

Abstract
Objective
Autoimmune encephalitis is most frequently associated with anti‐NMDAR autoantibodies. Their pathogenic relevance has been suggested by passive transfer of patients' cerebrospinal fluid (CSF) in mice in vivo. We aimed to analyze the intrathecal plasma cell repertoire, identify autoantibody‐producing clones, and characterize their antibody signatures in recombinant form.
Methods
Patients with recent onset typical anti‐NMDAR encephalitis were subjected to flow cytometry analysis of the peripheral and intrathecal immune response before, during, and after immunotherapy. Recombinant human monoclonal antibodies (rhuMab) were cloned and expressed from matching immunoglobulin heavy‐ (IgH) and light‐chain (IgL) amplicons of clonally expanded intrathecal plasma cells (cePc) and tested for their pathogenic relevance.
Results
Intrathecal accumulation of B and plasma cells corresponded to the clinical course. The presence of cePc with hypermutated antigen receptors indicated an antigen‐driven intrathecal immune response. Consistently, a single recombinant human GluN1‐specific monoclonal antibody, rebuilt from intrathecal cePc, was sufficient to reproduce NMDAR epitope specificity in vitro. After intraventricular infusion in mice, it accumulated in the hippocampus, decreased synaptic NMDAR density, and caused severe reversible memory impairment, a key pathogenic feature of the human disease, in vivo.
Interpretation
A CNS‐specific humoral immune response is present in anti‐NMDAR encephalitis specifically targeting the GluN1 subunit of the NMDAR. Using reverse genetics, we recovered the typical intrathecal antibody signature in recombinant form, and proved its pathogenic relevance by passive transfer of disease symptoms from man to mouse, providing the critical link between intrathecal immune response and the pathogenesis of anti‐NMDAR encephalitis as a humorally mediated autoimmune disease.
Introduction
Encephalitis associated with anti‐N‐methyl‐d‐asparate receptor (anti‐NMDAR) antibodies (“antibodies”) can be triggered by the presence of peripheral tumors1, 2 or preceding viral CNS infection or occur in the absence of such obvious events.3, 4 It typically presents with neurocognitive deficits variably accompanied by psychiatric symptoms, epileptic seizures, and disorders of movement and consciousness. Antibodies against NMDAR can be detected both in peripheral blood (PB) and cerebrospinal fluid (CSF), and patients typically exhibit intrathecal synthesis of anti‐NMDAR antibodies.1
The pathogenic relevance of these antibodies is supported by the response of clinical symptoms to immunotherapy and the correlation between antibody titers and neurological outcome.5 The intrathecal source of pathogenic antibodies in anti‐NMDAR encephalitis has been suggested to originate from CD138+ plasma cells identified in perivascular and interstitial spaces in biopsy and autopsy studies.6, 7
In vitro, IgG antibodies contained in serum/CSF have been shown to bind to the N368/G369 region of the GluN1 subunit of NMDARs and reversibly decrease postsynaptic NMDAR surface density and synaptic localization in inhibitory as well as excitatory cultured rat hippocampal neurons by selective antibody‐mediated cross‐linking and internalization.8, 9 These effects are associated with a disruption of the normal interaction between the NMDAR and Ephrin‐B2 (EphB2) receptor at the synapse.10, 11 Consistently, anti‐NMDAR antibodies selectively decreased NMDAR‐mediated miniature excitatory postsynaptic currents (mEPSC) without affecting α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPA‐R)‐mediated mEPSCs in cultured rat hippocampal neurons.8, 9 Once internalized, antibody‐bound NMDARs traffic through both recycling endosomes and lysosomes, but do not induce compensatory changes in glutamate receptor gene expression.8, 12 Although anti‐NMDAR antibodies are mainly of the complement‐activating IgG1 and IgG3 subtypes, complement deposition was not detected in brain biopsy specimens from treatment‐naïve patients with anti‐NMDAR encephalitis, probably due to the low levels of complement present in CSF and relative preservation of the blood–brain barrier (BBB).7, 13 Consistently, anti‐NMDAR IgG antibodies did not reduce the number of synapses, dendritic spines, dendritic complexity, or cell survival in cultured rat hippocampal neurons,9 and neuronal damage was sparse in brain biopsy specimens.13, 14 Passive infusion of patients' CSF into the cerebroventricular system of mice via osmotic minipumps induced reversible memory impairment and behavioral changes consistent with symptoms found in patients with anti‐NMDAR encephalitis in vivo.15
It has been postulated that NMDAR expressed in nervous tissue contained in peripheral tumors, or released by viral‐induced neuronal destruction, is transported to the regional lymph nodes either in soluble form or loaded in antigen‐presenting cells.16 Naïve cognate antigen‐specific B cells exposed to NMDAR in cooperation with antigen‐specific CD4+ T cells become activated and thus capable of entering the CNS crossing the BBB or the choroid plexus. In the brain, these NMDAR‐specific B cells are thought to undergo further antigen stimulation with clonal expansion, affinity maturation of their antigen receptors, and differentiation into anti‐NMDAR antibody‐producing plasma cells.16 Indeed, a recombinant human anti‐NMDAR monoclonal antibody was recently cloned from intrathecal B cells and showed pathogenic effects in in vitro systems that were similar to those previously reported using CSF of patients.9, 17 However, whether the GluN1‐reactive human monoclonal antibody indeed exerted any pathogenic effects in vivo was not investigated.
