

Abstract
Objective
Patients with intracerebral hemorrhage (ICH) may elaborate varying degrees of perihematomal edema (PHE), requiring closer monitoring and a higher intensity of treatment. Here, we explore whether the soluble form of CD163, a scavenger receptor responsible for hemoglobin sequestration, can serve as a prognostic biomarker of PHE development and poor outcome after ICH.
Methods
Our study cohort was comprised of 51 primary age‐ and sex‐matched ICH patients with moderate‐sized, hypertensive deep hemorrhages. Patients were part of a prospective ICH registry cataloguing admission data along with functional outcomes. We measured sCD163 levels in serial serum and cerebrospinal fluid (CSF) samples obtained at prespecified timepoints. Descriptive statistics, including a generalized estimating equation for longitudinal data, were used to analyze sCD163 in relation to ICH outcomes.
Results
Acute serum sCD163 (<48 h postictus) was significantly elevated in ICH patients compared to both acute neurological event controls (P = <0.001) and healthy controls (P = 0.003). As predicted, acute serum sCD163 levels were significantly associated with both hematoma volume expansion (P = 0.009) and PHE expansion (P = 0.002). Further examination determined that patients with high PHE expansion had poorer modified Rankin Scale scores at discharge (P = 0.024), and circulating sCD163 levels were found to be significantly lower in patients with high‐level PHE expansion.
Interpretation
Acute sCD163 levels may be a useful biomarker for the acute identification of patients at risk for hematoma expansion, perihematomal edema expansion and poorer short‐term outcomes.
Introduction
Spontaneous intracerebral hemorrhage (ICH) is a devastating disease, with 30‐day mortality rates as high as 50%.1 Of the patients who survive, 80% will be unable to function independently at 6 months poststroke.2, 3 The initial tissue injury of ICH activates numerous secondary injury pathways, including the release of toxic hemolysis products, enhanced oxidative stress, and robust proinflammatory responses. On a macroscopic level, these processes can result in blood‐brain‐barrier (BBB) disruption, tissue shifts and perihematomal edema (PHE) formation that can result in further neurological deterioration.4, 5, 6 As these secondary injury pathways can evolve over hours to days, they represent attractive targets for medical intervention. Importantly, it remains largely unknown why patients with similar initial injuries develop variable degrees of PHE expansion, which has been associated with poorer functional outcomes.7, 8 The discovery of novel biomarkers to help identify those patients predisposed to greater secondary injury and PHE expansion is a key step in potentially modifying functional outcome after ICH.9
CD163 is a scavenger receptor that facilitates binding and endocytosis of hemoglobin‐haptoglobin complexes by monocytic cells, decreasing the cytotoxic load of free hemoglobin that occurs after tissue hemorrhage.10, 11 Recent evidence has shown that CD163 is also upregulated acutely in neurons following ICH both in vivo and in vitro.12 By sequestering toxic hemolysis products, CD163 may play a key role in limiting inflammation and suppressing secondary injury following hemorrhagic injury.13, 14 CD163 expression by glia and infiltrating immune cells in perihematomal tissue was recently demonstrated in ICH patients, with expression peaking 72 h poststroke.15 Unfortunately, histological evaluation of CD163 in brain tissue is not a practical tool for clinical prediction of ICH outcome. However, CD163 has been shown to undergo cleavage from the cell surface, resulting in a soluble form that circulates in plasma.16 Therefore, we hypothesized that this soluble form of CD163 (sCD163) may represent a promising prognostic marker for PHE expansion and functional outcome in ICH.
In this study, we used a well‐characterized cohort of age‐ and sex‐matched patients with similar ICH volume, location, and etiology to (1) measure the temporal changes in the levels of circulating and cerebrospinal (CSF) sCD163 in ICH patients; and (2) investigate whether sCD163 was associated with the degree of PHE expansion and functional outcome after ICH.
Methods
Study population
Our study cohort was composed of 51 primary ICH patients with relatively homogeneous ICH characteristics. All hemorrhages were “deep” supratentorial hemorrhages originating from the basal ganglia or thalamus, and hypertensive in etiology as per stroke specialist consensus, including an independent review by an experienced neurologist (NJE). Coagulopathic patients, patients with subacute hematoma expansion (defined as continued hematoma volume increases greater than 48 h postictus), and patients with secondary ICH were excluded. All patients were part of a prospective ICH registry whereby relevant laboratory and radiologic data throughout the inpatient stay was catalogued and assessed. Patients in the registry also had short‐ and long‐term functional neurologic outcome previously validated telephone assessments performed by research assistants trained in assessment of modified Rankin Scale Score, Barthel Index, GOSE, and Euroqol. The registry enrolled patients from 2014 through 2016 at a comprehensive stroke center (University of Texas Health Science Center at Houston).
