each of the 49 proteins in all 47 patient samples were derived from the SOMAscan and Discovery assays and tested for correlation (Spearman’s rank correlation coefficient; r = 0.3165, p = 0.0267). Twelve of the 49 proteins (represented in red) showed significant positive correlation between the two assays when tested individually using Spearman’s rank correlation coefficient (results of the statistical analysis shown in Table 4)
As shown in Fig. 3b, the Discovery and SOMAscan median expression data for the 49 shared proteins in the PRE and EDT clinical samples were weakly correlated (r = 0.309, p = 0.0308, n = 47 plasma samples). However, when compared individually, only 12 of the 49 shared proteins showed significant positive correlation between these two assays (both PRE and EDT samples were compared; Table 4). The lack of correlation did not appear to reflect relative fluorescence readings as both high (CTACK and Eotaxin-1) and low level (IL-7 and I-309) proteins were not significantly correlated in these assays (Additional file 1: Figure S3). We noted that nearly all of the 37 proteins that did not correlate between the two assays showed one of two features. The proteins had low RFU in the Discovery assay (p < 0.01, Fig. 4a) and/or the proteins showed limited range of RFU in the SOMAscan assay when compared to the equivalent RFU range of the Discovery assay (Fig. 4b). For instance, 30 out of the 37 proteins (81%) that were not correlated had a median RFU of less than 100 in the Discovery assay, and in the SOMAscan assay, 27 out of the 37 proteins (73%) that were not correlated showed limited RFU distribution compared to the Discovery assay.
Table 4.
Correlation analysis of Discovery and SOMAScan assays
| Cytokine | r value | P valuea | Cytokine | r value | P valuea | Cytokine | r value | P valuea |
|---|---|---|---|---|---|---|---|---|
| CTACK | 0.0242 | 0.8719 | IL-4 | 0.1467 | 0.3252 | MCP-4 | 0.1200 | 0.4218 |
| EGF | 0.2815 | 0.0553 | IL-5 | 0.1427 | 0.3387 | MDC | 0.5461 | <0.0001 |
| ENA-78 | 0.2833 | 0.0536 | IL-6 | 0.2487 | 0.0918 | MIP-1α | −0.0028 | 0.9852 |
| Eotaxin-1 | 0.2312 | 0.1179 | IL-7 | −0.0071 | 0.9618 | PDGF-AA | 0.7951 | <0.0001 |
| Eotaxin-2 | 0.1752 | 0.2388 | IL-8 | 0.4526 | 0.0014 | PDGF-BB | 0.6449 | <0.0001 |
| Eotaxin-3 | 0.5295 | 0.0001 | IL-9 | 0.1587 | 0.2866 | RANTES | −0.0445 | 0.7664 |
| Flt-3 Ligand | 0.4585 | 0.0012 | IL-10 | −0.2023 | 0.1727 | sCD40L | 0.4882 | 0.0005 |
| Fractalkine | −0.4095 | 0.0043 | IL-13 | 0.0454 | 0.7619 | SDF-1 | 0.2765 | 0.0599 |
| G-CSF | 0.0897 | 0.5486 | IL-16 | −0.2042 | 0.1686 | TARC | 0.8669 | <0.0001 |
| GM-CSF | 0.1330 | 0.3727 | IL-17A | 0.1628 | 0.2743 | TNFα | 0.3373 | 0.0204 |
| GROpan | 0.4460 | 0.0017 | IL-20 | −0.3942 | 0.0061 | TNFβ | 0.3215 | 0.0275 |
| IFNγ | −0.0659 | 0.6595 | IL-23 | −0.1467 | 0.3252 | TPO | 0.1433 | 0.3366 |
| IL-1A | −0.0624 | 0.6767 | IP-10 | 0.5764 | <0.0001 | TSLP | −0.0460 | 0.7587 |
| IL-1B | 0.1263 | 0.3975 | I-309 | −0.1289 | 0.3877 | VEGF | −0.1383 | 0.3539 |
| IL-1RA | 0.0253 | 0.8661 | MCP-1 | 0.0614 | 0.6818 | 6CKine | 0.1104 | 0.4602 |
| IL-2 | −0.467 | 0.0009 | MCP-2 | −0.2492 | 0.0912 | |||
| IL-3 | 0.1065 | 0.4761 | MCP-3 | −0.2023 | 0.1728 |
aSpearman’s rank correlation coefficient test. Only 12 (shown in bold) of the 49 common proteins showed significant positive correlation between the Discovery and SOMAScan assays (p < 0.05)
Fig. 4.
