

Abstract
Purpose of review
Visceral venous congestion of the gut may play a key role in the pathogenesis of right-sided heart failure (HF) and cardiorenal syndromes. Here we review the role of right ventricular (RV) dysfunction, visceral congestion, splanchnic hemodynamics, and the intestinal microenvironment in the setting of right-sided HF. We review recent literature on this topic, outline possible mechanisms of disease pathogenesis, and discuss potential therapeutics.
Recent Findings
There are several mechanisms linking RV-gut interactions via visceral venous congestion which could result in (1) hypoxia and acidosis in enterocytes, which may lead to enhanced sodium-hydrogen exchanger 3 (NHE3) expression with increased sodium and fluid retention; (2) decreased luminal pH in the intestines which could lead to alteration of the gut microbiome which could increase gut permeability and inflammation; (3) alteration of renal hemodynamics with triggering of the cardiorenal syndrome; and (4) altered phosphate metabolism resulting in increased pulmonary artery stiffening, thereby increasing RV afterload. A wide variety of therapeutic interventions that act on the RV, pulmonary vasculature, intestinal microenvironment, and the kidney could alter these pathways and should be tested in patients with right-sided HF.
Summary
The RV-gut axis is an important aspect of HF pathogenesis that deserves more attention. Modulation of the pathways interconnecting the right heart, visceral congestion, and the intestinal microenvironment could be a novel avenue of intervention for right-sided HF.
Keywords: heart failure, right ventricle, venous congestion, intestine, sodium-hydrogen exchanger 3, microbiome
Introduction
Right-sided heart failure (RHF) is a major public health problem that affects between one-third to one-half of all heart failure (HF) patients regardless of underlying left ventricular (LV) ejection fraction [1, 2]. Evidence of right ventricular (RV) dysfunction or signs of RHF (i.e., venous congestion) portend a poor prognosis when present in patients with HF, with multiple studies demonstrating worse outcomes in HF patients who have evidence of abnormalities of RV structure/function, pulmonary hypertension (PH), and/or venous congestion [3–5]. Thus, the right heart plays a critical role in the pathogenesis of HF. Despite this fact, our understanding of the effects of RHF on HF pathogenesis and the effect of venous congestion on systemic organs is still in its infancy. Furthermore, unlike the left ventricle (LV), there are few available therapies that specifically target the RV, right-sided HF, or its downstream consequences (i.e., venous congestion).
One specific organ system that may play an essential role in the pathogenesis of RHF is the gut. Visceral venous congestion of the intestines may be central to the pathogenesis of right-sided HF and cardiorenal syndromes, via the interaction between the right heart, the splanchnic venous circulation, the liver/kidney, and the intestinal microenvironment. Here we review the role of RV dysfunction, visceral congestion, splanchnic hemodynamics, and the intestinal microenvironment in the setting of RHF. We review recent literature on this topic, and discuss possible mechanisms of disease pathogenesis which could elucidate novel therapeutic targets for RHF.
The clinical importance of venous congestion
Elevated jugular venous pressure is one of the hallmarks of HF, but it is not universally present in all HF patients. The prognostic value of this physical exam finding was tested in a study by Drazner et al., who performed a retrospective analysis of the Studies of Left Ventricular Dysfunction (SOLVD) treatment trial [4]. SOLVD enrolled 2569 patients with HF and reduced ejection fraction (HFrEF) who underwent comprehensive physical examination at the time of enrollment, and these patients were followed for HF hospitalization, HF-related death, or a composite end-point of death or hospitalization. The investigators found that elevated jugular venous pressure was associated with increased the risk of HF hospitalization (relative risk [RR], 1.32; 95% confidence interval [CI], 1.08–1.62; P<0.01), death or HF hospitalization (RR, 1.30; 95% CI, 1.11–1.53; P<0.005), and death from pump failure (RR, 1.37; 95% CI, 1.07–1.75; P<0.05) [4]. This study underscores the importance of venous congestion in the prognosis—and possible progression—of HF.
Historically speaking, worsening renal function (WRF) in hospitalized patients has been attributed to over-diuresis and/or poor perfusion due to reduced cardiac output. However, a study by Mullens et al. challenged this notion. These investigators prospectively enrolled 145 patients with acute HF admitted to a dedicated intensive care unit for HF patients (mean LV ejection fraction = 20±8%), where they underwent pulmonary artery (PA) catheter-guided therapy [6]. WRF was defined as an increase of serum creatinine ≥ 0.3 mg/dL during the hospitalization, and 58/145 (40%) of the study participants met this criteria. Invasive hemodynamic data were obtained from an experienced HF cardiologist upon insertion of the PA catheter, and again prior to its removal once therapy goals were achieved. Comparing those who developed WRF vs. those who did not, the prevalence of moderate-to-severe RV dysfunction was similar between those with vs. without WRF (60% in both groups; p = NS), as was B-type natriuretic peptide (median [interquartile range]: 1,100 [497, 1,921] pg/ml vs. 874 [333, 1,430] pg/ml; p = NS). Statistically significant correlations between baseline cardiac index and baseline creatinine (r = 0.32, p = 0.001) and glomerular filtration rate (r = –0.3, p = 0.002) were observed, but no correlation between baseline cardiac index and central venous pressure (CVP) was observed. Compared to those who did not develop WRF, participants who developed WRF were no different in terms of heart rate, systolic blood pressure, PA systolic pressure, pulmonary capillary wedge pressure, or cardiac index (p>0.05 for all comparisons). Only elevated CVP at baseline (p < 0.001), and follow-up (p < 0.04), was observed to be predictive of WRF. This study yielded several interesting conclusions: (1) worse baseline renal function was associated with a higher, not lower, cardiac index; (2) there is incremental risk of WRF with increasing CVP; (3) renal perfusion pressure (mean arterial pressure – CVP) was similar between patients with and without WRF; (4) higher CVP at the time of hospital admission was predictive of incident WRF during the hospitalization. This study suggests there may be mechanisms of WRF directly related to venous congestion. Thus, venous congestion may not simply be a physical exam finding that denotes a high risk patient; rather it may be integral in the pathogenesis of WRF and HF. Furthermore, these findings suggest that increased CVP may prove to be a pathogenic cause of other organ dysfunction, such as the gastrointestinal tract.