Biospecimen collection
Serum and CSF samples were obtained from our study cohort at prespecified timepoints postictus: timepoint 1 (0–24 h), timepoint 2 (24–48 h); timepoint 3 (3–5 days), timepoint 4 (6–8 days) and timepoint (10 days). Serial samples were collected from each patient when possible, for a total of 136 serum and 38 CSF samples. In order to minimize timing bias, all biosamples were timed from the onset of ICH ictus and collected by a biospecimen collection team who were blinded to the clinical status of the patient. Samples were processed within 1 h of collection and stored at −80 degrees Celsius for future use. CSF was acquired in ICH patients with external ventricular drains (EVD) inserted into the lateral ventricle(s), as deemed necessary by the clinical team. Each CSF specimen was drawn from the buretrol of the EVD using sterile technique. Patients or their families were consented for both sample collection and involvement in the data registry. All patients in this study reached peak hematoma volumes within 48 h of ictus. The study was approved by the Institutional Review Board at the University of Texas Health Science Center at Houston.
Healthy controls
To compare sCD163 expression in ICH patients to relatively healthy patients, we obtained serum samples from “healthy control” patients (n = 10 patients). These patients were patients admitted to the neurosurgical service for elective trigeminal nerve decompression or pineal cyst resection.
Acute neurological event controls
To compare sCD163 expression in ICH patients, we obtained serum samples from transient ischemic attack (TIA) patients to serve as acute neurological controls (n = 24 patients). These patients were admitted for focal, stroke‐like symptoms, with neurological injury subsequently ruled out based on negative MRI imaging. TIA sample collection was approved by the Institutional Review Board at Hartford Hospital and conducted at Hartford Hospital in Hartford, Connecticut.
Radiologic/clinical outcomes
Our primary radiologic outcome was perihematomal edema formation, as quantified via computer‐based analysis. Every head CT scan obtained as a part of routine clinical care throughout the inpatient stay was assessed using the Medical Image Processing, Analysis, and Visualization (MIPAV) software; imaging analysis was performed by two independent researchers (NJE and DS). Patients were subsequently divided into PHE‐Low versus PHE‐High subgroups based on their peak PHE volume. Previous studies have correlated a greater than 10 cm3 increase in absolute PHE with poorer functional outcome.17 Similarly, we defined our PHE‐High population as those patients whose peak PHE was >10 cm3 higher than their initial PHE estimate, whereas the PHE‐Low population had a less than 10 cm3 increase in edema. Patients with borderline PHE‐expansion levels were excluded from this analysis, resulting in n = 47.
Our primary outcome was the modified Rankin Scale (mRS) score, as assessed at discharge and at 90 days post‐ICH. Outcome assessments were performed prospectively as a part of the ICH registry and the researcher conducting the mRS evaluations (DS) was blinded to the results of sCD163 performed on patient biosamples.
Soluble CD163 and albumin measurement
Serum and CSF levels of soluble CD163 were assessed by a blinded investigator (MR) via enzyme‐linked immunosorbent assay (sCD163/M130 human ELISA, ThermoFisher Scientific). Serum and CSF albumin levels were also quantified by ELISA (Albumin Human ELISA Kit, Abcam). Samples were thawed and diluted per manufacturer recommendations before ELISA assay and analysis on a Perkin Elmer Enspire Multimode Plate Reader.
Intrathecal index quantification
The intrathecal index was determined by dividing the CSF to serum ratio of sCD163 (Q sCD163) by the CSF to serum ratio of albumin (Q Albumin), as previously described.18
Statistical analysis
Descriptive statistics were provided for demographic and clinical variables for all ICH patients (Table 1). Peak serum levels of sCD163 within 48 h of ICH onset were plotted and compared between ICH and acute neurological control (TIA) patients by Wilcoxon rank‐sum test (Fig. 1). Serum and CSF levels of sCD163 in ICH patients were plotted and modeled on time by the generalized estimating equation (GEE) method (Fig. 2). For intrathecal experiments, sign test was used to compare the CSF/Serum Ratio of sCD163 to the CSF/Serum Ratio of albumin (Fig. 3). Spearman correlation method or Wilcoxon rank‐sum test were used, for continuous or dichotomous variables, respectively, to evaluate the association of acute peak serum sCD163 levels within 48 h and clinic‐radiologic outcomes after ICH (Table 2). Descriptive statistics were provided for demographic and clinical variables for PHE‐Low and PHE‐High patients. Two‐sample t test or Wilcoxon rank‐sum test was used to compare continuous variables between the two groups, and the chi‐square test or Fisher's exact test was used for categorical variables (Table 3). The GEE method was used to compare the longitudinal serum and CSF sCD163 levels between PHE‐low and PHE‐high patients, using PHE group, time, and their interactions in the model (Fig. 4A, B). All statistical analyses were performed in SAS 9.4 software (Cary, NC).