In this study, we focused on the analysis of clonally expanded intrathecal plasma cells (cePc), the cell population previously shown to be the main source of intrathecal immunoglobulin, often detectable as “oligoclonal bands.”18, 19 We determined whether cePc are responsible for anti‐NMDAR‐specific autoantibody production in patients with acute treatment‐naïve anti‐NMDAR encephalitis and whether these autoantibodies functionally interact with NMDAR in vitro and are indeed capable of eliciting core clinical symptoms of the disease in mice in vivo.
Materials and Methods
Patients and rhuMab synthesis
Patients
Four patients with anti‐NMDAR encephalitis and controls were recruited and analyzed using flow cytometry at the Department of Neurology, Westfälische Wilhelms University of Münster, Germany. The study was approved by the local ethics committee of the Medical Faculty of the University of Münster, Germany (Az 2013‐350‐f‐S). All participants (in retrospect after clinical recovery) and their nearest relatives (in prospect) gave written informed consent to the study including scientific evaluation and publication of all clinical, paraclinical, and scientific data obtained. Clinical and paraclinical assessments of patients (MRI, neuropsychological assessment, multiparameter flow cytometry of PB and CSF, antineuronal antibody testing, and histopathological studies) were essentially performed as described previously.20
Single cell RT‐PCR analysis
Cryopreserved CSF cells were thawed in RPMI 1640 cell culture medium, washed, resuspended in FACS buffer (2% FBS in PBS), and incubated with PerCp‐Cy5.5‐labeled anti‐human‐CD19 antibody (BD Biosciences), APC‐labeled anti‐human‐CD138 antibody (Miltenyi Biotec), and 300 nmol/L of DAPI for 30 min on ice. After washing, single‐cell sorting was performed on a MoFlo XDP High Speed Cell Sorter (Beckman Coulter) at the flow cytometer facility of the Biomedical Research Center of the Heinrich Heine University Düsseldorf. Single CD138+ plasma cells were sorted in individual microtiter wells and stored at −80°C until use. RT‐PCR amplification of IgG heavy‐ and light‐chain transcripts from single cells was performed as described previously21 with minor modifications. PCR amplicons were purified from 1% agarose gel using QIAquick PCR purification kit and subsequently sequenced with the respective constant region primers at GATC Biotech (Germany). All Ig‐VH, Ig‐Vκ, and Ig‐Vλ sequences were analyzed by IgBLAST in comparison with IMGT germline gene entry (http://www.ncbi.nlm.nih.gov/igblast/) to identify closest germline V(D)J gene segments with highest identity. Plasma cells from which sequences were amplified sharing more than 95% nucleotide identity, having highly similar or identical CDR3 regions, and belonging to the same V(D)J germline families were considered clonally expanded (cePc). Sequences that appeared to be clonally expanded were manually cross‐checked to confirm the identity among them and accordingly processed for cloning and production of recombinant human monoclonal antibodies (rhuMab). The Ig‐VH and Ig‐VL complementarity‐determining region 3 (CDR3) lengths were determined as indicated in IgBlast by counting the amino acid residues following framework region 3 (FR3) up to the conserved WG (tryptophan–glycine) motif in all JH segments or up to the conserved FG (phenylalanine–glycine) motif in JL segments.22
Ig–gene repertoire analysis