The relationship between RV dysfunction and the gastrointestinal system has been examined previously. A study by Valentova et al. observed that RV dysfunction, not LV structural abnormalities or reduced LV ejection fraction, was correlated with increased liver enzymes and cardiac cachexia in HF patients [7]. Furthermore, these investigators found impaired RV function to be strongly associated with cardiac cachexia (area under the receiver operating characteristic curve = 0.89; 95% CI [0.83–0.94]), characterized by gastrointestinal discomfort, postprandial fullness, and pro-inflammatory activation [8]. Sandek et al. performed a comprehensive analysis in patients with chronic HF compared to controls, measured intestinal permeability, ischemia, absorption, and bacterial growth [9]. The findings were striking. The investigators observed significant differences in bowel wall thickness, altered absorption, increased permeability, increased bowel wall ischemia, and increased mucus-adherent bacterial growth in patients with chronic HF compared to controls [9]. Together, these findings suggest that RHF-induced hepatic and gastrointestinal dysfunction may be an important source of chronic inflammation in HF patients, as the gut becomes more permeable and susceptible to bacterial translocation is the setting of RHF [10]. The clinical implications of such findings may be considerable for patients with HF and RV dysfunction.
Normal and abnormal gut physiology: Implications for patients with right-sided heart failure
NHE3 and sodium balance
The gastrointestinal tract, once thought of as an innocent bystander, is garnering increasing attention for its role in cardiovascular disease [11–23]. The intestines hold tremendous surface area for absorption of nutrients and fluids. The gastrointestinal system secretes and reabsorbs several liters of fluid on a daily basis. Disruptions in these absorptive mechanisms are well observed in diarrheal illness, a testament of the incredible fluctuations in fluid balance possible with slight perturbations in this tightly controlled system. This fluid balance is maintained by several families of ion channels, one of which is the sodium-hydrogen exchanger 3 (NHE3). Several isoforms exist, but NHE3 appears to be most important in Na+ and H+ regulation for the epithelial cells [24–27]. Activity of NHE3 is the primary mechanism of sodium balance in the intestines, and is therefore closely regulated. Several different signaling pathways up-regulate NHE3 function, including aldosterone, glucocorticoids, and increased concentrations of intracellular H+ [27, 28, 24, 29, 25]. Upregulation of NHE3 leads to extrusion of H+ from the gastrointestinal epithelial cells (enterocytes) into the gut lumen with concomitant absorption of Na+. This electrolyte exchange appears to have at least 2 direct consequences: (1) increased Na+ absorption from the gastrointestinal lumen conceivably results in an increase in fluid absorption, delivering the cardiovascular system an acute salt and water load; (2) extrusion of H+ from the enterocytes into the gut lumen may cause a local alteration in the pH and environmental conditions, thereby affecting the gut microbiome and altering the micro-organismal landscape within the gut lumen. NHE3 has also been observed to be closely related to phosphate and calcium absorption [30], which could have further implications in the pathogenesis of cardiorenal syndrome, RHF, and PH, as described below.
Short chain fatty acids in barrier function
Short-chain fatty acids (SCFA) are small molecules supplied to the body from 2 major sources: (1) a healthy diet high in fiber and leafy green vegetables; and (2) symbiotic bacteria in the gut. Several studies have shown bacteria in the gut lumen produce signaling molecules which exert their effects on the gut epithelia. An example of such is butyrate, an SFCA secreted by gut flora and subsequently absorbed by the intestinal epithelial cells. Butyrate is believed to act on the mitochondria to alter cell metabolism in a slight way to keep the concentration of oxygen in the epithelial cell in a range which is below normal for other cells [31]. This slightly hypoxic environment stabilizes hypoxia-inducible factor (HIF), a polypeptide signaling molecule which is necessary for the production and maintenance of several epithelial cell barrier proteins [31]. Loss of butyrate production in the gut lumen has been associated with reduced intestinal barrier function, and improvement in available butyrate has been the target of some therapies for inflammatory bowel disease [31]. In patients with RHF, changes in the gut microbiome due to increased acidity in the gut lumen (due to increased NHE3 activity) or poor diet may result in the decreased production of butyrate, which may increase gut permeability and lead to increased low-grade endotoxinemia and inflammation.
Short chain fatty acids and arteriolar vasodilation
SFCAs also play a role in several downstream hemodynamic regulatory functions. Pluznick and colleagues identified 2 novel receptors (G protein-coupled receptor 41 [Glfr41] and olfactory receptor 78 [Olfr78]) that are linked to SCFAs and alter blood pressure homeostasis [32]. These investigators found that these receptors, regulated by SCFAs, play important roles in arteriolar vasoconstriction and influence the regulatory pathways of the renin-angiotensin-aldosterone system [32, 33]. They also found SCFA-inducible response to Glfr41 in the juxtaglomerular apparatus, which is partially responsible for renin activation [33, 32]. Glfr41 and Olfr78 are also found on vascular smooth muscle cells, and stimulation by SCFA induces smooth muscle relaxation [33, 34].
Trimethylamine N-oxide (TMAO)
TMAO is another small gut-derived biomolecule with known cardiovascular effects. Several studies have found that elevated serum TMAO is associated with increased incidence of myocardial infarction, HF, and cardiovascular mortality [35–40]. The mechanisms underlying these adverse associations between TMAO and cardiovascular have been partially described. For example, TMAO functions as a pro-thrombotic molecule [41, 42]. In one study, Organ et al. examined pressure-overloaded mice (created by transverse aortic constriction) and compared a control, choline-, or TMAO-supplemented diet for 12 weeks. These investigators found that the mice fed TMAO or choline had significantly worse pulmonary edema, lower LV ejection fraction, and higher cardiac mass and cardiac fibrosis when compared to control diet [35]. These findings suggest TMAO or choline-supplemented diets enhanced HF susceptibility and may accelerate HF progression.
Bacterial invasion
The human gastrointestinal tract is a key element of the body’s host defenses. There are multiple layers of protection from potentially pathogenic bacteria, protozoa, and viral infectious agents which pass through or reside in the gut lumen. In advanced cirrhosis, portal venous pressures are increased due to an outlet obstruction imposed by a cirrhotic liver. In these patients, studies have demonstrated improved hemodynamics and reduced progression to cardiorenal syndrome with the administration of rifaximin, an oral, non-absorbable antibiotic [43, 44]. The mechanism of renal protection in this setting is believed to be from the eradication of pathologic microorganisms in the gut lumen, with reduced endotoxinemia and improved splanchnic hemodynamics [45]. Supporting this hypothesis are studies that have found increased serum endotoxin concentrations have been observed in patients with chronic HF [46], though the clinical implications of these findings require further investigation. It is believed that in patients with reduced gut-barrier function, RHF-induced liver and venous congestion cause poor clearance of toxic bacterial products and activation of the inflammatory cascade [10, 47, 48].