Table 1.
Overall patient demographics table
| Variable | Measure (n = 51) |
|---|---|
| Age (mean ± SD) | 56.51 ± 12.98 |
| Gender (% male) | 37 (74%) |
| Race | |
| Black | 21 (41.18%) |
| White | 19 (37.25%) |
| Asian | 4 (7.84%) |
| Hispanic/Other | 7 (13.73%) |
| Hypertension | 46 (90.20%) |
| Diabetes mellitus | 12 (23.53%) |
| Ischemic stroke | 2 (3.92%) |
| Chronic kidney disease | 3 (5.88%) |
| Statin use | 13 (26.53%) |
| Admission GCS (Mean ± SD) | 11.06 ± 3.63 |
| ICH Score | |
| 0 | 9 (17.65%) |
| 1 | 17 (33.33%) |
| 2 | 19 (37.25%) |
|
3 4 |
5 (9.80%) 1 (1.96%) |
| Baseline glucose | 167.37 ± 63.71 |
| Admission SBP (Mean ± SD) | 197.63 ± 39.50 |
| Admission DBP (Mean ± SD) | 111.46 ± 27.56 |
| Hematoma expansion (%) | 14 (28%) |
| Peak hematoma volume (Mean ± SD) | 21.04 ± 13.61 |
| Peak PHE (Mean ± SD) | 17.37 ± 14.75 |
| IVH (%) | 33 (64.71%) |
| IVH Score (Mean ± SD) | 7.92 ± 7.35 |
| END (%) | 16 (31.37%) |
| DND (%) | 8 (15.69%) |
| mRS at discharge (Mean ± SD) | 4.16 ± 1.07 |
| mRS at 90 days (Mean ± SD) | 3.68 ± 1.71 |
SBP, Systolic Blood Pressure; DBP, Diastolic Blood Pressure; GCS, Glasgow Coma Scale; ICH, Intracerebral Hemorrhage; PHE, Perihematomal Edema; IVH, Intraventricular Hemorrhage; END, Early Neurological Deterioration; DND, Delayed Neurological Deterioration.
Figure 1.

Peak serum levels (ng/ml) of sCD163 in intracerebral hemorrhage patients (n = 46) with sCD163 levels measured in the first 48 h versus transient ischemic attack (TIA) acute neurological controls (n = 24). ****p < 0.0001.
Figure 2.
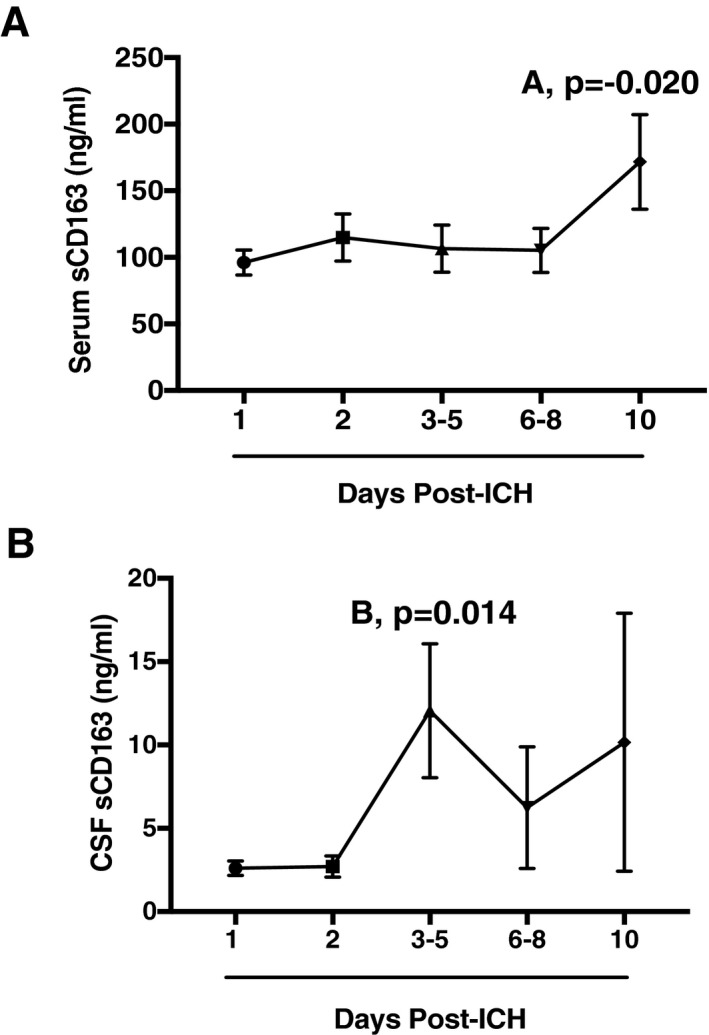
(A) Serum levels of sCD163 in ICH patients over time: 1 day (n = 46), 2 days (n = 27), 3–5 days (n = 29), 6–8 days (n = 18), and 10 days (n = 15). (B) CSF levels of sCD163 in ICH patients over time: 1 day (n = 11), 2 days (n = 8), 3–5 days (n = 11), 6–8 days (n = 5), and 10 days (n = 3). (A)*p = −0.020, (B)*p = 0.014.