For Ig–gene repertoire analysis and phylogenetic tree construction, all sequences were trimmed before FR1 and after J regions, aligned in Ig‐VH, Ig‐Vκ, and Ig‐Vλ groups, and analyzed using an online multiple alignment program for amino acid and nucleotide sequences called “MAFFT version 7” (http://mafft.cbrc.jp/alignment/server/). To construct phylogenetic trees of Ig‐V sequences, “Newick file format” was generated by using default neighbor‐joining (NJ) phylogeny method23 on MAFFT (version 7) software. FigTree v1.4.2 software was used to construct unrooted phylogenetic trees. The bootstrap support values for each branch were calculated based on 100 resamplings of the original dataset. High values of the bootstrap (more than 70) represent a better statistical support for the topology in the tree.24
Ig expression vector cloning
Clonally expanded paired Ig heavy‐ and light‐chain variable region amplicons identified and selected for rhuMab production were reamplified with primers containing restriction sites and cloned in frame into mammalian expression vectors containing the respective human Igγ1, Igκ, or Igλ2 constant gene regions. In brief, restriction digestion of PCR products and vectors were carried out with respective restriction enzymes, namely EcoRI, NheI, and AvrII (all from New England Biolabs), followed by 5′ dephosphorylation of the digested vectors using shrimp alkaline phosphatase (New England Biolabs). All digested products were purified from 1% agarose gel. The ligation reactions between restriction enzymes digested PCR products and corresponding vectors were performed using LigaFast™ Rapid DNA Ligation System (Promega), overnight at 4°C. Cloning of the second round PCR products was carried out into mammalian expression vectors containing a hEF1‐HTLV promotor, an IL2 signal peptide sequence, and a multiple cloning site upstream of the human Ig constant regions (Invivogen). One Shot®TOP10 chemically competent E. coli cells were transformed with the ligated products at 42°C following the instruction protocol of the supplier (Invitrogen). The transformed Ig heavy‐chain colonies were selected on Zeocin (Invitrogen) agar plates, whereas Ig light‐chains colonies (Igκ or Igλ) were selected on Blasticidine (Invitrogen) agar plates. Mini‐prep plasmid preparations (Qiagen miniprep kit), and subsequent sequencing, were carried out on the selected bacterial colonies to confirm the presence of original Ig variable regions. All cloned expression vectors were sequenced to confirm the presence of Ig‐V region inserts with 100% identity to the respective original PCR products.
rhuMab production and purification
FreeStyle™ 293‐F cells (Invitrogen) were cultured in TC‐175 cm2 flask (Greiner Bio‐One) under standard conditions in Gibco® RPMI 1640 medium, supplemented with 10% heat‐inactivated fetal calf serum (FCS), 100 μg/mL streptomycin, 100 U/mL penicillin (all from Invitrogen). Cotransfections of Ig heavy‐chain and matching Ig light‐chain expression vectors (20 μg each/flask) were performed using linear polyethylenimine (PEI) as a transfection reagent (Polysciences, Inc.)25. In brief, 1:3 concentration ratio of plasmids:PEI were mixed in 500 μL Opti‐MEM‐reduced serum media (Invitrogen), 30 min at RT, and subsequently incubated on 90% confluent cells in a total volume of 20 mL/flask for 24 h. The transfection medium was replaced with antibody‐harvesting medium containing RPMI 1640 supplemented with 1× Nutridoma‐SP (Roche), 100 μg/mL streptomycin, 100 U/mL penicillin, and cultured. The antibody harvesting medium was collected after 1 week of incubation, and transfected cells were either reincubated with fresh harvesting medium or stored in cell freezing medium (10% DMSO in FBS) at −80°C for future use.
