

Abstract
The tumor microenvironment (TME) is a relevant target for novel biological therapies. MV-m-uPA and MV-h-uPA are fully retargeted, species-specific, oncolytic measles viruses (MVs) directed against murine or human urokinase receptor (PLAUR/uPAR), expressed in tumor and stromal cells. The effects of stromal selective targeting by uPAR retargeted MVs were investigated. In vitro infection, virus-induced GFP expression and cytotoxicity by MV-h-uPA and MV-m-uPA were demonstrated in human and murine cancer cells and cancer associated fibroblasts (CAFs) in a species-specific manner. In a murine fibroblast/human breast cancer 3D co-culture model, selective fibroblast targeting by MV-m-uPA inhibited breast cancer cell growth. Systemic administration of murine specific MV-m-uPA in mice bearing human MDA-MB 231 xenografts was associated with a significant delay in tumor progression and improved survival compared to controls. Experiments comparing tumor (MV-h-uPA) vs. stromal (MV-m-uPA) vs. combined virus targeting showed that tumor and stromal targeting was associated with improved tumor control over the other groups. Correlative studies confirmed in vivo viral targeting of tumor stroma by MV-m-uPA, increased apoptosis, and virus induced differential regulation of murine stromal genes associated with inflammatory, angiogenesis and survival pathways, as well as indirect regulation of human cancer pathways, indicating viral induced modulation of tumor-stromal interactions. These data demonstrate the feasibility of stromal selective targeting by an oncolytic MV, virus-induced modulation of tumor-stromal pathways, and subsequent tumor growth delay. These findings further validate the critical role of stromal uPAR in cancer progression and the potential of oncolytic viruses as anti-stromal agents.
Keywords: Urokinase receptor, measles virus, stromal targeting, breast cancer progression
INTRODUCTION
The tumor stroma plays a critical role in cancer biology [1, 2]. Tumor fibroblasts [3], macrophages [4], endothelial [5] and inflammatory/immune cells [6] contribute to different steps of cancer initiation, angiogenesis, progression and metastasis. Strategies targeting the tumor microenvironment have been investigated in the preclinical and clinical setting, and several stromal directed agents are currently approved for clinical use, such as antiangiogenic and immunotherapeutic drugs [7, 8]. Active investigation is being pursued to develop agents targeting macrophages, fibroblasts or immune suppressive cells [9–12].
The oncolytic virotherapy field has expanded exponentially in the last decade, with an increasing number of viral vectors being moved from the bench to clinical trials, and recently, to clinical use [13]. Among the novel oncolytic viruses under development, the Edmonston vaccine strain of measles virus (MV-Edm) is a promising one, whose in vitro and in vivo preclinical safety and efficacy are well established [14]. Recombinant, non-targeted oncolytic MVs are currently being evaluated in ovarian cancer, brain tumors, multiple myeloma and mesothelioma [13, 15], with promising preliminary reports of antitumor efficacy [16].
The urokinase receptor (uPAR) is a GPI anchored cell surface receptor, whose clinical and biological relevance are well established. It plays a critical role in tumor-stromal interactions, tumor progression and metastasis [17, 18]. Murine and human cancers overexpress uPAR, and it has been shown to be abundantly expressed in stromal cells, including tumor fibroblasts, macrophages and endothelial cells, as well as tumor infiltrating lymphocytes [19–22]. Therefore, the uPAR is an attractive target for tumor and stromal directed biological therapies.
In an effort to develop stromal directed oncolytic viral vectors, our group has engineered and rescued oncolytic measles viruses fully retargeted against murine (MV-m-uPA) or human (MV-h-uPA) urokinase receptor (uPAR) [23]. Recombinant oncolytic MVs retargeted against human or mouse uPAR induce antitumor effects in primary (orthotopic) and metastatic tumors, in xenograft or syngeneic cancer models, respectively [23–26]. As MV-m-uPA and MV-h-uPA are species specific, the above viral agents represent invaluable tools to dissect the contribution of stromal targeting on an oncolytic virus’ antitumor effects. The current study is the first to characterize the in vitro, in vivo and molecular effects of stromal selective targeting by an oncolytic measles virus.
MATERIALS AND METHODS
Virus preparation, characterization, and cell culture
MV-GFP, MV-m-uPA and MV-h-uPA were generated as previously reported [23, 27]. Murine 4T1 (mammary carcinoma, ATCC #2539, purchased in 2007), CT-26 (colon carcinoma, ATCC #2638, purchased in 2009), NIH-3T3 (immortalized fibroblasts, ATCC #1658, purchased in 2009) and NMuMG (murine mammary epithelial, ATCC #1636, purchased in 2009), as well as human breast (MDA-MB-231, ATCC #HTB-26, purchased in 2007; T47D, ATCC #HTB-133; purchased in 2009) and colon cancer cells (HT-29, ATCC #HTB-38, purchased in 2012) were purchased from ATCC (Manassas, VA), and maintained in Dulbecco’s modified eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin and streptomycin at 37 °C and 5% CO2. HMEC (human mammary epithelial cells) were purchased from Lonza (Walkersville, MD) in 2009 and maintained in mammary epithelial cell growth medium (MEGM) according to manufacturer’s instructions, at 37 °C and 5% CO2. Cancer associated fibroblasts CAF19 (from a luminal A tumor) and CAF23 (from a basal like tumor) were isolated and characterized from primary breast tumors [28] in 2016, and maintained in Iscove’s modified dulbecco’s medium (IMDM).
In vitro infection, assessment of virus induced cytotoxicity, and in vitro viral replication were performed as previously described [23, 26].
uPAR Immunocytochemistry
Cells were plated in chamber slides (VWR, Radnor, PA) at 5×104 cells per chamber overnight, washed with PBS and fixed with 4% paraformaldehyde for one hour at room temperature. Following three times PBS washes, cells were blocked (3% BSA with 0.1% TritonX-100 in PBS for one hour at room temperature) and probed with FITC conjugated anti-human uPAR (1:100; Abcam, Cambridge, MA) or FITC conjugated anti-mouse uPAR antibody (1:100; Abcam), and incubated overnight at 4° C. This was followed by three times PBS washes; slides were mounted with anti-fade mounting medium (Molecular Probes, Eugene, OR) and analyzed by fluorescent microscopy.