Visceral congestion, the gut, and the pathogenesis of heart failure
The detrimental effects of visceral congestion in HF are poorly understood, though several studies have linked RHF to the gastrointestinal system and associated adverse outcomes such as cachexia [49–51, 9, 52, 8, 7, 53] (Table 1). Here we propose several potential mechanisms through which visceral congestion may worsen the condition of the HF patient (summarized in Figure 1). Elevated CVP reduces the arterial-venous pressure gradient across the intestinal capillary network. The microstructure of the intestinal villus forms a plexus, an ideal structure to optimize nutrient absorption, but also susceptible to shunting of oxygenated blood through the base of the villus, putting the villus tip at risk of a relative ischemia. This relative ischemia creates a unique microenvironment for enterocytes in RHF patients. First, the anaerobic conditions imposed by visceral congestion may produce an intracellular and regional acidosis. Intracellular acidosis is a known activator of NHE3, which, as described above, is a major source of Na+ absorption in the gut. Increased absorption of Na+ would be counter-productive in the setting of RHF, increasing volume overload, thereby adding further stress on the cardiovascular system, and worsening venous congestion.
Table 1.
Studies linking right-sided heart failure to the gastrointestinal tract and associated adverse outcomes
| Author | Year | N | HF patient population | Main findings |
|---|---|---|---|---|
| Mullens et al. | 2008 | 40 | Patients admitted to cardiac intensive care unit for acute HF medical management; mean LVEF = 19±9% | ⇑ Intra-abdominal pressure was common, and most closely associated with worsening renal function. |
| Nikolaou et al. | 2013 | 1134 | Acute HF patients from SURVIVE cohort; LVEF <30% requiring inotropes | ⇑ Alkaline phosphatase and hepatic transaminases were associated with increased short-term mortality. |
| Pasini et al. | 2016 | 60 HF patients, 20 controls | 30 NYHA class I–II, mean LVEF 39±1.2%; 30 NYHA class III-IV, mean LVEF 35±1.4% | ⇑ Intestinal overgrowth of pathogenic bacteria + candida associated with disease severity, venous congestion, and inflammation. |
| Poelzl et al | 2013 | 1290, 253 with hemodynamic measurements | Ambulatory HF patients, mean LVEF 29%, NYHA class I (25%), II (47%), III/IV (27%) | Impaired renal and hepatic function is associated with poor prognosis. Venous congestion, not reduced cardiac output is associated with hepatic and renal congestion. |
| Sandek et al. | 2014 | 65 HF patients, 25 controls | LVEF ≤ 40%; mean NYHA 2.5±0.5 | ⇓ Intestinal blood flow in patients with cachexia. |
| Sandek et al. | 2007 | 22 HF patients, 22 controls | LVEF ≤ 40% | ⇑ Bowel wall thickness, ⇑ gut permeability, ⇑ bowel wall ischemia, ⇑ adherent gut bacteria. |
| Valentova et al. | 2016 | 165 | Outpatients, LVEF ≤ 40% | ⇑ RAP is best predictor of cardiac cachexia. |
| Valentova et al. | 2013 | 118 | LVEF ≤ 40%, NYHA class II, III & III with cachexia | ⇑ RV function is associated with cachexia. |
Figure 1. Conceptual diagram: Right-sided heart failure, splanchnic hemodynamics, and the gut microenvironment in relation to the cardiorenal syndrome, pulmonary hypertension, and adverse outcomes.
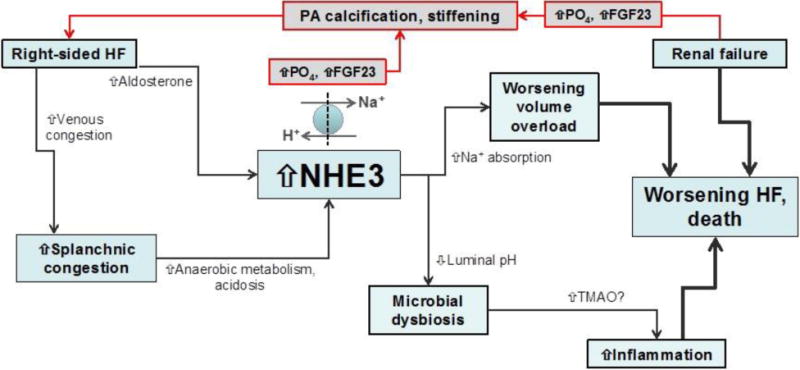
It is well known that right-sided heart failure is associated with the cardiorenal syndrome, worsening renal failure, worsening heart failure, and death. However, the mechanisms by which these processes are inter-related are less clear. Here we propose a conceptual overview of potential pathophysiologic mechanisms underlying the aforementioned associations. Right-sided heart failure results in venous congestion, which leads to splanchnic congestion. Venous congestion of the intestines results in reduced blood flow to the gut enterocytes thereby resulting in hypoxia within these cells. Anaerobic metabolism ensues, with build-up of lactate and acidosis. In an effort to extrude hydrogen ions into the gut lumen, the NHE3 channel gets upregulated and this results in increased sodium absorption, which exacerbates the fluid overload. Right-sided heart failure may also directly result in increased aldosterone secretion, which also stimulates NHE3. Meanwhile, the increased hydrogen ions in the gut lumen result in decreased pH in the gut lumen; this may result in microbial dysbiosis, which is known to increase TMAO and may trigger inflammation which is involved in cardiac cachexia and poor outcomes in heart failure. Renal failure results in increased retention of phosphates and increased production of the phosphoturic hormone FGF23. Both of these factors may be playing a role in the promotion of PA stiffening which is associated with increased PA pressure and increased load on the right ventricle, thereby worsening right-sided heart failure.
HF = heart failure; NHE3 = sodium-hydrogen exchanger 3; Na+ = sodium; H+ = hydrogen; TMAO = trimethylene N-oxide; PA = pulmonary artery; PO4 = phosphate; FGF23 = fibroblast growth factor 23.
Second, acidotic conditions within the enterocytes are extended to the intestinal brush border through normal mechanisms of homeostasis and shifting the H+ load to the gut lumen. The reduced pH in the gut may alter the bacterial flora of the brush border in a way that is deleterious to SCFA-producing bacteria, thereby promoting the growth of species which secrete TMAO. The shift in these novel regulatory biomolecules may prove deleterious to the decompensated RHF patient. TMAO has been shown to be associated with higher mortality in HF patients [38], and has been shown to be associated with a pro-atherosclerotic state [37]. Moreover, SCFAs have recently been demonstrated to function as vasodilators and reduce peripheral vascular resistance [34, 32]. Reduction in serum SCFA is associated with worsening HF [19]. Identifying how HF patients become predisposed to these adverse biochemical profiles may lead to the development of therapies that could slow the progression of HF and renal dysfunction.