Figure 3.
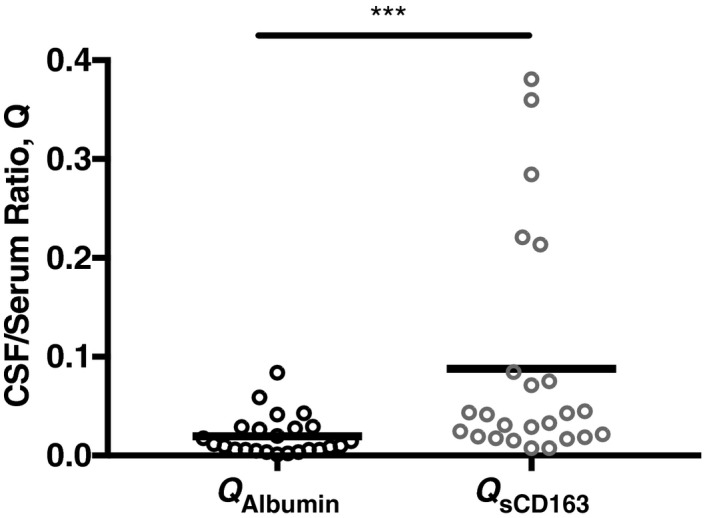
CSF/serum ratio of albumin (QA lbumin) and of sCD163 (Qs CD 163) in ICH patients (n = 25). p = 0.004.
Table 2.
Association of acute peak serum sCD163 levels within 48 h and clinic‐radiologic outcomes after ICH
| Variable | Peak Serum sCD163 |
|---|---|
| Hematoma volume increase | P = 0.009 |
| PHE volume increase | P = 0.002 |
| Relative PHE | P = 0.452 |
| END | P = 0.380 |
| DND | P = 0.890 |
| mRS at discharge | P = 0.126 |
| Discharge disposition | P = 0.072 |
| Mortality at discharge | P = 0.268 |
| mRS at 90 Days | P = 0.388 |
| Mortality at 90 days | P = 0.892 |
SBP, Systolic Blood Pressure; DBP, Diastolic Blood Pressure; GCS, Glasgow Coma Scale; ICH, Intracerebral Hemorrhage; PHE, Perihematomal Edema; IVH, Intraventricular Hemorrhage; END, Early Neurological Deterioration; DND, Delayed Neurological Deterioration.
Table 3.
Clinical characteristics of PHE‐Low versus PHE‐High patients
| Variable | PHE‐High (n = 20) | PHE‐Low (n = 27) | P‐value |
|---|---|---|---|
| Age (years) | 57.0 ± 14.0 | 55.2 ± 12.8 | 0.789 |
| Gender (Male) | 15 (78.9%) | 19 (70.4%) | 0.735 |
| Race | |||
| Black | 9 (45.0%) | 11 (40.7%) | 0.969 |
| Caucasian | 6 (30.0%) | 10 (37.0%) | |
| Asian | 2 (10.0%) | 2 (7.4%) | |
| Hispanic/Other | 3 (15.0%) | 4 (14.8%) | |
| Hypertension | 18 (90.0%) | 25 (92.6%) | 1.000 |
| Diabetes mellitus | 4 (20.0%) | 6 (22.2%) | 1.000 |
| Ischemic stroke | 0 (0.0%) | 1 (3.7%) | 1.000 |
| Chronic kidney disease | 0 (0.0%) | 3 (11.1%) | 0.251 |
| Statin use | 5 (25.0%) | 7 (25.9%) | 1.000 |
| Admission GCS (Mean ± SD) | 10.8 ± 2.9 | 11.9 ± 3.8 | 0.137 |
| ICH Score | |||
| 0 | 3 (15.0%) | 6 (22.2%) | 0.174 |
| 1 | 4 (20.0%) | 12 (44.4%) | |
| 2 | 11 (55.0%) | 7 (25.9%) | |
|
3 4 |
2 (10.0%) | 2 (7.4%) | |
| Baseline glucose | 180.5 ± 67.9 | 157.1 ± 61.9 | 0.223 |
| Admission SBP (Mean ± SD) | 199.1 ± 34.0 | 201.5 ± 39.0 | 0.623 |
| Admission DBP (Mean ± SD) | 109.9 ± 26.5 | 114.5 ± 28.0 | 0.475 |