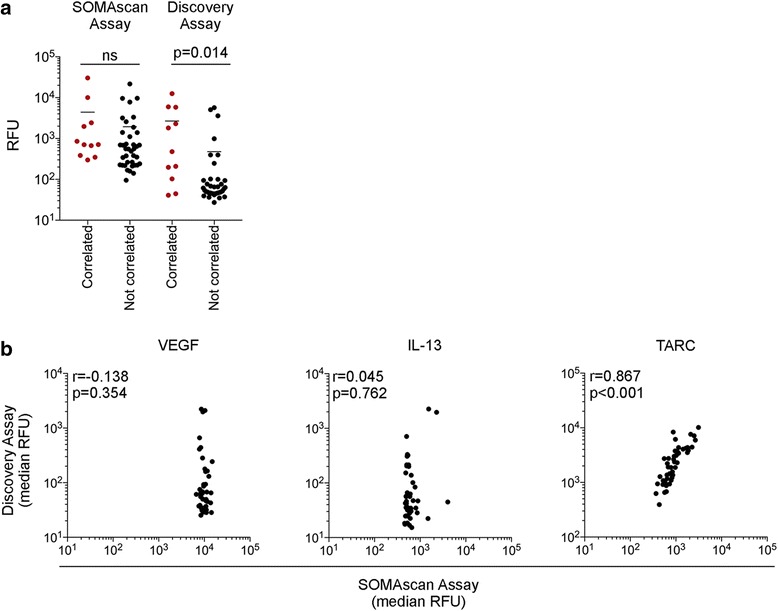
Sensitivity and range of detection of the protein assays. a Scatter plot of RFU values for the 49 shared proteins comparing correlated and non-correlated proteins in the Discovery and SOMAscan assays. The RFU values between the correlated and non-correlated proteins in each assay were compared using a Mann-Whitney test (b). Median RFU of VEGF, IL-13 and TARC from the SOMAscan and Discovery assays were tested for correlation using Spearman’s rank correlation coefficient; only TARC showed significant positive correlation (r = 0.867, p < 0.001). Correlation graphs of the non-correlated, high abundance VEGF and low abundance IL-13 showed limited range of RFU in the SOMAscan assay compared to the Discovery assay, in contrast to the correlated TARC protein
To validate the protein data, we assessed expression of four proteins (IL-1RA, IL-1A, TNFα and IL-6) that were measured in both the Discovery and SOMAscan assays, and showed variable expression and correlation data (i.e. only TNFα was highly expressed in the Discovery and SOMAscan assays, whereas IL-1RA, IL-1A and IL-6 were not correlated and showed moderate to low expression). These four circulating proteins were assessed in 28 plasma samples from a subset of the same patients (n = 15) using the MAP Human Cytokine/Chemokine Milliplex assay. Fluorescence intensity values for each of the 4 proteins were analyzed for correlation between all three assays. Only TNFα was significantly correlated between all assays while IL-1RA, IL-1A and IL-6 showed significant positive correlation between the Milliplex assay and the Discovery assay but not the SOMAscan assay (Fig. 5a).
Fig. 5.