RhuMab were purified from supernatants by using either HiTrap Protein G HP columns (GE Healthcare) on an FPLC system (ÄKTA Prime) or Magne™ Protein G Beads (Promega). In brief, harvested supernatants were centrifuged at 8000g for 10 min, filtered using 0.2‐μm vacuum filters (Sarstedt), and adjusted to pH 7.4 using a 1 mol/L Tris‐HCl buffer (pH 8.8). Supernatants were either pumped through protein G HP columns (columns pre‐equilibrated with 20 mmol/L Tris‐HCl, pH 7.4), or incubated with Magne™ protein G beads overnight at 4°C. Bound rhuMab were eluted with 0.1 mol/L glycine‐HCl buffer (pH 2.7) and neutralized to pH 7.4 with 1 mol/L Tris‐HCl buffer (pH 8.8). Buffer exchange was performed with PBS either by dialysis using 10 K MWCO Slide‐A‐Lyzer Dialysis Cassettes (Thermo Scientific) or using 30 K Amicon Ultra‐15 centrifugal Filter Units (Merck Millipore). The protein concentrations of purified antibodies were determined by BC assay kit (Uptima).
Purity of the eluted rhuMab was determined by SDS‐PAGE gel. Semidenatured samples were prepared by mixing 5 μg of eluted antibodies with 1X‐NuPAGE sample reducing agent (Invitrogen), heated 10 min at 60°C, and loaded on 4–16% PreciseTM Protein gel (Thermo Scientific), along with Novex Sharp prestained protein standard (Invitrogen). The samples were run in NuPage‐MOPS SDS running buffer (Invitrogen) for 30 min at 120 V and subsequently stained with Gel codeTM blue stain reagent (Thermo Scientific) to visualize the complete antibody bands along with separate Ig heavy‐ and light‐chain bands. Purified rhuMab were aliquoted and stored at −80°C. Initial screening of rhuMab for antineuronal reactivity was performed by Euroimmun, Lübeck, Germany (http://www.euroimmun.de).
Characterization of rhuMab specificity
To demonstrate the specificity of rhuMab, we used techniques including immunoprecipitation, western blot, immunohistochemistry with rat brain, immunocompetition assays, and immunocytochemistry with transfected HEK cells essentially as previously reported.26, 27 As isotype control, a recombinant human antibody (12D7) with – in this context – irrelevant antigen specificity (cancer/testis antigen NY‐ESO‐1) was used as indicated.
Functional analysis of anti‐NMDAR‐specific rhuMab SSM5 in vitro and in vivo
To assess functional effects of rhuMab in vitro, we used two‐electrode voltage‐clamp analysis of GluN1‐1a‐ and GluN2B‐transfected Xenopus laevis oocytes together with rhuMab incubation in primary cultures of hippocampal neurons with quantification of the effects of rhuMab on total cell‐surface and synaptic NMDAR clusters as described previously.9, 28
To examine functional consequences of rhuMab in vivo, we used behavioral testing of mice receiving cerebroventricular infusion of patients' CSF or monoclonal antibodies together with subsequent determination of rhuMab binding to brain tissue and quantification of the effects of rhuMab on total cell‐surface and synaptic NMDAR clusters as described previously.11, 15
Statistical analysis
If not explicitly stated otherwise, all statistical analyses were performed using Sigma Plot 11 (Systat Software, Germany) or GraphPad Prism Version 6 (GraphPad Software, San Diego, CA). Data were tested for normality using the Shapiro–Wilk test. Depending on the result, unpaired Student's t test or Mann–Whitney test was used to compare means between two independent groups, whereas paired Student's t test or Wilcoxon matched pairs signed‐rank test was used for different treatments within the same group. More than two datasets were compared using ANOVA with Bonferroni post hoc correction. All normally distributed data are given as mean together with the standard deviation (SD) or standard error of the mean (SEM). All not normally distributed data are given as median together with the interquartile range. If not explicitly stated otherwise, the prechosen significance level for all confirmatory tests was set to P < 0.05.
Results
Intrathecal B‐cell and plasma cell accumulation in patients with anti‐NMDAR encephalitis corresponds to the clinical disease course
We identified four therapy‐naive female Caucasian patients with recent onset typical anti‐NMDAR encephalitis (for details, see Tables S1–S7). Cerebral MRI (1.5 Tesla, Suppl. Fig. 1) showed either frontal or temporal cortical lesions and was otherwise normal. All patients underwent multiparameter flow cytometry analysis of PB and CSF to characterize the systemic and intrathecal autoimmune response in detail before initiation of immunotherapy (Fig. 1, Table S6). Twenty‐five patients that had PB and CSF analyses for suspected neurological disease but turned out to suffer from somatization disorders served as controls.