Generation of RFP expressing cells
cDNA encoding RFP (pDsRed-Express Vector, Clontech, Mountain View, CA) were subcloned into the BamHI-NotI site of the lentiviral vector pHR-SIN-CSGWd1NotI (gift of Y. Ikeda, Mayo Clinic, Rochester, MN). Lentiviral packaging and virus transduction of cancer cells were performed as previously reported [25, 29, 30]. Briefly, Lentiviral packaging was performed by cotransfection of the vector plasmid with pCMV-Gag-Pol vector and pCMV-VSVG-poly-A vector into 293T cells using CaCl2 transfection kit (Promega, Madison, WI). After 48 hours, lentivirus-containing supernatant was harvested, and stored at −80°C. Cells were infected with RFP expressing lentiviral vector in a 6-well plate. 24 hours post-infection, fresh growth media were replaced.
In vitro fibroblast-to-cancer cell viral transfer
Fibroblasts (CAF19, CAF23, NIH-3T3)were incubated with species specific uPAR retargeted MVs, which express eGFP [23] (MOI of 1) in Opti-MEM for 2 h at 37 °C. Free viruses were then removed and cells were maintained in the appropriate medium. To rule out persistence of cell free virions, 100 μl of the last wash was inoculated on Vero monolayers. Cells were cultured in the presence of 80 μg/ml fusion inhibitory peptide (Bachem California, Torrance, CA) to block syncytia formation. Virus infected fibroblasts were overlaid on RFP-expressing tumor cells. Mixed (double-color) syncytia were demonstrated at different periods of incubation by fluorescent microscopy.
In vitro 3-D collagen co-culture model
Three-dimensional collagen gel co-culture was established based on a method described previously [31]. Briefly, 3T3 cells were infected with MV-m-uPA at MOI = 3 in Opti-MEM for 2 hours at 37°C, followed by virus removal. RFP expressing T47D cells and 3T3 cells were mixed at a ratio of 2:1 and cultured on collagen type I gel (concentration: 1.3mg/ml). At different time points, cells were released from the collagen gel by collagenase treatment (2mg/ml, Worthington). After washing with PBS, samples were analyzed on a FACSCalibur benchtop cytometer (BD Biosciences) for quantification of RFP expressing cells.
Animal studies
Animal studies were approved by the University of Miami Institutional Animal Care and Use Committee. Eight to 10 week old female NSG mice were injected with 2×106 MDA-MB-231 cells into the 5th mammary fat pad. When tumors reached 0.4–0.5 cm, mice (n = 8/group) were treated with PBS or 1.5×106 TCID50 of MV-m-uPA, via tail vein 3 times a week for 4 weeks. Tumor volume was measured/calculated twice a week as reported [23]. Clinical signs of toxicity were monitored. Animals were followed until they reached sacrifice criteria (when tumor burden reached 10% of body weight, tumor ulceration occurred or mice became moribund). For correlative studies, tumor bearing mice (n = 3/group) were treated with two intravenous injections of 1.5×106 TCID50 of MV-m-uPA or vehicle, and tumors were resected after 24 or 72 hours of the last injection, and snap frozen.
In additional experiments, NOD/SCID were implanted with MDA-MB-231 cells as above. When tumors reached a diameter of 0.4–0.5 cm, animals (n = 8/group) were treated with 1.5×106 TCID50 of MV-m-uPA, MV-h-uPA, combination of MV-m-uPA and MV-h-uPA (1.5×106 TCID50 for each of virus) or equal volumes of PBS via tail vein every other day for a total of 3 treatments. Tumor evaluation was conducted as above. For correlative studies, tumor bearing mice (n = 3) were given two intravenous injections of 1.5×106 TCID50 of MV-h-uPA, MV-m-uPA or vehicle. Tumors were resected at day 2 and 5 after the last injection, and snap frozen.
Virus Recovery
Tissues were weighed and homogenized in three volumes (w/v) of Opti-MEM utilizing mechanical crushing and a single freeze thaw cycle. The supernatant was clarified by centrifugation and ten-fold serial dilutions of samples were prepared in Opti-MEM. Aliquots (50 μL) of each dilution were placed in 96 well plates containing Vero-his cells and TCID50 titrations were performed [32]. Viral titers are displayed as TCID50/ml and were normalized for gram of tissue. Lower limit of detection was 84 TCID50/ml.
Viral RNA quantification
Total RNA was extracted from frozen specimens using the RNeasy tissue mini kit (Qiagen, Valencia, CA). qRT-PCR for MV-N mRNA was performed as previously reported [24, 26].
Hematology and chemistry analysis
Whole blood and serum were harvested from mice at specific timepoints, and host toxicity was evaluated by assessment of changes in hematological and biochemical parameters using a Hemavet 1700 multispecies hematology analyzer and an Ortho Vitros 250 analyzer, respectively.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
To detect and visualize apoptosis, slides were washed twice with PBS (after fixation), permeabilized with 0.2% TritonX-100 for 20 minutes at room temperature and after 2 additional washes with PBS, sections were probed with label solution (for negative controls) or TUNEL reaction mix, following manufacturer’s instructions (In Situ Cell Death Detection Kit-TMR red; Roche Applied Science, Indianapolis, IN) [24, 26].
In situ hybridization
In situ RNA hybridization was performed from cryopreserved (8–10 μm) tumor sections using RNAscope technology, using RNA probes for murine uPAR (RNAscope Probe- Mm-Plaur) and measles virus N gene (RNAscope Probe-V-MEV-N-C2) from Advanced Cell Diagnostics (Newark, California), following manufacturer’s protocol. Fluorescent signal was visualized and captured using an open-field Nikon Eclipse TE2000-U microscope. According Advanced Cell Diagnostics, each mRNA molecule hybridized to a probe appears as a separate small fluorescent dot.
Immunofluorescence studies
Tumor bearing mice (n = 3) were treated with two intravenous injections of 1.5×106 TCID50 of PBS, MV-m-uPA, MV-h-uPA. Mice were sacrificed and tumors resected after 48 hours of the last injection for immunofluorescence studies. Tissue samples were collected and frozen, and cryostat sections were fixed in cold acetone for 10 min. The slides were washed in PBS and incubated with anti-MV-nucleoprotein-FITC antibody (1:100; Chemicon International), or anti-Ki 67 antibody-FITC (1:100; Abcam) for 30 min at 37°C. Followed by three PBS washes, slides were mounted with anti-fade mounting medium (Molecular Probes). For fibroblast co-immunofluorescence staining, the MV nucleoprotein positive slides were washed in PBS and incubated with anti-ER-TR7 primary antibody (1:100; Santa Cruz, Dallas, TX) overnight at 4°C. After two washes, Alexa Fluor 594 anti-rat IgG secondary antibody (Invitrogen) was applied (1:500; for 1 hour at room temperature). For costaining FAP, the MV nucleoprotein positive slides were incubated with anti-FAP antibody(1:100; Abcam) overnight at 4°C. After two washes, Alexa Fluor 594 anti-goat IgG secondary antibody (Invitrogen) was applied (1:500; for 1 hour at room temperature). Slides were mounted with anti-fade mounting medium after three PBS washes and analyzed by fluorescent microscopy.