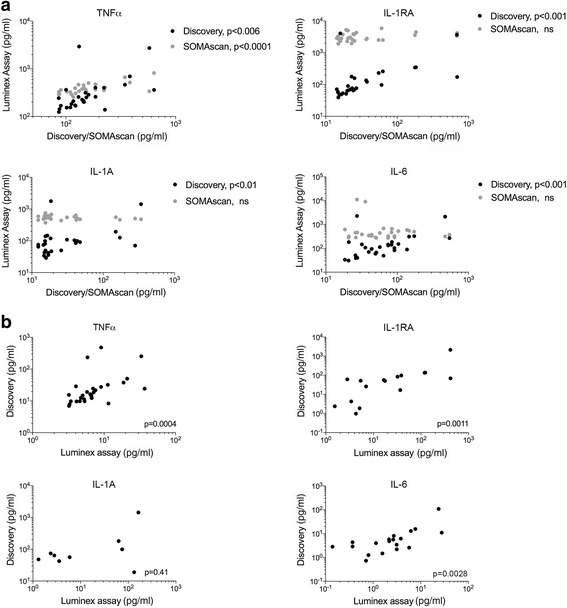
Correlation of four protein targets in bead-based and aptamer-based assays. a The relative fluorescence units (RFU) of each of the 4 proteins (IL-1A, IL-1RA, TNFα and IL-6) in 28 plasma samples were derived from the Milliplex, SOMAscan and Discovery assays and tested for correlation (Spearman’s rank correlation coefficient). b The absolute protein concentrations (pg/ml) of each of the 4 proteins (IL-1A, IL-1RA, TNFα and IL-6) in 28 plasma samples were derived from the Milliplex and Discovery assays and tested for correlation (Spearman’s rank correlation coefficient)
Because absolute protein concentrations could be derived for the four proteins from the Discovery and Milliplex assays, we additionally correlated these values and found significant positive correlation for TNFα, IL-1RA and IL-6 (Fig. 5b). Absolute concentration values of IL-1A from the Discovery and Milliplex assays were not significantly correlated, however, it is important to point out that these values could not be calculated from the Milliplex assay in more than 65% of plasma samples as their fluorescence intensities were below the standard curve.
Identification of differentially expressed proteins in immunotherapy response
We performed differential expression analyses to identify circulating proteins predictive of response to immunotherapy. The PRE and EDT plasma samples were grouped into either response (n = 12 for PRE, n = 12 for EDT samples) or non-response (n = 11 for PRE, n = 12 for EDT samples) groups and differential expression between the two groups was analyzed using LimmaGP. From the 1310 proteins in the SOMAscan assay, 178 were differentially expressed in EDT plasma samples (q < 0.25, p < 0.05), whereas no proteins were differentially expressed in the baseline samples between the two response groups. 175 out of the 178 differentially expressed proteins were expressed higher in the non-response group and PCA analysis of the differentially expressed proteins indicated clear separation of the EDT plasma samples between the two groups (Fig. 6a). In contrast, comparison of the fluorescence intensity values of the 65 cytokines from the Discovery assay did not reveal any significant differences at baseline or EDT. Of the 178 differentially expressed proteins in EDT samples from the SOMAscan assay, five were part of the Discovery assay panel. However, the five proteins (EGF, SDF-1, CTACK, IL-20 and IL-6) were not positively correlated between the two assays (Table 4).
Fig. 6.