Third, both ischemia and relative depletion of SCFAs can destabilize HIF, the aforementioned enterocyte regulatory protein that promotes intact barrier function of the gut. Loss of the gut-blood barrier allows gut-derived bacterial endotoxins to enter the blood, triggering a pro-inflammatory state, which has been observed in acute HF patients [54]. Indeed, endotoxinemia and sub-clinical septic hemodynamic vasodilation has been observed in acute decompensated HF. This period of vasodilation may actually improve the patient in the short term; however, once the gut recovers and the vasodilatory proteins clear, a rebound vasoconstriction can occur, increasing late-systolic aortic pulse-wave reflections, which contributes to additional cardiac compromise.
Furthermore, some studies have found increased levels of bacterial endotoxins within the blood of patients in acutely decompensated HF, and may function as a causal factor for renal dysfunction in these patients. Finally, some studies have shown that lipopolysaccharide can directly affect proteins in epithelial cells, including NHE3, and NHE8, a basolateral exchange protein involved in pH and Na+ balance [55].
Cardiorenal syndrome, phosphate balance, and pulmonary hypertension
PH is common in patients with left-sided HF due to passive elevation of pulmonary venous pressure [56]. However, in some patients with left-sided HF, superimposed pulmonary vascular remodeling occurs, and has been extensively reviewed elsewhere [57, 56, 58]. The cause of elevated pulmonary vascular resistance and/or diastolic pulmonary gradient (elevated PA diastolic pressure – pulmonary capillary wedge pressure gradient) in left-sided HF is likely multifactorial. However, one potential contributor may be chronic kidney disease (CKD). In a detailed study of 299 patients with HF and preserved ejection fraction (HFpEF), CKD was associated with higher PA systolic pressure and worse intrinsic RV systolic function (lower absolute RV free wall strain, a sensitive indicator of RV function) [59].
CKD could result in increased PA pressures due to volume overload. Elderly patients with left-sided HF often have stiff systemic vasculature, and a similar process is likely present in the central pulmonary vasculature. Volume loading of the stiff pulmonary arteries in the setting of CKD could itself raise PA pressure. In addition, elevated pulmonary venous pressure in patients with CKD (due to volume overload) could raise pressure in the central pulmonary arteries via increased reflected waves with pressure augmentation of the PA systolic pressure [60]. However, CKD may also increase PA stiffening due to direct actions of dysregulated mineral metabolism. CKD is associated with secondary hyperparathyroidism with increased phosphate and FGF23 levels [61, 62]. In a canine nephrectomy model, CKD was associated with increased PA pressure and pulmonary vascular calcification in the setting of secondary hyperparathyroidism [63]. These adverse effects of CKD on the pulmonary vasculature were abrogated by prophylactic parathyroidectomy, suggesting a potential role of mineral metabolism in the cardiorenal-pulmonary vascular-RHF interaction.
The gut plays a major role in mineral metabolism based on the absorption of phosphate. Furthermore, there may be interactions between NHE3 and phosphate absorption based on the observation that tenapanor, a gut NHE3 inhibitor also inhibits phosphate absorption [64]. Thus, the gut-kidney-pulmonary-RV interaction could possibly also involve deranged mineral metabolism.
Potential therapeutic interventions
In addition to standard of care decongestion with loop diuretics, further discovery of gut-related pathological mechanisms in the setting of RHF opens avenues for novel therapeuticss. Potential interventions for RHF-induced visceral congestion, gut dysfunction, and cardiorenal syndrome include treatments in the following broad categories: (1) direct improvement of RV function; (2) reduction of RV afterload (pulmonary vasodilation) and improvement of RV-PA coupling; (3) alteration of visceral hemodynamics; (4) NHE3 inhibition; (5) enhancement of gut barrier function; and (6) improvement of mineral metabolism with reduction of PO4 and FGF23. Below we briefly describe potential therapies in each of these categories (Figure 2).
Figure 2. Potential therapies for the maladaptive right heart-gut interaction in heart failure.
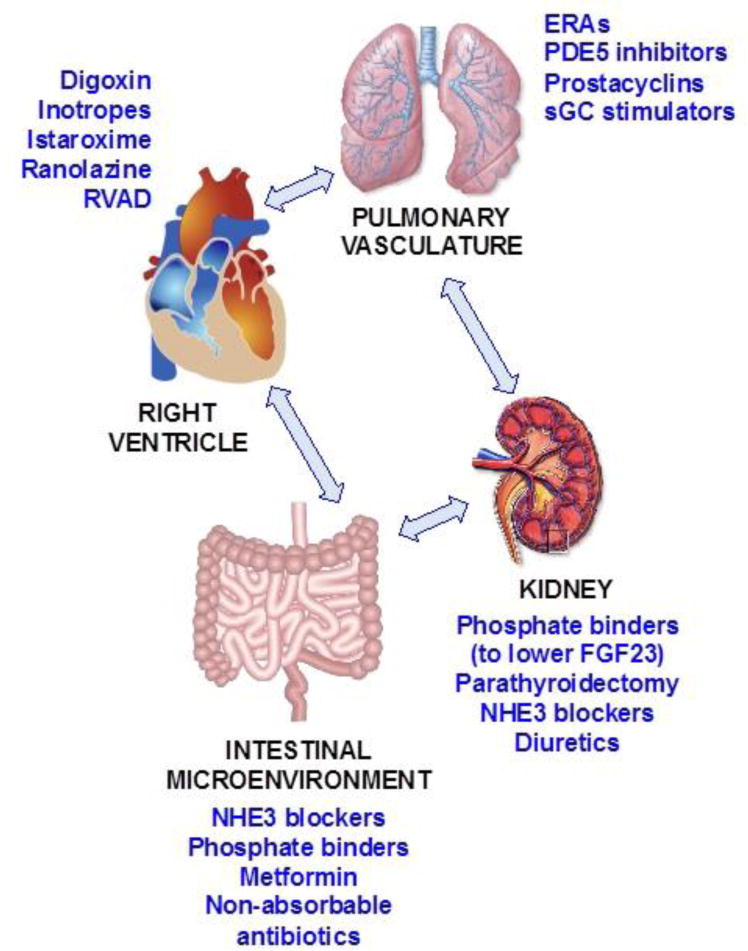
There are several potential therapies that could ameliorate the pathophysiological changes relevant to the right heart-gut axis in heart failure. Shown here are potential therapies that could improve the right ventricle, pulmonary vasculature, intestinal microenvironment, and the kidney. These treatments are theoretical for the most part, and still need to be thoroughly examined and proven effective.
RVAD = right ventricular assist device; ERAs = endothelin receptor antagonists; PDE5 = phosphodiesterase-5; sGC = soluble guanylate cyclase; FGF23 = fibroblast growth factor-23; NHE3 = sodium-hydrogen exchanger 3.