Nanostring analysis of Gene expression
Two commercially available 770-gene panels (nCounter Human PanCancer Patwhways and mouse nCounter PanCancer Immune Profiling were used to analyze human and murine genes. Assays were performed following manufacturer’s instructions, with standard input of 100 ng and the codeset with the current XT chemistry using a nanoString nCounter Gen2 scanner and Prep-Station (NanoString Technologies, Seattle, WA). Samples were verified by Agilent Bioanalyzer 2100 using Nano 6000 chips to determine RNA integrity number (RIN). Data were exported from NanoString’s nCounter Raw Code Count Collector tool and loaded into R environment (v3.2.4) for analysis. Data normalization was performed using R package NanoStringNorm (v1.1.21), with a combination of multiple normalization steps, as previously reported [33]. To minimize possible human-murine cross-hybridization, human (n=58) and mouse (n=10) orthologous genes with high sequence similarity (>=94%) were excluded from further downstream analysis.
For differential expression analysis, R package limma (v3.26.9) was used to perform two group comparison test [34], and raw p values based on empirical Bayes moderated t-statistics were adjusted for multiple testing with Benjamini & Hochberg false discovery rate (FDR) correction [35]. Pathway enrichment and functional analyses were generated using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity).
Statistical analysis
In vitro data are presented as means +/− SD. Results from in vivo studies are shown as means +/− SEM. All in vitro experiments were performed in triplicate unless otherwise specified. Statistical analysis among groups was performed by ANOVA, and sub-group comparisons were made the Student t-test, Tukey-Kramer, Fisher’s or Wilcoxon rank sum test, as appropriate. Overall survival was analyzed by the Kaplan–Meier method and differences were analyzed by the log-rank test. Statistical significance was set at p = 0.05, with adjustements for multiple comparisons as appropriate. All statistical tests were two-sided.
RESULTS
Differential in vitro tumor and species selectivity of human vs. murine uPAR retargeted measles viruses
Human and murine cancer cells known to express uPAR [23, 36, 37], as well as (human) HMEC and (murine) NMuMG non-cancer cells were exposed to non-targeted MV (MV-GFP), MV-h-uPA and MV-m-uPA. As expected, from prior reports by us [23, 24], MV-h-uPA and MV-m-uPA infected human MDA-MB 231 and murine 4T1 tumor cells in a species specific manner, while non-cancer epithelial cells (HMEC, NMuMG) were not susceptible to viral infection (Fig. 1). Similar species specific effects were observed in human HT-29 and T47D, as well as murine CT-26 cancer cells. As previously reported [24], MV-h-uPA was more selective to human cancer than non-cancer epithelial cells (HMEC) than MV-GFP.
Figure 1. In vitro tumor and species specificity of uPAR retargeted Measles Viruses.
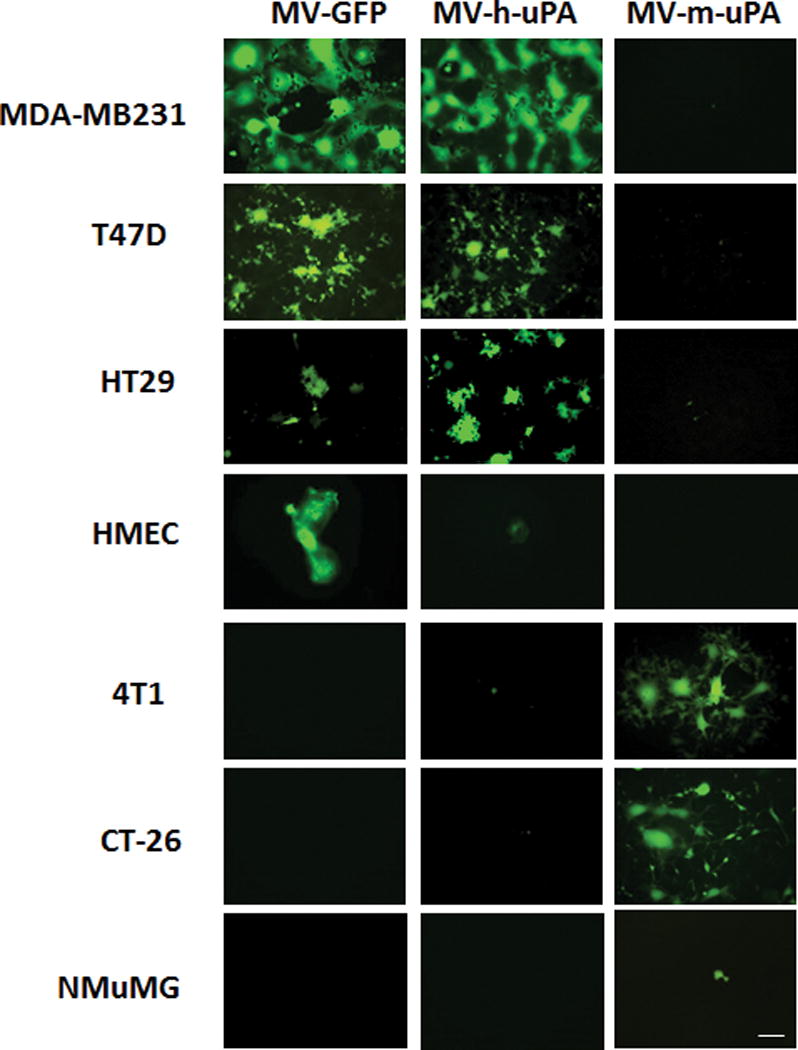
Human cancer cells (MDA-MB-231, T47D, HT29), human mammary eptithelial cells (HMEC), murine cancer cells (4T1, CT-26), and murine mammary epithelial cells (NMuMG) were infected with MV-GFP control virus, MV-h-uPA, or MV-m-uPA at an MOI= 1 and photographed 48 h after infection using a fluorescent microscope. Scale bar = 100 μm.