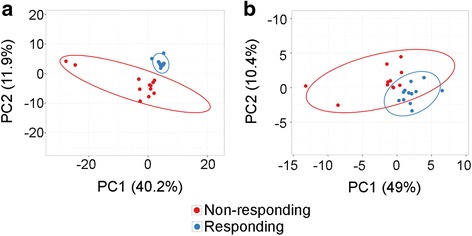
Principal Component Analysis (PCA) of differentially expressed proteins. PCA plots of the (a) 178 differentially expressed protein in EDT plasma samples and (b) 32 differentially expressed proteins altered in response to immunotherapy showed good separation between the responding (blue) and non-responding (red) patients
We also compared changes in proteins in response to therapy and assessed whether these changes predicted response. For this analysis, EDT RFU values were subtracted from the matched baseline RFU data. None of the 65 cytokines from the Discovery assay showed significant differences between the two patient groups in response to therapy. However, we found that 32 SOMAscan proteins were differentially altered on therapy in the response versus non-response groups (q < 0.25, p < 0.05). These 32 proteins were all higher (i.e. upregulated on therapy) in the non-response group and PCA analysis of the differentially regulated proteins showed some separation between the two response groups (Fig. 6b). Of the 32 SOMAscan proteins that were significantly altered in response to therapy, MIP-1α and IL-3 were included in the Discovery assay panel, but expression data of these from the two assays did not show significant correlation (Table 4).
Discussion
Despite advances in proteomic technologies and high-throughput analyte detection systems, successful identification and validation of new biomarkers for cancer diagnosis, and for predicting treatment response has been poor. A significant challenge lies in the limitations of current proteomic techniques. For example, mass spectrometry, although offering more accurate identification of proteins, is limited by interference from high abundance proteins. Other technologies such as multiplex bead-based and aptamer-based assays also have limitations imposed by the specificity and potential cross-reactivity of the capture antibodies or aptamers.
In this study, we compared two proteomic techniques widely used in biomarker discovery. We found that the Discovery and SOMAscan assays showed poor correlation in fluorescence data for 49 shared proteins, and as such, these assays did not detect any proteins in common that identified melanoma patients likely to respond to immunotherapy. The lack of correlation appears to be associated with low detection levels in the Discovery assay and/or restricted range of detection for the SOMAscan platform. To gain further insights into the variability between these assays, we re-quantitated four common cytokines using the Milliplex assay, which also utilizes the bead-based Luminex technology. As expected, all four re-tested cytokines were significantly correlated between the Milliplex and Discovery assays. In contrast, three of four re-tested cytokines (i.e. IL-1RA, IL-1α and IL-6) were not correlated between the Luminex and SomaLogic platforms, and importantly these three cytokines showed a limited range of RFU in the SOMAscan assays. These data strongly suggest that the variability between the Discovery and SOMAscan assays reflect poor specificity and sensitivity of many of the capture antibodies or aptamers.
Our analysis of the data from the Discovery assay did not yield any proteins reflective of response to immunotherapy. However, from the SOMAscan assay, 178 proteins were differentially expressed early during therapy in plasma of patients who responded to treatment compared to those who did not respond, and 32 proteins were upregulated upon treatment in patients who did not respond to immunotherapy. These differentially expressed proteins could represent potential predictive biomarkers of immunotherapy response but given the poor concordance between the Discovery and SOMAscan assays, these targets need to be further validated using separate proteomic approaches and in a larger patient cohort. The complete list and analysis of differentially expressed proteins will be reported as part of another study.
It is also worth noting that there may be more value in using the fluorescence intensity data to identify potential targets during differential expression analysis. To support this, we observed that the mean fluorescence values of the lower standards (S1 and S2, Additional file 1: Figure S1) of some cytokines (i.e. SDF-1 and MIP-1a) did not separate well. This suggests decreased sensitivity in detection of these low-level cytokines, and in turn, may contribute to errors in absolute concentration calculation for each of these cytokines as their standard curve will be skewed. However, this effect will not impact analysis using fluorescence values alone.
Conclusions
Our study highlights inadequacies in two proteomic platforms commonly used for biomarker discovery, which up till now, have not been evaluated extensively side by side. We show that each technique had specific limitations including sensitivity and specificity of the aptamers and antibodies, which may impede biomarker discovery, and this is particularly relevant as these two assays are routinely applied for this purpose. Our findings underscore the critical need for sensitive, accurate and reproducible protein detection systems, and although the application of multiple, independent detection platforms could be beneficial for discovery, this is not always possible or practical. In particular, the proteomic approaches described in this report are expensive and it is not always possible to apply multiple detection methods when analyzing limited and unique patient biopsies. Further, discordant results between assays, as shown in this study, may necessitate additional measurements of the target proteins.