Direct improvement of RV function
Several potential methods for augmentation of RV function are available, though there has been limited testing of these therapies in patients with RHF. Traditional inotropes such as dobutamine, milrinone, and dobutamine can augment RV contractile function at the expense of increasing myocardial oxygen demand. Direct myosin activators, such as omecamtiv mecarbil, directly increase LV contractility without increasing oxygen demand [65]; however, they have not been studied systematically in patients with RV failure. Ranolazine, a late inward Na+ current inhibitor, may improve RV diastolic function by decreasing the amount of Na+ in RV cardiomyocytes, thereby decreasing calcium overload in these cells [66]. Theoretically, a reduction in cardiomyocyte calcium concentration in diastole would decrease diastolic tension in these cells thereby improving diastolic microvascular coronary blood flow with improved oxygen delivery to the failing RV. In a small, open-label pilot study, ranolazine was associated with improve RV contractile function during exercise in patients with pulmonary arterial hypertension and evidence of RV failure [66]. Istaroxime, a novel combined Na/K-ATPase and SERCA2a agonist [67] could theoretically increase RV contractility and relaxation, but has not been specifically examined in the setting of RHF. Partial and full support RV assist devices, including the RV Impella, TandemHeart, and continuous flow ventricular assist devices, could provide short- and long-term unloading of the RV [68, 69], but these therapeutic modalities have been understudied in the setting of RHF.
Reduction of RV afterload and improvement of RV-pulmonary artery coupling
Theoretically, pulmonary vasodilators could decrease RV afterload and improve RV-PA coupling in patients with RHF and high pulmonary vascular resistance. However, these drugs have not fared well in patients with left-sided heart failure with multiple failed trials of prostacyclins, endothelin receptor antagonists, phosphodiesterase-5 (PDE5) inhibitors, and soluble guanylate cyclase (sGC) stimulators [56]. However, it is possible that trials specifically in left-sided HF patients with significant RHF would benefit from these drugs, as seen in small studies of PDE5 inhibitors [70, 71] and sGC stimulators [72].
There are 2 multi-center, randomized controlled trials that are underway to examine pulmonary vasodilators in patients with HFpEF and evidence of superimposed pulmonary vascular disease and/or RHF. These include the SERENADE trial (macitentan) and SOUTHPAW trial (oral treprostinil). The inclusion/exclusion criteria for both trials are designed to specifically target patients with RHF. Furthermore, both studies are designed with safety in mind given the theoretical possibility that pulmonary vasodilation could result in pulmonary edema in these patients. In the SERENADE trial, a 1-month observation/stabilization period, followed by a 1-month open-label treatment run-in period with macitentan will be required in all patients to ensure clinical stability and lack of sodium/fluid retention prior to randomization to macitentan vs. placebo. In the SOUTHPAW trial, low starting doses and capped maximum dose titration will be required in the initial patients enrolled in the trial, with subsequent gradual escalation of maximum allowable doses. This strategy will allow careful safety monitoring and will prevent rapid titration to high doses of oral treprostinil prior to determining whether low doses are safe in patients with left-sided HF and pulmonary vascular disease (i.e., combined post- and pre-capillary PH).
NHE3 inhibition
Tenapanor is a non-absorbable oral NHE3 inhibitor recently FDA-approved for the treatment of hyperphosphatemia in patients with late-stage CKD. In studies of tenapanor compared with placebo, humans treated with tenapanor had greater stool sodium concentrations, and lower urinary sodium excretion, suggesting reduced sodium absorption by the intestines [73, 74]. In a CKD rat model (nephrectomy plus high-salt diet), which exhibits volume overload, cardiac hypertrophy, and arterial stiffening, treatment with tenapanor decreased extracellular fluid volume, LV hypertrophy, albuminuria, and blood pressure in a dose-dependent manner. [74]. It has been proposed that sodium-glucose co-transporter (SGLT) inhibitors may antagonize NHE3 in the gut and the kidney [75, 76]. These drugs, particularly combined SGLT-1/2 inhibitors, may be a novel method to improve RHF given their actions on NHE3 in the gut and kidney.
Enhancement of gut barrier function
As described above, rifaximin, a non-absorbable antibiotic, has been studied previously and found to improve splanchnic hemodynamics in cirrhosis via alteration of the gut microbiome, which could help enhance gut barrier function [44, 45]. Metformin may also improve gut barrier function through tightening of gut epithelial cell junctions via the AMP-activated protein kinase (AMPK) pathway [77]. Compared to other anti-diabetic drugs, metformin is associated with decreased risk for sepsis, which supports this potential therapeutic effect of metformin [78]. However, the effects of metformin in patients with RHF remains to be determined.
Improvement of mineral metabolism with reduction of PO4 and FGF23
Given the potential for dysregulated mineral metabolism as an underlying cause of PH in patients with left-sided HF, reduction in phosphate via the use of phosphate binders or parathyroidectomy (for secondary hyperparathyroidism in patients with cardiorenal syndrome) may reduce PA calcification and stiffening either directly or via lowering of FGF23. As described above, in a canine nephrectomy model of secondary hyperparathyroidism, parathyroidectomy rescued the animals from the development of PH [63]. Whether these therapies could help improve pulmonary vascular function and secondarily result in RV unloading in humans remains to be seen.
Future directions
Despite the clear role of RV dysfunction and RHF in the pathogenesis and progression of HF, the RV, visceral congestion, and RHF in general remain underappreciated therapeutic targets. Both acute and chronic HF studies should focus on visceral congestion, particularly in advanced HF patients, and especially in patients with HFpEF, for which there are very limited treatment options. Furthermore, translational studies of RHF and visceral congestion are needed. Better animal models of RHF and detailed mechanistic human studies of RHF and visceral congestion are additional unmet needs. Instead of continually testing LV-targeted therapies in HF, we advocate for improved targeting of the RV and its downstream consequences (e.g., visceral congestion) in clinical trials. As mentioned above, there are numerous potential treatments that could be studied in patients with RHF. Given the poor prognosis of patients with RHF, finding adequate treatments for these patients is essential.
Conclusions
RV dysfunction in left-sided HF is a common problem that is often unrecognized. Detrimental effects of increased visceral congestion as a result of RHF and its relation to the gut is likely multifactorial and involves several potential mechanisms: (1) visceral congestion may affect the sodium and phosphate transport metabolism of the gut; (2) increased venous pressure imposes relative ischemia on the intestinal microvillus, leaving enterocytes at the villus tip susceptible to ischemic injury, acidosis, and modulation of the sodium transporter NHE3; (3) relative ischemic conditions in the gut may cause reduced barrier function and translocation of potentially pathogenic microorganisms and bacterial endotoxin into the bloodstream; (4) visceral congestion and generation of relative ischemic conditions may impose environmental alterations in the bacterial microbiome of the intestinal lumen and potentially predisposing the host to colonization with TMAO-producing or SCFA-deficient bacteria. Taken together, these conditions may play an incremental but important role in the progression of HF and the cardiorenal syndrome that warrants further investigation.