In vitro effects of uPAR retargeted viruses on cancer associated fibroblasts
Cancer associated fibroblasts (CAFs) express uPAR and play a critical role in tumor progression and metastasis [3, 38, 39]. Patient derived CAF 19 and CAF 23 [28], as well as murine 3T3 fibroblasts, express human and murine uPAR, respectively (Fig. 2. A). Human CAFs were permissive to MV-h-uPA but not to MV-m-uPA infection (Fig. 2. A). On the other hand, 3T3 cells were permissive only to murine, but not human uPAR retargeted viral vector (Fig 2. A). Viral replication was demonstrated in both human and murine fibroblasts at 24, 48 and 72 hours (Fig. 2. B). Human and murine uPAR retargeted MVs induced significant cytotoxitiy to species specific fibroblasts (Fig. 2. C).
Figure 2. In vitro viral infection, cytopathic effects and replication of MV-uPA on human and murine fibroblast.
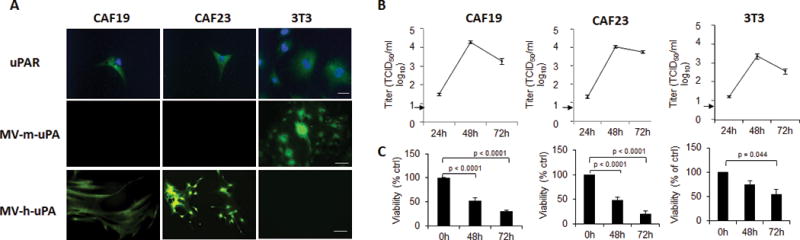
(A) Upper panel: uPAR expression in human primary cancer associated fibroblast CAF19, CAF23 and NIH-3T3 cells (immunocytochemistry). Scale bar = 20 μm. Mid and lower panels: CAF19, CAF23 and 3T3 species specific MV-uPA infection and virus induced GFP expression. Cells were photographed 48 h after infection (MOI=1). Scale bar = 100 μm. (B), In vitro viral replication (24, 48, 72 hours) in human CAF19, CAF23 and mouse 3T3 fibroblasts. Results are displayed as the average (+/− SD) of triplicate experiments. Lower limit of detection was 8 TCID50/ml. (C), Time dependent, MV-h-uPA induced cytotoxicity in CAF19, CAF23, and MV-m-uPA induced cytotoxicity in murine 3T3 cells. Virus dose: MOI=1. Results are presented as percentage of controls. Bars represent averages +/− SD of triplicate experiments.
Effects of fibroblast targeting by MV-uPA on fibroblast-tumor cell interactions
Next, the ability of MV-uPA infected fibroblasts to transfer viral infection to tumor cells was determined. Human CAF 19 were infected by MV-h-uPA and subsequently overlaid onto RFP expressing MDA-MB 231 (Fig 3. A. I) or HT 29 (Fig. 3. A, II). At 48 hours, yellow fluorescent syncytia (GFP and RFP on overlay images) were observed, indicating successful viral transfer from CAFs to tumor cells via heterofusion. Similar results were observed when murine 3T3 cells were infected with species specific MV-m-uPA and overlaid onto RFP expressing 4T1 (Fig. 3. B, I) or CT-26 cells (Fig. 3. B, II).
Figure 3. Effects of uPAR mediated fibroblast targeting on fibroblast-tumor cell interactions in vitro.
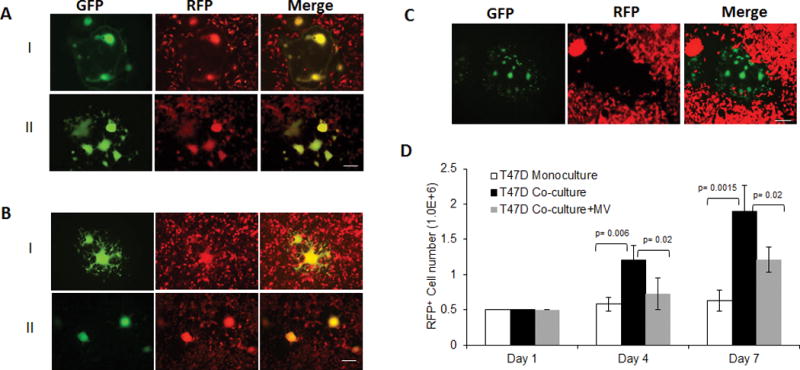
CAF19 (A) were infected with MV-h-uPA (MOI=1) as in methods, and then overlaid on RFP-expressing MDA-MB-231 (A, I) and HT-29 (A, II) cancer cells. Mixed (double-color) syncytia were demonstrated at 48h incubation by fluorescent microscopy. (B) In a similar way, MV-m-uPA infected 3T3 cells were overlaid on RFP-expressing murine 4T1 (B, I) or CT-26 (B, II) cells and yellow syncytia were demonstrated at 48h incubation by fluorescent microscopy. Scale bar = 100 μm. (C). MV-m-uPA infected murine 3T3 cells were overlaid onto human T47D, RFP expressing T47D breast cancer cells. Note that no yellow syncitia was observed in the merged image. Scale bar = 100 μm. (D) Effects of murine fibroblast specific viral targeting on human T47D cancer cell growth in vitro using an in vitro 3-D collagen co-culture model (see methods). Bars represent averages +/− SD of triplicate experiments.
To evaluate the effects of fibroblast targeting by MV-uPA on cancer cell growth, a murine fibroblast/human cancer cell 3D coculture model was used [31], where permissive murine 3T3 cells were infected by MV-m-uPA and then co-cultured with RFP expressing human T47D cells, resistant to the murine virus (Fig 1, and 3. C). Growth of T47D mono- and co-cultures was quantitated by FACS of RFP positive cancer cells at days 1, 4, and 7. Growth of T47D in co-culture with 3T3s was significantly increased at day 4 and 7, compared to mono-cultures (Fig. 3. D). In contrast, MV-m-uPA infection of 3T3 cells led to significant inhibition of co-cultured T47Ds at similar time points.
In vivo effects of stromal selective targeting by oncolytic MV-uPA in breast cancer xenografts
To test the hypothesis that stromal selective targeting by MV-m-uPA is associated with tumor growth delay, the human MDA-MB 231 orthotopic breast cancer model was used, where human tumor cells are resistant to MV-m-uPA infection (Fig. 1), while murine stromal cells are expected to be susceptible to the murine virus via mouse uPAR (Figs. 2, 3). Tumor bearing mice were treated and followed as in methods. MV-m-uPA treatment was associated with a significant delay in tumor progression, compared to vehicle treated mice. Tumor growth in MV-m-uPA treated mice was inhibited by 53% at day 28 (p < 0.0001, Fig. 4. A). These effects were associated with a significant prolongation of survival (p= 0.001, Log rank test) in MV-m-uPA treated mice, compared to controls (60-day survival: 90% vs. 30%, Fig. 4. B).