Method development and validation is essential for the field of biomarker discovery. For instance, the identification of all plasma proteins bound to each SOMAmer reagent by liquid chromatography and tandem mass spectroscopy is an ongoing process that will validate the specificity of the aptamers. Indeed, this has recently resulted in the removal of five aptamer reagents due to non-specific protein enrichment from human plasma and protein preparations. Similarly, although individual detection antibodies used in bead-based assays are tested for cross-reactivity, this is limited to reactivity against a restricted panel of antigens. At present, we rely on a series of modern proteomic methods each with significant limitations that hamper the rapid and accurate identification of novel biomarkers. Investment in improving and advancing these technologies is critical to increasing the effectiveness and value of proteomic biomarker discovery.
Acknowledgements
We would like to thank the biobanking staff at the Melanoma Institute Australia, Royal Prince Alfred and Westmead Hospitals.
Funding
This work was supported by funding from NHMRC project grants #1130423 and #1093017.
SYL is supported by grant #1123911, awarded through the Priority-driven Collaborative Cancer Research Scheme and co-funded by Cancer Australia and Cure Cancer Australia Foundation. AMM is supported by a Cancer Institute NSW Fellowship. RS is supported by an NHMRC Practitioner Fellowship. GVL and HR are supported by NHMRC Research Fellowships.
Availability of data and materials
All data generated or analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- AJCC
American Joint Committee on Cancer
- CEA
Carcinoembryonic antigen
- CR
Complete response
- ctDNA
circulating tumor DNA
- CTLA-4
Cytotoxic T lymphocyte antigen 4
- EDT
Early during therapy
- IL
Interleukin
- PCA
Principle component analysis
- PD
Progressive disease
- PD-1
Programmed cell death 1
- PR
Partial response
- PRE
Baseline
- PSA
Prostate specific antigen
- RECIST
Response evaluation criteria in solid tumors
- RFU
Relative fluorescence unit
- SD
Stable disease
- TNF
Tumor necrosis factor
Additional file
Figure S1. Distribution plots of the 65 cytokines targeted by the Discovery assay. Boxplot graphs showing relative fluorescence intensity (RFU; y axis) of the plasma samples (X) for (a), 60 of the 65 cytokines that were within the range of the external standards (from known low (S1) to high (S7) concentrations) while (b). 5 of the 65 cytokines were out of the standard curve range. Blank (B) values were also included in the assay. Figure S2. Distribution plots of five cytokines detected below the standard curve range in the Discovery assay. Histogram graphs showing distribution of the relative fluorescence units (RFU) of 47 plasma samples for Eotaxin-3, IL-21, IL-3, IL-9 and TSLP. Figure S3. Correlation of high and low abundance proteins. Median RFU of highly abundant proteins CTACK and Eotaxin-1, and low abundance proteins IL-7 and I-309 from the SOMAscan and Discovery assays were plotted; each point corresponds to a different patient sample (PRE and EDT plasmas). Proteins that are high and low abundance show poor correlation between the two assays. (DOCX 1640 kb)
Authors’ contributions
SYL and HR analyzed and interpreted the protein and clinical data and wrote the manuscript. JHL, MSC, AMM, RFK, RAS and GVL provided the clinical samples and data. SBA, EB and AG acquired and processed protein data. All authors read, revised and approved the final manuscript.
Ethics approval and consent to participate
Informed consent was obtained from all patients under approved human research ethics committee protocols from the Royal Prince Alfred Hospital.
Consent for publication
Not applicable.