Acknowledgments
Grant Support
National Institutes of Health R01 HL107577 and R01 HL 127028; and American Heart Association #16SFRN28780016 and #15CVGPSD27260148.
Sanjiv J. Shah has received grant support from Actelion, AstraZeneca, Corvia, and Novartis; and consulting fees from Actelion, AstraZeneca, Bayer, Boehringer-Ingelheim, Cardiora, Ironwood, Merck, Novartis, Pfizer, and Sanofi.
Footnotes
Conflict of Interest
Vincenzo B. Polsinelli and Arjun Sinha each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Spinarova L, Meluzin J, Toman J, Hude P, Krejci J, Vitovec J. Right ventricular dysfunction in chronic heart failure patients. Eur J Heart Fail. 2005;7(4):485–9. doi: 10.1016/j.ejheart.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Puwanant S, Priester TC, Mookadam F, Bruce CJ, Redfield MM, Chandrasekaran K. Right ventricular function in patients with preserved and reduced ejection fraction heart failure. Eur J Echocardiogr. 2009;10(6):733–7. doi: 10.1093/ejechocard/jep052. [DOI] [PubMed] [Google Scholar]
- 3.Burke MA, Katz DH, Beussink L, Selvaraj S, Gupta DK, Fox J, et al. Prognostic importance of pathophysiologic markers in patients with heart failure and preserved ejection fraction. Circ Heart Fail. 2014;7(2):288–99. doi: 10.1161/CIRCHEARTFAILURE.113.000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drazner MH, Rame JE, Stevenson LW, Dries DL. Prognostic importance of elevated jugular venous pressure and a third heart sound in patients with heart failure. N Engl J Med. 2001;345(8):574–81. doi: 10.1056/NEJMoa010641. [DOI] [PubMed] [Google Scholar]
- 5.Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. J Am Coll Cardiol. 2009;53(13):1119–26. doi: 10.1016/j.jacc.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53(7):589–96. doi: 10.1016/j.jacc.2008.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valentova M, von Haehling S, Krause C, Ebner N, Steinbeck L, Cramer L, et al. Cardiac cachexia is associated with right ventricular failure and liver dysfunction. Int J Cardiol. 2013;169(3):219–24. doi: 10.1016/j.ijcard.2013.08.134. [DOI] [PubMed] [Google Scholar]
- 8*.Valentova M, von Haehling S, Bauditz J, Doehner W, Ebner N, Bekfani T, et al. Intestinal congestion and right ventricular dysfunction: a link with appetite loss, inflammation, and cachexia in chronic heart failure. Eur Heart J. 2016;37(21):1684–91. doi: 10.1093/eurheartj/ehw008. This is a recent article that found that visceral (intestinal) congestion is the strongest factor associated with cardiac cachexia in heart failure patients. [DOI] [PubMed] [Google Scholar]
- 9.Sandek A, Bauditz J, Swidsinski A, Buhner S, Weber-Eibel J, von Haehling S, et al. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol. 2007;50(16):1561–9. doi: 10.1016/j.jacc.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Valentova M, von Haehling S, Anker SD, Sandek A. Cardiac hepatopathy versus end-stage liver disease: two different entities. J Am Coll Cardiol. 2014;63(17):1809–10. doi: 10.1016/j.jacc.2013.08.1654. [DOI] [PubMed] [Google Scholar]
- 11.Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J Am Heart Assoc. 2017;6(7) doi: 10.1161/JAHA.116.004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koutsos A, Tuohy KM, Lovegrove JA. Apples and cardiovascular health–is the gut microbiota a core consideration? Nutrients. 2015;7(6):3959–98. doi: 10.3390/nu7063959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li D, Kirsop J, Tang WH. Listening to Our Gut: Contribution of Gut Microbiota and Cardiovascular Risk in Diabetes Pathogenesis. Curr Diab Rep. 2015;15(9):63. doi: 10.1007/s11892-015-0634-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mafra D, Lobo JC, Barros AF, Koppe L, Vaziri ND, Fouque D. Role of altered intestinal microbiota in systemic inflammation and cardiovascular disease in chronic kidney disease. Future Microbiol. 2014;9(3):399–410. doi: 10.2217/fmb.13.165. [DOI] [PubMed] [Google Scholar]
- 15.Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31(6):817–44. doi: 10.1210/er.2009-0030. [DOI] [PubMed] [Google Scholar]
- 16.Miele L, Giorgio V, Alberelli MA, De Candia E, Gasbarrini A, Grieco A. Impact of Gut Microbiota on Obesity, Diabetes, and Cardiovascular Disease Risk. Curr Cardiol Rep. 2015;17(12):120. doi: 10.1007/s11886-015-0671-z. [DOI] [PubMed] [Google Scholar]
- 17.Sanduzzi Zamparelli M, Compare D, Coccoli P, Rocco A, Nardone OM, Marrone G, et al. The Metabolic Role of Gut Microbiota in the Development of Nonalcoholic Fatty Liver Disease and Cardiovascular Disease. Int J Mol Sci. 2016;17(8) doi: 10.3390/ijms17081225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serino M, Blasco-Baque V, Nicolas S, Burcelin R. Far from the eyes, close to the heart: dysbiosis of gut microbiota and cardiovascular consequences. Curr Cardiol Rep. 2014;16(11):540. doi: 10.1007/s11886-014-0540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124(10):4204–11. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang WH, Kitai T, Hazen SL. Gut Microbiota in Cardiovascular Health and Disease. Circ Res. 2017;120(7):1183–96. doi: 10.1161/CIRCRESAHA.117.309715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuohy KM, Fava F, Viola R. ‘The way to a man’s heart is through his gut microbiota’–dietary pro- and prebiotics for the management of cardiovascular risk. Proc Nutr Soc. 2014;73(2):172–85. doi: 10.1017/S0029665113003911. [DOI] [PubMed] [Google Scholar]
- 22.Ussher JR, Lopaschuk GD, Arduini A. Gut microbiota metabolism of L-carnitine and cardiovascular risk. Atherosclerosis. 2013;231(2):456–61. doi: 10.1016/j.atherosclerosis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita T, Kasahara K, Emoto T, Matsumoto T, Mizoguchi T, Kitano N, et al. Intestinal Immunity and Gut Microbiota as Therapeutic Targets for Preventing Atherosclerotic Cardiovascular Diseases. Circ J. 2015;79(9):1882–90. doi: 10.1253/circj.CJ-15-0526. [DOI] [PubMed] [Google Scholar]