Figure 4. In vivo effects of murine uPAR retargeted MV on human breast cancer xenografts.
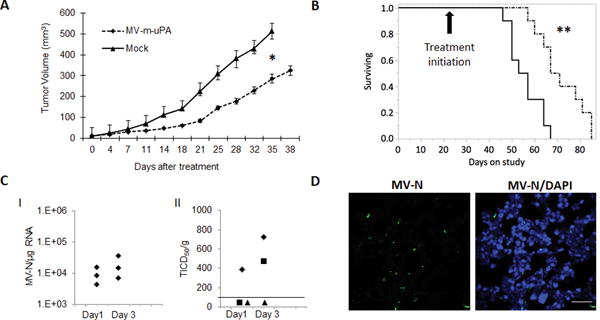
(A). Effects of MV-m-uPA on MDA-MB-231 tumor progression. Tumor bearing NSG mice were treated with vehicle (PBS) or MV-m-uPA intravenously, as in methods. (B). Kaplan-Meier analysis of survival of mice treated with vehicle or MV-m-uPA. Mice were monitored until they reached sacrifice criteria (methods). Arrow represents the day of treatment initiation (day 21 of tumor inoculation). (C). Quantitative analysis of MV-N-RNA (qRT-PCR) (C.I) and infectious viral particles (C.II) from treated tumors (n=3/group) at days 1 and 3 after MV administration (see methods). Viral titers are displayed as TCID50/gram of tissue. Line indicates lower limit of detection (LLD). Data points below the LLD represent samples with undetectable viral particles (D). Immunostaining for MV-N (using FITC labeled anti MV-N antibody) protein in tumor tissues obtained at day 3 after virus administration. Scale bar = 20 μm.
Tumor studies performed in additional (3 per group) mice treated with 2 doses of the virus demonstrated increasing viral RNA (Fig. 4.C.I) as well as viable viral particles (Fig. 4. C. II) over time. While viral RNA was detected in all tumor samples (3/3) in each time point, viable viral particles were identified only in 1/3 (day 1) and 2/3 (day3) tumor samples after virus administration. Moreover, MV-N protein (Fig. 4. D). was identified in tumors treated with intravenous MV-m-uPA. As MV-m-uPA does not bind to human tumor cells, the above findings strongly suggest successful in vivo stromal targeting by MV-m-uPA after systemic administration.
Next, to determine the in vivo effects of targeting cancer vs stromal cell components of the tumor, MDA-MB231 bearing mice were treated (n=8 per group) with either vehicle, MV-m-uPA, MV-h-uPA or the combination (MV-m-uPA+MV-h-uPA). All treatment groups were associated with significant delay in tumor growth compared to controls (Fig. 5. A; p< 0.0001). While treatment with MV-h-uPA led to better effects than MV-m-uPA, the combination group was associated with improved tumor control compared to MV-h-uPA (35.5% reduction in tumor volume by day 28; p= 0.0332). No treatment related deaths were observed, and virus treatment was not associated with significant changes in hematology or biochemical parameters (table S1).
Figure 5. In vivo effects of tumor vs stromal targeting by oncolytic uPAR retargeted measles viruses.
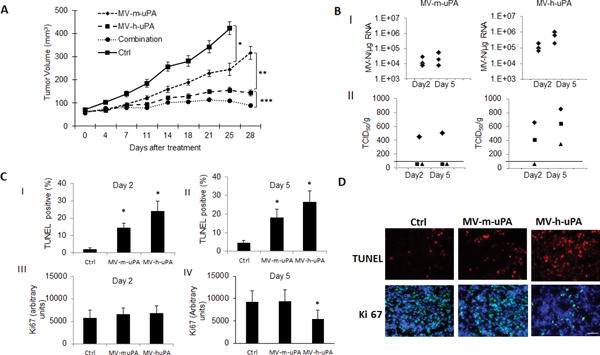
(A). MDA-MB-231 tumor bearing NOD/SCID mice were treated with either vehicle, MV-m-uPA, MV-h-uPA or both, and tumor volume was assessed as in methods (n=8 mice per group). (B). I. qRT-PCR analysis of MV-N mRNA at days 2 and 5 after virus treatment. II. Infectious virus titers from tumors after MV-m-uPA and MV-h-uPA administration. Viral titers are displayed as TCID50/gram of tissue. Line indicates lower limit of detection. Data points below the LLD represent samples with undetectable viral particles (C) Analysis of TUNEL–positive nuclei (C. I, II) and Ki 67 (C.III, IV) staining in control, MV-m-uPA and MV-h-uPA treated tumors at day 2 (I, III) and 5 (II, IV) after treatment. Bars represent averages +/− SEM of triplicate experiments (n=3 per group). (D). Representative pictures of TUNEL and Ki67 antibody staining of treated and untreated tumors from mice at day 5. Scale bar = 20 μm.
In correlative tumor studies (n=3 per group) performed at days 2 and 5 after treatment with MV-h-uPA and MV-m-uPA, viral RNA was detected in all treated mice at both time points (Fig. 5,B), with higher copy numbers observed in tumors treated with the human uPAR retargeted virus (Fig. 5. B. I). Likewise, titers of viable virus were higher in the MV-h-uPA treated group (Fig 5. B. II). More tumors treated with MV-h-uPA had viable viral particles recovered (2/3 at day 2 and 3/3 at day 5) compared to MV-m-uPA treated tumors (1/3 in both days 2 and 5).
Apoptosis (TUNEL) was significantly increased at days 2 (Fig. 5. C. I) and 5 (Fig. 5. C. II) in tumors treated with both MV-m-uPA and MV-h-uPA, with a higher percentage of TUNEL positive nuclei observed in tumors treated with the human retargeted virus. While tumor proliferation (Ki67) was not significantly altered by either virus at day 2, a significant decrease in Ki67 staining was observed in tumors treated by MV-h-uPA only at day 5 (Fig 5. C. IV, 5.D).
To further characterize MV-m-uPA distribution within the tumor, in situ hybridization was performed to co-localize viral (MV-N) and stromal (murine uPAR) RNA. Immunofluorescence was also performed to evaluate the distribution of MV-N in relation with murine tumor fibroblasts. As shown in figure 6 A (and Figure S-1), Viral RNA (MV-N, red) co-localized with murine uPAR RNA (green) in MV-m-uPA treated tumors, but not in controls. Immunofluorescence studies showed co-localization of MV-N protein in murine FAP+ and ER-TR7+ stromal cells (Fig. 6. B, C).