Competing interests
MSC has been on advisory boards for MSD, Novartis, BMS, Amgen and Pierre Fabre. AMM has been on advisory boards for MSD, Novartis, Pierre Fabre and Chugai and has received honoraria from BMS and Roche. RFK has been on advisory boards for Roche, Amgen, BMS, Merck, Novartis and TEVA and has received honoraria from Merck, BMS and Novartis. GVL has been on advisory boards for Array, Amgen, BMS, Novartis, Merck and Pierre Fabre and has received honoraria from Roche and Merck. The remaining authors declare no conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40364-017-0112-9) contains supplementary material, which is available to authorized users.
Contributor Information
Su Yin Lim, Email: esther.lim@mq.edu.au.
Jenny H. Lee, Email: jenny.lee@mq.edu.au
Sarah J. Welsh, Email: sarah7welsh@gmail.com
Seong Beom Ahn, Email: Charlie.ahn@mq.edu.au.
Edmond Breen, Email: ebreen@proteome.org.au.
Alamgir Khan, Email: alamgir.khan@me.edu.au.
Matteo S. Carlino, Email: matteo.carlino@sydney.edu.au
Alexander M. Menzies, Email: alexander.menzies@sydney.edu.au
Richard F. Kefford, Email: Richard.kefford@mq.edu.au
Richard A. Scolyer, Email: richard.scolyer@health.nsw.gov.au
Georgina V. Long, Email: georgina.long@melanoma.org.au
Helen Rizos, Phone: +61 (02) 9850 2762, Email: Helen.rizos@mq.edu.au.
References
- 1.Holderfield M, Deuker MM, McCormick F, McMahon M, Targeting RAF. Kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–467. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol. 2014;32:2248–2254. doi: 10.1200/JCO.2013.52.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 5.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–1304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 7.Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol. 2014;8:1132–1139. doi: 10.1016/j.molonc.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270–1271. doi: 10.1056/NEJMoa1504030. [DOI] [PubMed] [Google Scholar]
- 9.Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, Linette GP, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558–1568. doi: 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez J, Thompson IM. Prostate-specific antigen: a review of the validation of the most commonly used cancer biomarker. Cancer. 2004;101:894–904. doi: 10.1002/cncr.20480. [DOI] [PubMed] [Google Scholar]
- 11.Grunnet M, Sorensen JB. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer. 2012;76:138–143. doi: 10.1016/j.lungcan.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Alix-Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479–491. doi: 10.1158/2159-8290.CD-15-1483. [DOI] [PubMed] [Google Scholar]
- 13.Lee JH, Long GV, Boyd S, Lo S, Menzies AM, Tembe V, Guminski A, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol. 2017; [DOI] [PubMed]
- 14.Bast RC, Jr., Badgwell D, Lu Z, Marquez R, Rosen D, Liu J, Baggerly KA, et al. New tumor markers: CA125 and beyond. Int J Gynecol Cancer 2005;15 Suppl 3:274–281. [DOI] [PubMed]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan A. Detection and quantitation of forty eight cytokines, chemokines, growth factors and nine acute phase proteins in healthy human plasma, saliva and urine. J Proteome. 2012;75:4802–4819. doi: 10.1016/j.jprot.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 18.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 19.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015;43:W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Discovery Assay - Eve Technologies. Eve Technologies Corporation; 2017.
- 21.Breen EJ, Polaskova V, Khan A. Bead-based multiplex immuno-assays for cytokines, chemokines, growth factors and other analytes: median fluorescence intensities versus their derived absolute concentration values for statistical analysis. Cytokine. 2015;71:188–198. doi: 10.1016/j.cyto.2014.10.030. [DOI] [PubMed] [Google Scholar]
- 22.Breen EJ, Tan W, Khan A. The statistical value of raw fluorescence signal in Luminex xMAP based multiplex immunoassays. Sci Rep. 2016;6:26996. doi: 10.1038/srep26996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.SOMAscan Assay: An unprecedented protein biomarker discovery and development tool. SOMALOGIC, INC.; 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are available from the corresponding author on reasonable request.