| Hematoma Expansion (%) | 7 (35.0%) | 5 (19.2%) | 0.314 |
| Peak Hematoma Volume (Mean ± SD) | 26.2 ± 15.2 | 16.7 ± 10.6 | 0.029 |
| Peak PHE (Mean ± SD) | 27.3 ± 16.1 | 8.7 ± 4.9 | <.0001 |
| IVH (%) | 12 (60.0%) | 18 (66.7%) | 0.638 |
| IVH Score (Mean ± SD) | 7.6 ± 7.8 | 8.1 ± 7.2 | 0.742 |
| END (%) | 8 (40.0%) | 6 (22.2%) | 0.188 |
| DND (%) | 5 (25.0%) | 2 (7.4%) | 0.118 |
| mRS at discharge (Mean ± SD) | 4.6 ± 0.9 | 3.8 ± 1.1 | 0.024 |
| mRS at 90 days (Mean ± SD) | 3.8 ± 1.6 | 3.5 ± 1.8 | 0.721 |
SBP, Systolic Blood Pressure; DBP, Diastolic Blood Pressure; GCS, Glasgow Coma Scale; ICH, Intracerebral Hemorrhage; PHE, Perihematomal Edema; IVH, Intraventricular Hemorrhage; END, Early Neurological Deterioration; DND, Delayed Neurological Deterioration.
Figure 4.
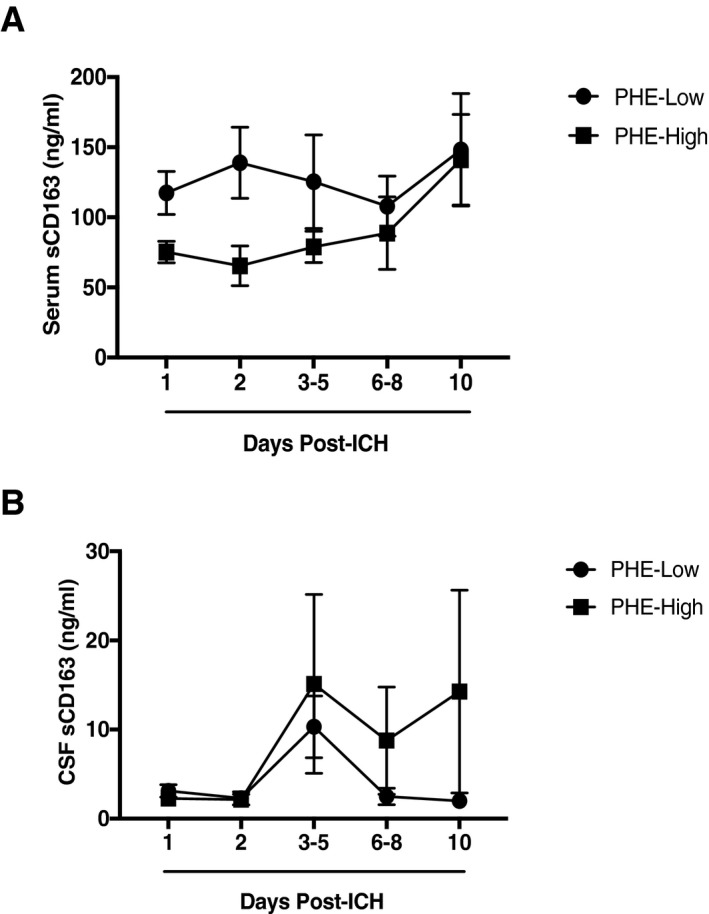
(A) Serum sCD163 levels in PHE‐Low as compared to PHE‐High patients. (B) CSF sCD163 levels in PHE‐Low as compared to PHE‐High patients.
Results
sCD163 is elevated in the serum of ICH patients compared to healthy controls
To begin, we investigated the levels of sCD163 in the serum of patients after ICH (for cohort demographics, see Table 1). ICH patients had significantly higher peak serum levels of sCD163 ((106.37 ± 76.39 ng/mL) vs. TIA controls (34.65 ± 23.5 ng/ml) within 48 h of ICH (P = >0.0001) (Fig. 1). Importantly, no sex effect was seen on serum sCD163 levels in either the ICH patients (P = 0.421) or the TIA patients (P = 0.546). Serum levels of sCD163 were also analyzed in healthy control patients; these patients were also found to have significantly lower levels of sCD163 (mean = 46.24 ± 20.27 ng/mL) than ICH patients (mean = 106.37 ± 35.22 ng/mL), P = .0003. sCD163 levels in ICH patients rose over time, with significant elevations of serum sCD163 from baseline seen at 10 days in ICH patients (P = 0.020) (Fig. 2 A). sCD163 in the CSF also increased over time, peaking significantly at 3–5 days (P = 0.014) (Fig. 2B).