Figure 6. Stromal targeting by MV-m-uPA.
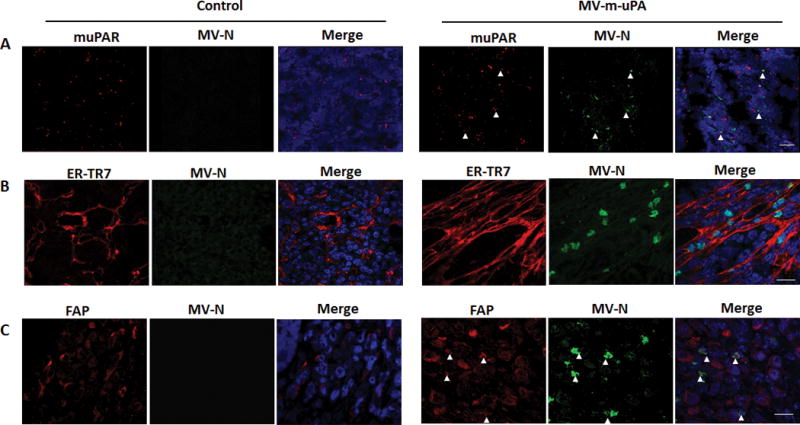
(A). In situ hybridization for mouse uPAR and MV-N RNA (RNAscope). Viral RNA (green) signals co-localized with murine uPAR RNA (red) signals in MV-m-uPA treated tumors, but not in controls at day 5 after virus treatment. Scale bar = 20 μm (B, C) Immunofluorescence staining for MV-N protein (green) and fibroblast marker ER-TR7 (B, red) and FAP (C, red) in untreated and treated mice. Tumor sections were processed as in methods. (Fig. C). Scale bar = 20 μm.
Effects of MV-m-uPA on murine and human gene expression
Analysis was performed on tumor RNA at days 2 and 5 after virus treatment, using the nCounter Pancancer pathways (human) and (murine) Pancancer immune profiling panels (which encompass murine inflammation and stromal related genes). Time dependent differential expression of murine and human genes were observed in tumors treated by MV-m-uPA. Figure 7 shows heat maps of the top 100 significantly (p ≤ 0.05) differentially expressed murine (Fig 7.A, B) and human (Fig. 7. C, D) genes, as well as fold change of the top 20 genes, comparing MV-m-uPA treated tumors vs controls. Pathway analysis of significantly differentially expressed (≥ 1.2 fold and p ≤ 0.05) murine (stromal) genes showed that MV-m-uPA significantly modulated genes associated with inflammatory responses, angiogenesis, as well as survival pathways (Table S-2 and S-3). Examples of differentially upregulated stromal genes at day 2 include Fez1[40, 41] and Pycard [42], which negatively regulate cell proliferation/differentiation and mediate inflammation induced apoptosis, respectively. Angiogenesis related mouse genes were found to be down regulated, such as DLL-4, Angiopoietin 2, PECAM and Tie-1 (Fig 7.A and Table S-2). Other significantly down regulated genes that play a role in regulation of stromal cell components include FOS, EGR2, CREB-5, IL-6, Map2K1, among others (table S-1). At day 5, genes such as CCL22 [43], TIMD4 [44] and CCL19 [45], associated with macrophage networks were significantly down regulated.
Figure 7. MV-m-uPA induced differential regulation of murine (stromal) and human (cancer) gene expression.
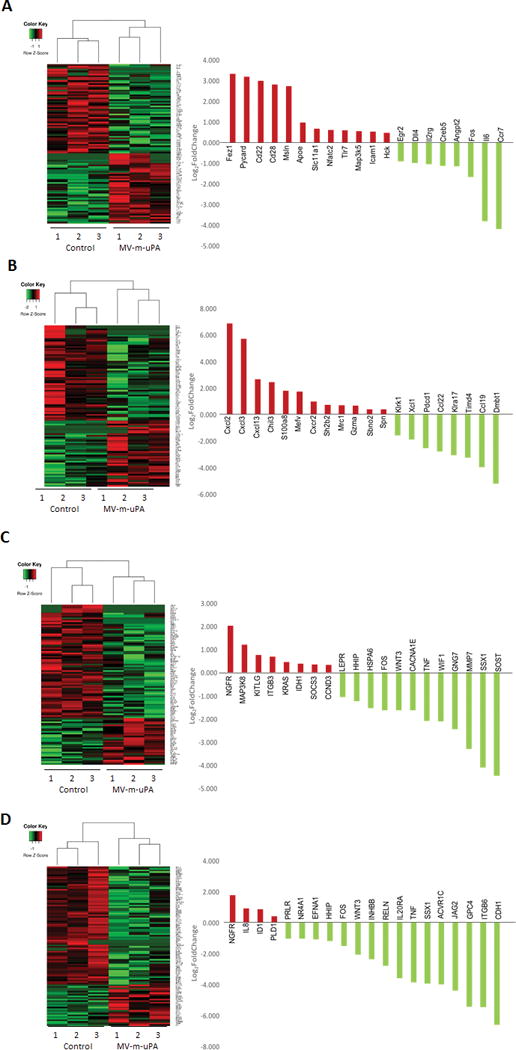
Heat map of expression of top 100 differentially expressed genes (left) and fold change (treated versus control) of top 20 genes (right) are shown for (A) murine genes measured at day 2, (B) murine genes measured at day 5, (C) human genes measured at day 2, and (D) human genes measured at day 5. Intensities of gene expression are row-standardized to reflect high (red) relative to low gene expression (green). Samples (columns) are segregated by unsupervised clustering.
Among the top differentially regulated human genes at day 2, down regulation of Wnt/β-catenin pathway genes (TCF-3, DKK1, WNT3 and WIF1) was observed, as well as down regulation of proliferation and survival genes, such as FOS, SSX1, TNF, leptin receptor, among others (Table S-4). NGFR (p75), which has been shown to function as a tumor suppressor in multiple cancers [46–48] was among the most upregulated human genes at days 2 and day 5. At day 5, significant down regulation of Wnt-β-catenin (GPC4, WNT 3, DKK1, WNT 7B), TGF beta (ACVR1C, INHBB, BMPR1B, SMAD3), JAK/Stat (JaK3, IL20RA), and Notch (JAG2) pathway genes was observed (figure 7. D, and Table S-5).