- 24.Bookstein C, DePaoli AM, Xie Y, Niu P, Musch MW, Rao MC, et al. Na+/H+ exchangers, NHE-1 and NHE-3, of rat intestine. Expression and localization. J Clin Invest. 1994;93(1):106–13. doi: 10.1172/JCI116933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiela PR, Guner YS, Xu H, Collins JF, Ghishan FK. Age- and tissue-specific induction of NHE3 by glucocorticoids in the rat small intestine. Am J Physiol Cell Physiol. 2000;278(4):C629–37. doi: 10.1152/ajpcell.2000.278.4.C629. [DOI] [PubMed] [Google Scholar]
- 26.Broere N, Chen M, Cinar A, Singh AK, Hillesheim J, Riederer B, et al. Defective jejunal and colonic salt absorption and alteredNa(+)/H (+) exchanger 3 (NHE3) activity in NHE regulatory factor 1 (NHERF1) adaptor protein-deficient mice. Pflugers Arch. 2009;457(5):1079–91. doi: 10.1007/s00424-008-0579-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, et al. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2002;282(5):G776–84. doi: 10.1152/ajpgi.00297.2001. [DOI] [PubMed] [Google Scholar]
- 28.Musch MW, Lucioni A, Chang EB. Aldosterone regulation of intestinal Na absorption involves SGK-mediated changes in NHE3 and Na+ pump activity. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G909–19. doi: 10.1152/ajpgi.90312.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucioni A, Womack C, Musch MW, Rocha FL, Bookstein C, Chang EB. Metabolic acidosis in rats increases intestinal NHE2 and NHE3 expression and function. Am J Physiol Gastrointest Liver Physiol. 2002;283(1):G51–6. doi: 10.1152/ajpgi.00529.2001. [DOI] [PubMed] [Google Scholar]
- 30.Giral H, Cranston D, Lanzano L, Caldas Y, Sutherland E, Rachelson J, et al. NHE3 regulatory factor 1 (NHERF1) modulates intestinal sodium-dependent phosphate transporter (NaPi-2b) expression in apical microvilli. J Biol Chem. 2012;287(42):35047–56. doi: 10.1074/jbc.M112.392415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glover LE, Lee JS, Colgan SP. Oxygen metabolism and barrier regulation in the intestinal mucosa. J Clin Invest. 2016;126(10):3680–8. doi: 10.1172/JCI84429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pluznick J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. 2014;5(2):202–7. doi: 10.4161/gmic.27492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110(11):4410–5. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G-protein coupled receptor 41. Physiol Genomics. doi: 10.1152/physiolgenomics.00089.2016. 2016: physiolgenomics.00089.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Organ CL, Otsuka H, Bhushan S, Wang Z, Bradley J, Trivedi R, et al. Choline Diet and Its Gut Microbe-Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload-Induced Heart Failure. Circ Heart Fail. 2016;9(1):e002314. doi: 10.1161/CIRCHEARTFAILURE.115.002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senthong V, Wang Z, Li XS, Fan Y, Wu Y, Tang WH, et al. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J Am Heart Assoc. 2016;5(6) doi: 10.1161/JAHA.115.002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, et al. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J Am Coll Cardiol. 2016;67(22):2620–8. doi: 10.1016/j.jacc.2016.03.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64(18):1908–14. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–55. doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang WH, Wang Z, Li XS, Fan Y, Li DS, Wu Y, et al. Increased Trimethylamine N-Oxide Portends High Mortality Risk Independent of Glycemic Control in Patients with Type 2 Diabetes Mellitus. Clin Chem. 2017;63(1):297–306. doi: 10.1373/clinchem.2016.263640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165(1):111–24. doi: 10.1016/j.cell.2016.02.011. This paper describes the mechanistic link between the gut-derived microbial metabolite TMAO, enhanced platelet reactivity, and thrombosis risk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu W, Wang Z, Tang WHW, Hazen SL. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation. 2017;135(17):1671–3. doi: 10.1161/CIRCULATIONAHA.116.025338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlachogiannakos J, Saveriadis AS, Viazis N, Theodoropoulos I, Foudoulis K, Manolakopoulos S, et al. Intestinal decontamination improves liver haemodynamics in patients with alcohol-related decompensated cirrhosis. Aliment Pharmacol Ther. 2009;29(9):992–9. doi: 10.1111/j.1365-2036.2009.03958.x. [DOI] [PubMed] [Google Scholar]
- 44.Dong T, Aronsohn A, Gautham Reddy K, Te HS. Rifaximin Decreases the Incidence and Severity of Acute Kidney Injury and Hepatorenal Syndrome in Cirrhosis. Dig Dis Sci. 2016 doi: 10.1007/s10620-016-4313-0. [DOI] [PubMed] [Google Scholar]
- 45.Ponziani FR, Gerardi V, Pecere S, D’Aversa F, Lopetuso L, Zocco MA, et al. Effect of rifaximin on gut microbiota composition in advanced liver disease and its complications. World J Gastroenterol. 2015;21(43):12322–33. doi: 10.3748/wjg.v21.i43.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandek A, Bjarnason I, Volk HD, Crane R, Meddings JB, Niebauer J, et al. Studies on bacterial endotoxin and intestinal absorption function in patients with chronic heart failure. Int J Cardiol. 2012;157(1):80–5. doi: 10.1016/j.ijcard.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 47.Sandek A, Anker SD, von Haehling S. The gut and intestinal bacteria in chronic heart failure. Curr Drug Metab. 2009;10(1):22–8. doi: 10.2174/138920009787048374. [DOI] [PubMed] [Google Scholar]
- 48.Sandek A, Rauchhaus M, Anker SD, von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11(5):632–9. doi: 10.1097/MCO.0b013e32830a4c6e. [DOI] [PubMed] [Google Scholar]
- 49.Nikolaou M, Parissis J, Yilmaz MB, Seronde MF, Kivikko M, Laribi S, et al. Liver function abnormalities, clinical profile, and outcome in acute decompensated heart failure. Eur Heart J. 2013;34(10):742–9. doi: 10.1093/eurheartj/ehs332. [DOI] [PubMed] [Google Scholar]
- 50.Pasini E, Aquilani R, Testa C, Baiardi P, Angioletti S, Boschi F, et al. Pathogenic Gut Flora in Patients With Chronic Heart Failure. JACC Heart Fail. 2016;4(3):220–7. doi: 10.1016/j.jchf.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Poelzl G, Ess M, Von der Heidt A, Rudnicki M, Frick M, Ulmer H. Concomitant renal and hepatic dysfunctions in chronic heart failure: clinical implications and prognostic significance. Eur J Intern Med. 2013;24(2):177–82. doi: 10.1016/j.ejim.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 52.Sandek A, Swidsinski A, Schroedl W, Watson A, Valentova M, Herrmann R, et al. Intestinal blood flow in patients with chronic heart failure: a link with bacterial growth, gastrointestinal symptoms, and cachexia. J Am Coll Cardiol. 2014;64(11):1092–102. doi: 10.1016/j.jacc.2014.06.1179. [DOI] [PubMed] [Google Scholar]