sCD163 is synthesized intrathecally in ICH patients, accumulating during the subacute period
We then explored whether the sCD163 measured in the CSF represented intrathecal synthesis of the protein versus accumulation of blood‐derived sCD163 via a disrupted BBB. Using matched serum and CSF values within ICH patients, we found that the CSF to serum ratio of sCD163 (QsCD163) was significantly higher than that of albumin (QAlbumin), (P = 0.004; Fig. 3). Then, utilizing a previously described technique (see Methods), we compared the intrathecal ratio of sCD163 (QsCD163) and intrathecal ratio of albumin (QAlbumin) to calculate the intrathecal index for each patient. Across the ICH cohort, the average intrathecal index for sCD163 was 13, indicating that only 7% of the CSF sCD163 was derived from the circulation, whereas greater than 93% was produced intrathecally.
Low early serum sCD163 levels are associated with hematoma and edema expansion after ICH
After characterizing sCD163 in our ICH patients, we then examined whether early levels of sCD163 were predictive of ICH progression and/or patient outcome. Specifically, we assessed whether serum sCD163 measurements within 48 h were associated with hematoma expansion, perihematomal edema, early or delayed neurological deterioration, mRS and mortality at discharge and at 90 days, and discharge disposition (Table 2). Of note, all patients within our study reached peak hematoma volumes prior to 48 h from the time of onset. Peak serum sCD163 levels within 48 h of ICH were significantly associated with both hematoma volume increase (P = 0.009) and PHE volume increase (P = 0.002). While there was a trend toward an association of peak serum sCD163 and discharge disposition, this did not reach statistical significance.
sCD163 patterns are unique in patients who develop high versus low edema
Given the significant association of acute serum sCD163 with PHE, we further investigated the relationship between serum and CSF sCD163 levels and the development of PHE in our ICH patients. Specifically, our ICH cohort was divided into PHE‐High patients and PHE‐Low patients (see Methods). These patients were age‐and sex‐matched, with all patients having similar initial hemorrhage volume and location (ganglionic or thalamic). An overall comparison of the PHE‐High versus ‐Low patients is summarized in Table 3. PHE‐High patients were found to have a mean mRS at discharge of 4.6, versus 3.8 in the PHE‐Low patients (P = 0.0243), though this relationship was not seen at 90 days.
We then compared sCD163 levels over time in the PHE‐Low versus PHE‐High patients. Overall, serum sCD163 was significantly higher in the PHE‐low group than in the PHE‐high patients over time (P = .0119) (Fig. 4A). No significant overall differences were seen in CSF sCD163 in PHE‐low versus PHE‐high patients (Fig. 4B).
Discussion
This is the first study to describe the temporal profile of soluble CD163 in the serum and CSF of ICH patients across both acute and subacute phases. In comparison to both acute neurological event controls and healthy controls, early (<48 h) serum levels of sCD163 were significantly elevated in ICH patients. Serum sCD163 levels in ICH patients remained elevated over time, peaking significantly from baseline values at Day 10 post‐ICH. An increase in circulating sCD163 has been previously described in many acute inflammatory conditions, including sepsis and acute coronary syndrome.19, 20 Ectodomain shedding of soluble CD163 from the cell surface of monocytic cells is believed to be the direct result of upregulated CD163 and TNF‐α expression by macrophages.21 While TNF‐α is fairly rapidly cleared, soluble CD163 may be a long‐lived surrogate marker for macrophage activation following tissue injury.16 Aside from the utility of sCD163 as a biomarker of inflammation, the biological function of the soluble form of CD163 is not yet well established, although some studies suggest that sCD163 retains the capacity to bind haptoglobin‐hemoglobin complexes and exert anti‐inflammatory effects.22, 23
Interestingly, sCD163 levels in the CSF became significantly elevated from baseline at 3–5 days post‐ICH, revealing a differential, delayed pattern compared to sCD163 levels in serum. Our findings demonstrated that the intrathecal index of sCD163 to albumin in the CSF was 13, suggesting that the vast majority of sCD163 detected in the CSF was largely intrathecally derived. These studies are consistent with previous work that has shown that CD163 expression increases over time within the ICH‐affected brains of both human patients and rodent models.23
After determining that circulating levels of sCD163 are significantly upregulated in ICH patients compared to controls, we then conducted further studies to examine the relationship of acute (<48 h) levels of sCD163 with ICH outcomes. We demonstrated lower peak serum sCD163 levels within 48 h of ICH onset were significantly associated with both hematoma volume expansion and PHE expansion. Interestingly, there was a trend toward an association between low acute serum sCD163 levels and poorer mRS and disposition at discharge. The association between low sCD163 and functional clinical outcome may not have reached statistical significance due to (1) relatively small patient numbers and (2) the examination of a relatively homogenous group of ICH patients, but the trend certainly warrants further examination of sCD163 as a potential novel biomarker for functional outcome.