DISCUSSION
Oncolytic viral agents offer great hope as novel strategies for cancer treatment, both alone, or in combination with other treatment modalities. An important, but not well studied area in the field is the understanding of the biological effects of oncolytic viruses in the tumor microenvironment, which plays a critical role in cancer progression. While it has been previously shown that tumor-stromal cross-talk modulates the sensitivity of cancer cells to oncolytic virues [49], ours is the first report, to our knowledge, to investigate the direct effect(s) of a stromal selective oncolytic viral vector on in vivo tumor progression. The current study differs from previous publications in that other reports have investigated the additional direct or indirect effects of targeted or armed oncolytic viruses (which primarily infect cancer cells) on other stromal components, especially the tumor vasculature [50].
The novel findings presented in the current study include the demonstration of successful viral infection, replication and cytotoxicity in human cancer associated fibroblasts and mouse 3T3 cells, as well as successful fibroblast to tumor cell viral transfer. Importantly, 3T3 selective targeting by MV-m-uPA inhibited paracrine growth of (virus insensitive) cancer cells in co-cultures. These results, which indicate that stromal specific targeting by an oncolytic virus regulates cancer cell growth, were validated in vivo, where intravenous administration of the murine specific MV-m-uPA (which does not infect human cancer cells) led to significant tumor growth delay and improved survival. In vivo tumor targeting was demonstrated by detection of viable virus, viral RNA and viral protein in treated tumors. Targeting of murine stroma by MV-m-uPA was confirmed by demonstrating co-localization of MV and mouse specific uPAR (the target of MV-m-uPA) RNA by in situ hybridization of treated tumors, as well as by co-localization of fibroblast markers and MV-N in treated tumors (by immunofluorescence). The above findings, and the fact that MV-m-uPA specifically infects murine, and not human cells, lead us to conclude that the in vivo antitumor effects of MV-m-uPA in MDA-MB 231 xenografts are due to stromal selective targeting, and that murine tumor fibroblasts are a target of the virus. Previously, we have demonstrated tumor endothelial targeting by the uPAR retargeted virus after systemic administration [23].
Experiments comparing the effects of (murine) stromal versus human (cancer cell), versus combined targeting showed that targeting both tumor and stroma was associated with improved tumor control. This further validates the concept that stromal targeting represents an important strategy for biological therapies in general, and oncolytic viruses in particular. The improved effects by the human uPAR redirected virus compared to the murine targeted virus could -in part- be explained by the fact that in xenograft models, the (human) cancer cells are the predominant component of the tumor, while host stromal cells represent a smaller fraction, therefore reducing the reach and efficacy of the stromal targeted virus. This was supported by the observation of increased viable virus and increased viral RNA titers in tumors treated by the human uPAR retargeted virus than the murine retargeted virus. However, it also suggests that stromal targeting by itself (at least in this model) is not sufficient to achieve long term antitumor effects. The above results open the door for further research combining stromal targeted viruses with standard or experimental antitumor agents that target the cancer cell component for therapeutic gain.
Correlative tumor studies showed that the antitumor effects of the stromal targeted virus were associated with increased apoptosis, as shown by TUNEL assay. Tumor cell proliferation, as measured by Ki67 determination was significantly decreased only in tumors treated by the human, but not the mouse uPAR retargeted virus. Targeted analysis of gene expression showed that important murine stromal pathways were affected, including pathways associated with angiogenesis, inflammation, viral infection, as well as survival/cell death. This provides insight into the mechanisms explaining how in vivo specific targeting of the tumor stromal component induces tumor delaying effects. Specifically, the data suggest that endothelial and fibroblast targeting contribute to the in vivo effects. We cannot, however, rule out the possibility that other stromal cells that express uPAR, such as macrophages or other inflammatory cells may not play an important role in the in vivo observations. Studies elucidating the degree to which virus targeting of specific stromal components (fibroblasts, endothelial cells, macrophages) contribute to the in vivo effects observed in the current model, as well as characterizing the stromal cell-virus-tumor cell interactions are underway.
The observation that MV-murine-uPA indirectly regulates human cancer pathways, provides important knowledge into the molecular mechanisms explaining how virus mediated stromal targeting directly affects stromal components, and indirectly modulates tumor cell behavior, resulting in antitumor effects. Studies aimed at further characterizing the mechanisms of stromal targeting on WNT, Jak-STAT and NOTCH pathways in cancer cells are under way, and will provide important information into the mechanisms of tumor stromal interactions that go beyond the oncolytic virus field.
In summary, this report demonstrates for the first time that in vivo stromal selective targeting by an oncolytic virus, after systemic administration, is feasible, associated with differential regulation of stromal associated genes, modulation of tumor-stromal interactions, and in vivo tumor delaying effects. The above results validate the concept of uPAR as a biologically relevant target for oncolytic virotherapy and the potential of oncolytic viruses as antistromal agents, both alone, and in combination with other antitumor agents.
Supplementary Material
Implications.
The current report demonstrates for the first time the biological, in vitro and in vivo anti-tumor and molecular effects of stromal selective targeting by an oncolytic virus.