- 53.Mullens W, Abrahams Z, Skouri HN, Francis GS, Taylor DO, Starling RC, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. 2008;51(3):300–6. doi: 10.1016/j.jacc.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 54.Kalogeropoulos AP, Tang WH, Hsu A, Felker GM, Hernandez AF, Troughton RW, et al. High-sensitivity C-reactive protein in acute heart failure: insights from the ASCEND-HF trial. J Card Fail. 2014;20(5):319–26. doi: 10.1016/j.cardfail.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 55.Cetin S, Dunklebarger J, Li J, Boyle P, Ergun O, Qureshi F, et al. Endotoxin differentially modulates the basolateral and apical sodium/proton exchangers (NHE) in enterocytes. Surgery. 2004;136(2):375–83. doi: 10.1016/j.surg.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 56.Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiery JL. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016;37(12):942–54. doi: 10.1093/eurheartj/ehv512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dixon DD, Trivedi A, Shah SJ. Combined post- and pre-capillary pulmonary hypertension in heart failure with preserved ejection fraction. Heart Fail Rev. 2016;21(3):285–97. doi: 10.1007/s10741-015-9523-6. [DOI] [PubMed] [Google Scholar]
- 58.Thenappan T, Prins KW, Cogswell R, Shah SJ. Pulmonary hypertension secondary to heart failure with preserved ejection fraction. Can J Cardiol. 2015;31(4):430–9. doi: 10.1016/j.cjca.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 59.Unger ED, Dubin RF, Deo R, Daruwalla V, Friedman JL, Medina C, et al. Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. Eur J Heart Fail. 2016;18(1):103–12. doi: 10.1002/ejhf.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Westerhof N, Westerhof BE. A review of methods to determine the functional arterial parameters stiffness and resistance. J Hypertens. 2013;31(9):1769–75. doi: 10.1097/HJH.0b013e3283633589. [DOI] [PubMed] [Google Scholar]
- 61.Pool LR, Wolf M. FGF23 and Nutritional Metabolism. Annu Rev Nutr. 2017;37:247–68. doi: 10.1146/annurev-nutr-071816-064620. [DOI] [PubMed] [Google Scholar]
- 62.Wahl P, Wolf M. FGF23 in chronic kidney disease. Adv Exp Med Biol. 2012;728:107–25. doi: 10.1007/978-1-4614-0887-1_8. [DOI] [PubMed] [Google Scholar]
- 63.Akmal M, Barndt RR, Ansari AN, Mohler JG, Massry SG. Excess PTH in CRF induces pulmonary calcification, pulmonary hypertension and right ventricular hypertrophy. Kidney Int. 1995;47(1):158–63. doi: 10.1038/ki.1995.18. [DOI] [PubMed] [Google Scholar]
- 64.Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Astrand M, Johansson S, Knutsson M, et al. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017;28(6):1933–42. doi: 10.1681/ASN.2016080855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu LC, Dorhout B, van der Meer P, Teerlink JR, Voors AA. Omecamtiv mecarbil: a new cardiac myosin activator for the treatment of heart failure. Expert Opin Investig Drugs. 2016;25(1):117–27. doi: 10.1517/13543784.2016.1123248. [DOI] [PubMed] [Google Scholar]
- 66.Khan SS, Cuttica MJ, Beussink-Nelson L, Kozyleva A, Sanchez C, Mkrdichian H, et al. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: a pilot study. Pulm Circ. 2015;5(3):547–56. doi: 10.1086/682427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah SJ, Blair JE, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J, et al. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am Heart J. 2009;157(6):1035–41. doi: 10.1016/j.ahj.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 68.Craig ML. Management of right ventricular failure in the era of ventricular assist device therapy. Curr Heart Fail Rep. 2011;8(1):65–71. doi: 10.1007/s11897-010-0043-3. [DOI] [PubMed] [Google Scholar]
- 69.Kapur NK, Bader YH. Percutaneous Circulatory Assist Devices for Right Ventricular Failure. Interv Cardiol Clin. 2013;2(3):445–56. doi: 10.1016/j.iccl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 70.Guazzi M, Samaja M, Arena R, Vicenzi M, Guazzi MD. Long-term use of sildenafil in the therapeutic management of heart failure. J Am Coll Cardiol. 2007;50(22):2136–44. doi: 10.1016/j.jacc.2007.07.078. [DOI] [PubMed] [Google Scholar]
- 71.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4(1):8–17. doi: 10.1161/CIRCHEARTFAILURE.110.944694. [DOI] [PubMed] [Google Scholar]
- 72.Bonderman D, Ghio S, Felix SB, Ghofrani HA, Michelakis E, Mitrovic V, et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. 2013;128(5):502–11. doi: 10.1161/CIRCULATIONAHA.113.001458. [DOI] [PubMed] [Google Scholar]
- 73.Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2017;21(3):407–16. doi: 10.1007/s10157-016-1302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74*.Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. Spencer et al. describe the protective beneficial effect of gut NHE3 inhibition on cardiorenal damage in rats. [DOI] [PubMed] [Google Scholar]
- 75.Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients With Heart Failure: Proposal of a Novel Mechanism of Action. JAMA Cardiol. 2017;2(9):1025–9. doi: 10.1001/jamacardio.2017.2275. [DOI] [PubMed] [Google Scholar]
- 76.Turner JR, Black ED. NHE3-dependent cytoplasmic alkalinization is triggered by Na(+)-glucose cotransport in intestinal epithelia. Am J Physiol Cell Physiol. 2001;281(5):C1533–41. doi: 10.1152/ajpcell.2001.281.5.C1533. [DOI] [PubMed] [Google Scholar]
- 77.Ghosh P. The stress polarity pathway: AMPK ‘GIV’-es protection against metabolic insults. Aging (Albany NY) 2017;9(2):303–14. doi: 10.18632/aging.101179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shih CJ, Wu YL, Chao PW, Kuo SC, Yang CY, Li SY, et al. Association between Use of Oral Anti-Diabetic Drugs and the Risk of Sepsis: A Nested Case-Control Study. Sci Rep. 2015;5:15260. doi: 10.1038/srep15260. [DOI] [PMC free article] [PubMed] [Google Scholar]