As numerous preclinical studies have implicated perihematomal edema as a marker of secondary tissue injury and a driver of poor ICH outcome, the ultimate goal of our study was to examine the association of sCD163 levels and PHE expansion. To this end, our ICH cohort was composed of relatively homogeneous patients with deep hypertensive hemorrhages of moderate size and similar degrees of IVH. Within this homogeneous cohort, serum sCD163 was found to be predictive of whether a patient would experience a large or small amount of PHE expansion, despite the homogeneity of our PHE‐High to PHE‐low groups (no significant difference in age, gender, race, co‐morbidities, ICH score, IVH or IVH score). In addition, PHE‐High patients had significantly poorer discharge mRS, supporting previous findings that PHE expansion may indeed be associated with poor outcome.5
The biologic mechanisms underlying the association of low acute sCD163 levels with poor outcome, whether radiologic (PHE) or clinical (mRS), are unclear. CD163 functions as a hemoglobin scavenger receptor,24 and the association of serum sCD163 with both hematoma and edema expansion may be a biologic consequence of the role of CD163 in hematoma handling. For instance, patients with naturally high levels of macrophage/microglial CD163 may have faster rates of hematoma resorption, and/or less neuroinflammation due to rapid sequestration of toxic hemoglobin. In various experimental models of ICH, augmenting hematoma clearance or neutralizing hemoglobin toxicity generally result in less PHE, reduced tissue injury, and improved functional performance in the animals.4, 5, 25, 26
It is interesting to note that the PHE‐Low patients in our study had significantly higher serum sCD163 levels, particularly within the first several days post‐ICH. Furthermore, this subgroup had a larger proportion of patients with favorable outcomes at discharge. Multiple isoforms of CD163 have been seen in humans, suggesting that differences in CD163 expression and shedding after injury may be due to genetic differences between patients.27 However, further experimental animal and clinical studies are needed to delineate the genetic components of the CD163 pathway, and the effect of CD163 genotype on PHE development and outcome after ICH.
There are several limitations to this study. First, the sample size was small, due to our desire to focus on a highly homogeneous patient population and increase the overall number of samples by collecting serial bio‐specimens from each patient. However, this work should be repeated with a larger and more diverse ICH patient cohort. Second, levels of sCD163 during the chronic recovery phase (weeks to months poststroke) need to be assessed whether determine whether serum sCD163 may also be useful as a biomarker for long‐term functional recovery in ICH patients. Finally, the pathophysiologic role of sCD163 in ICH, including the evolution of PHE, remains unclear. We believe the sequestration of toxic blood products via CD163/sCD163 contributes to hematoma detoxification and resolution, thereby minimizing secondary processes such as PHE. The mechanism underlying the association between serum and reduced secondary injury like PHE expansion merits further investigation in animal models.
In conclusion, we have demonstrated that acute soluble CD163 levels may be a useful biomarker for the identification of patients who are at risk of both hematoma expansion and perihematomal edema expansion. Further studies to address correlation with long‐term functional recovery and to determine the biologic significance of the CD163 pathway are needed, as sCD163 may represent a promising future therapeutic target in ICH as both a prognostic biomarker and potential therapeutic target.
Conflict of Interest
The authors of this publication declare that they have no conflict of interest.
Acknowledgments
The Russell and Diana Hawkins Family Foundation Discovery Fellowships to the Graduate School at UTHealth (to MAR), the National Institutes of Health 1F30NS098628 (to MAR), and the National Institutes of Health 5R01NS094543‐02 (to LDM).
[Correction added on 7 November 2017 after first online publication: The name of the third author was changed from Louise Atajada to Louise Atadja.]
Funding Statement
This work was funded by Russell and Diana Hawkins Family Foundation grant ; National Institutes of Health grants 1F30NS098628 and 5R01NS094543‐02.