Acknowledgments
This work was supported by a research grant from the National Cancer Institute (1R01CA149659-01 to JRM, JZ), and by the Sylvester Comprehensive Cancer Center (JRM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Mbeunkui F, Johann DJ., Jr Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol. 2009;63(4):571–82. doi: 10.1007/s00280-008-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7(6):513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa H, et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23(44):7366–77. doi: 10.1038/sj.onc.1208013. [DOI] [PubMed] [Google Scholar]
- 4.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes FG, et al. Tumor angiogenesis and lymphangiogenesis: tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2013;92(2):101–7. doi: 10.1016/j.lfs.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33(17):1974–82. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2015.17. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, et al. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed Pharmacother. 2015;74:215–21. doi: 10.1016/j.biopha.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, et al. Inhibition of tumor growth and metastasis by photoimmunotherapy targeting tumor-associated macrophage in a sorafenib-resistant tumor model. Biomaterials. 2016;84:1–12. doi: 10.1016/j.biomaterials.2016.01.027. [DOI] [PubMed] [Google Scholar]
- 11.Ye C, et al. Targeting Ornithine Decarboxylase by alpha-Difluoromethylornithine Inhibits Tumor Growth by Impairing Myeloid-Derived Suppressor Cells. J Immunol. 2016;196(2):915–23. doi: 10.4049/jimmunol.1500729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara M, et al. Suppression of Cancer-associated Fibroblasts and Endothelial Cells by Itraconazole in Bevacizumab-resistant Gastrointestinal Cancer. Anticancer Res. 2016;36(1):169–77. [PubMed] [Google Scholar]
- 13.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161:355–64. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura T, Russell SJ. Oncolytic measles viruses for cancer therapy. Expert Opin Biol Ther. 2004;4(10):1685–92. doi: 10.1517/14712598.4.10.1685. [DOI] [PubMed] [Google Scholar]
- 15.Galanis E, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70(3):875–82. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell SJ, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic proceedings. 2014;89(7):926–33. doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 18.Jo M, et al. The urokinase receptor promotes cancer metastasis independently of urokinase-type plasminogen activator in mice. The American journal of pathology. 2009;175(1):190–200. doi: 10.2353/ajpath.2009.081053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, et al. Endothelial and macrophage upregulation of urokinase receptor expression in human renal cell carcinoma. Hum Pathol. 1997;28(2):206–13. doi: 10.1016/s0046-8177(97)90108-8. [DOI] [PubMed] [Google Scholar]
- 20.Hildenbrand R, Schaaf A. The urokinase-system in tumor tissue stroma of the breast and breast cancer cell invasion. Int J Oncol. 2009;34(1):15–23. [PubMed] [Google Scholar]
- 21.He Y, et al. Interaction between cancer cells and stromal fibroblasts is required for activation of the uPAR-uPA-MMP-2 cascade in pancreatic cancer metastasis. Clin Cancer Res. 2007;13(11):3115–24. doi: 10.1158/1078-0432.CCR-06-2088. [DOI] [PubMed] [Google Scholar]
- 22.Boonstra MC, et al. Expression of uPAR in tumor-associated stromal cells is associated with colorectal cancer patient prognosis: a TMA study. BMC Cancer. 2014;14:269. doi: 10.1186/1471-2407-14-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jing Y, et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69(4):1459–68. doi: 10.1158/0008-5472.CAN-08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing Y, et al. In vivo anti-metastatic effects of uPAR retargeted measles virus in syngeneic and xenograft models of mammary cancer. Breast Cancer Res Treat. 2015;149(1):99–108. doi: 10.1007/s10549-014-3236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jing Y, et al. Role of plasminogen activator inhibitor-1 in urokinase’s paradoxical in vivo tumor suppressing or promoting effects. Mol Cancer Res. 2012;10(10):1271–81. doi: 10.1158/1541-7786.MCR-12-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jing Y, et al. In vivo safety, biodistribution and antitumor effects of uPAR retargeted oncolytic measles virus in syngeneic cancer models. Gene therapy. 2014;21(3):289–97. doi: 10.1038/gt.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura T, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–14. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 28.Drews-Elger K, et al. Primary breast tumor-derived cellular models: characterization of tumorigenic, metastatic, and cancer-associated fibroblasts in dissociated tumor (DT) cultures. Breast Cancer Res Treat. 2014;144(3):503–17. doi: 10.1007/s10549-014-2887-9. [DOI] [PubMed] [Google Scholar]
- 29.Palmowski MJ, et al. Intravenous injection of a lentiviral vector encoding NY-ESO-1 induces an effective CTL response. J Immunol. 2004;172(3):1582–7. doi: 10.4049/jimmunol.172.3.1582. [DOI] [PubMed] [Google Scholar]
- 30.Demaison C, et al. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther. 2002;13(7):803–13. doi: 10.1089/10430340252898984. [DOI] [PubMed] [Google Scholar]
- 31.Su G, et al. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. 2007;282(20):14906–15. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- 32.Hadac EM, et al. Reengineering paramyxovirus tropism. Virology. 2004;329(2):217–25. doi: 10.1016/j.virol.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 33.Waggott D, et al. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics. 2012;28(11):1546–8. doi: 10.1093/bioinformatics/bts188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57(1):289–300. [Google Scholar]
- 36.Sliutz G, et al. Quantification of uPA receptor expression in human breast cancer cell lines by cRT-PCR. Breast cancer research and treatment. 1996;40(3):257–63. doi: 10.1007/BF01806814. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen DH, I, Hussaini M, Gonias SL. Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. The Journal of biological chemistry. 1998;273(14):8502–7. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- 38.Dublin E, et al. Immunohistochemical expression of uPA, uPAR, and PAI-1 in breast carcinoma. Fibroblastic expression has strong associations with tumor pathology. Am J Pathol. 2000;157(4):1219–27. doi: 10.1016/S0002-9440(10)64637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432(7015):332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishii H, et al. FEZ1/LZTS1 gene at 8p22 suppresses cancer cell growth and regulates mitosis. Proc Natl Acad Sci U S A. 2001;98(18):10374–9. doi: 10.1073/pnas.181222898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vecchione A, et al. Fez1/Lzts1 absence impairs Cdk1/Cdc25C interaction during mitosis and predisposes mice to cancer development. Cancer Cell. 2007;11(3):275–89. doi: 10.1016/j.ccr.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mhyre AJ, et al. Stroma-dependent apoptosis in clonal hematopoietic precursors correlates with expression of PYCARD. Blood. 2009;113(3):649–58. doi: 10.1182/blood-2008-04-152686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wertel I, et al. Macrophage-derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumour Biol. 2015;36(6):4811–7. doi: 10.1007/s13277-015-3133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finkernagel F, et al. The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget. 2016 doi: 10.18632/oncotarget.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xuan W, et al. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J Leukoc Biol. 2015;97(1):61–9. doi: 10.1189/jlb.1A0314-170R. [DOI] [PubMed] [Google Scholar]
- 46.Jin H, et al. p75 neurotrophin receptor inhibits invasion and metastasis of gastric cancer. Mol Cancer Res. 2007;5(5):423–33. doi: 10.1158/1541-7786.MCR-06-0407. [DOI] [PubMed] [Google Scholar]
- 47.Khwaja F, et al. The p75(NTR) tumor suppressor induces cell cycle arrest facilitating caspase mediated apoptosis in prostate tumor cells. Biochem Biophys Res Commun. 2006;341(4):1184–92. doi: 10.1016/j.bbrc.2006.01.073. [DOI] [PubMed] [Google Scholar]
- 48.Yuanlong H, et al. The inhibitory effect of p75 neurotrophin receptor on growth of human hepatocellular carcinoma cells. Cancer Lett. 2008;268(1):110–9. doi: 10.1016/j.canlet.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 49.Ilkow CS, et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015;21(5):530–6. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 50.Toro Bejarano M, Merchan JR. Targeting tumor vasculature through oncolytic virotherapy: recent advances. Oncolytic Virother. 2015;4:169–81. doi: 10.2147/OV.S66045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.