References
- 1. Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin 2008;26:871–895, vii. [DOI] [PubMed] [Google Scholar]
- 2. Agnihotri S, Czap A, Staff I, et al. Peripheral leukocyte counts and outcomes after intracerebral hemorrhage. J Neuroinflammation 2011;16:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broderick J, Connolly S, Feldmann E, et al. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke association stroke council, high blood pressure research council, and the quality of care and outcomes in research interdisciplinary working group. Circulation 2007;116:e391–e413. [DOI] [PubMed] [Google Scholar]
- 4. Huang FP, Xi G, Keep RF, et al. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg 2002;96:287–293. [DOI] [PubMed] [Google Scholar]
- 5. Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol 2012;11:720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 2011;42:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murthy SB, Moradiya Y, Dawson J, et al. Perihematomal edema and functional outcomes in intracerebral hemorrhage: influence of hematoma volume and location. Stroke 2015;46:3088–3092. [DOI] [PubMed] [Google Scholar]
- 8. Leasure A, Kimberly WT, Sansing LH, et al. Treatment of edema associated with intracerebral hemorrhage. Curr Treat Options Neurol 2016;18:9. [DOI] [PubMed] [Google Scholar]
- 9. Senn R, Elkind MS, Montaner J, et al. Potential role of blood biomarkers in the management of nontraumatic intracerebral hemorrhage. Cerebrovasc Dis 2014;38:395–409. [DOI] [PubMed] [Google Scholar]
- 10. Buechler C, Ritter M, Orso E, et al. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro‐ and antiinflammatory stimuli. J Leukoc Biol 2000;67:97–103. [PubMed] [Google Scholar]
- 11. Thomsen JH, Etzerodt A, Svendsen P, Moestrup SK. The haptoglobin‐CD163‐heme oxygenase‐1 pathway for hemoglobin scavenging. Oxid Med Cell Longev 2013;2013:523652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu R, Cao S, Hua Y, et al. CD163 expression in neurons after experimental intracerebral hemorrhage. Stroke 2017;48:1369–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schaer CA, Schoedon G, Imhof A, et al. Constitutive endocytosis of CD163 mediates hemoglobin‐heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res 2006;99:943–950. [DOI] [PubMed] [Google Scholar]
- 14. Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature 2001;409:198–201. [DOI] [PubMed] [Google Scholar]
- 15. Liu B, Hu B, Shao S, et al. CD163/hemoglobin oxygenase‐1 pathway regulates inflammation in hematoma surrounding tissues after intracerebral hemorrhage. J Stroke Cerebrovasc Dis 2015;24:2800–2809. [DOI] [PubMed] [Google Scholar]
- 16. Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal 2013;18:2352–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Appelboom G, Bruce SS, Hickman ZL, et al. Volume‐dependent effect of perihaematomal oedema on outcome for spontaneous intracerebral haemorrhages. J Neurol Neurosurg Psychiatry 2013;84:488–493. [DOI] [PubMed] [Google Scholar]
- 18. Reiber H. External quality assessment in clinical neurochemistry: survey of analysis for cerebrospinal fluid (CSF) proteins based on CSF/serum quotients. Clin Chem 1995;41:256–263. [PubMed] [Google Scholar]
- 19. Ilter A, Orem C, Balaban Yucesan F, et al. Evaluation of serum sTWEAK and sCD163 levels in patients with acute and chronic coronary artery disease. Int J Clin Exp Med 2015;8:9394–9402. [PMC free article] [PubMed] [Google Scholar]
- 20. Su L, Feng L, Song Q, et al. Diagnostic value of dynamics serum sCD163, sTREM‐1, PCT, and CRP in differentiating sepsis, severity assessment, and prognostic prediction. Mediators Inflamm 2013;2013:969875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Etzerodt A, Maniecki MB, Moller K, et al. Tumor necrosis factor alpha‐converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol 2010;88:1201–1205. [DOI] [PubMed] [Google Scholar]
- 22. Etzerodt A, Berg RM, Plovsing RR, et al. Soluble ectodomain CD163 and extracellular vesicle‐associated CD163 are two differently regulated forms of ‘soluble CD163’ in plasma. Sci Rep 2017;13:40286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garton T, Keep RF, Hua Y, et al. CD163, a hemoglobin/haptoglobin scavenger receptor, after intracerebral hemorrhage: functions in microglia/macrophages versus neurons. Transl Stroke Res 2017. https://doi.org/10.1007/s12975-017-0535-5. [DOI] [PubMed] [Google Scholar]
- 24. Roberts ES, Masliah E, Fox HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J Neuropathol Exp Neurol 2004;63:1255–1264. [DOI] [PubMed] [Google Scholar]
- 25. Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg 1998;89:991–996. [DOI] [PubMed] [Google Scholar]
- 26. Zhao X, Song S, Sun G, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci 2009;29:15819–15827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Law SK, Micklem KJ, Shaw JM, et al. A new macrophage differentiation antigen which is a member of the scavenger receptor superfamily. Eur J Immunol 1993;23:2320–2325. [DOI] [PubMed] [Google Scholar]