Figure 1.

Intrathecal B‐cell and plasma cell accumulation in patients with anti‐NMDAR encephalitis corresponds to the clinical disease course. (A–C) Representative multiparameter flow cytometry analysis of CD19+ B cells and CD19+ CD138+ plasma cells in peripheral blood (PB, left panels) and cerebrospinal fluid (CSF, right panels) in a healthy control (A) and a patient (Pt. 1) with anti‐NMDAR encephalitis before (B) and after (C) immunotherapy. (D) Representative CD138+ plasma cell staining of a biopsy specimen of a newly occurring cerebral MRI lesion in a patient with later on established anti‐NMDAR encephalitis (scale bar represents 50 μm; insert scale bar represents 10 μm). (E–H) Multiparameter flow cytometry analysis of absolute numbers (E) and relative fractions (F) of CD19+ B cells and absolute numbers (G) and relative fractions (H) of CD138+ CD19+ plasma cells within peripheral blood (PB, left panels) and cerebrospinal fluid (CSF, right panels) in four patients with anti‐NMD‐R encephalitis and 25 controls. Data are given as whisker plots. Statistical testing was performed using the Wilcoxon rank sum test with an α error of 0.05. Levels of significance are indicated by n.s. (not significant) for all P > 0.05, *P < 0.05 and **P < 0.01. (I–K) Time course of absolute numbers of CD19+ B cells (I), CD19+ CD138+ plasma cells (J), and NMDAR IgG titers (K) in cerebrospinal fluid of anti‐NMDAR encephalitis before, during, and after treatment. (L) Comprehensive results of detailed neuropsychological assessments (total rank sum of test of attention, memory and executive functions) of patients with anti‐NMDAR encephalitis 1–16 weeks after admission (pre) and after 8–48 months (post).
A representative flow cytometry of CD19+ and CD138+ cells in PB and CSF in anti‐NMDAR encephalitis (Pt. 1) and controls is shown in Figure 1A and B (PB left panels, CSF right panels). As compared to controls, the absolute numbers and relative fractions of CD19+ B cells (Fig. 1E and F) and CD138+ CD19+ plasma cells (Fig. 1G and H) were significantly elevated in CSF of patients with anti‐NMDAR encephalitis, but not in their PB (for details, see Table S6, PB left panels, CSF right panels). Notably, controls harbored some intrathecal CD19+ B cells but no intrathecal CD138+ CD19+ plasma cells at all (Table S6). Likewise, all patients showed positive NMDAR‐specific Ig indices (Table S2). These data demonstrate intrathecal B‐cell and plasma cell accumulation as a likely intrathecal source of antibody production in patients with anti‐NMDAR encephalitis. Consistently, prominent CD138+ plasma cells could be detected in perivascular, and interstitial spaces of a biopsy specimen from another patient with anti‐NMDAR encephalitis (Fig. 1D).6, 7
All patients received immunotherapy consisting of repeated cycles of protein A immunoadsorption combined with pulses of intravenous methylprednisolone (Pt. 1–4) and immunosuppression using azathioprine or methotrexate (Pt. 1–3) or radiochemotherapy for small cell lung cancer (Pt. 4). Under these treatment regimens, clinical symptoms, EEG, and MRI findings as well as inflammatory CSF changes regressed almost completely (Tables S1 and S2). Moreover, absolute numbers and relative fractions of CD19+ B cells (Fig. 1I, Table S7) and CD138+ CD19+ plasma cells (Fig. 1J, Table S7) as well as NMDAR IgG titers (Fig. 1K, Table S2) in CSF decreased rapidly. Neuropsychological performance (attention, memory, executive function) substantially improved in each patient upon follow‐up assessment after 8–48 months (Fig. 1L, Tables S4 and S5).
Hence, intrathecal B‐cell and plasma cell accumulation corresponded well to the clinical disease course of patients with anti‐NMDAR encephalitis.
Presence of clonally expanded plasma cell with hypermutated antigen receptors indicates an intrathecal antigen‐driven immune response in patients with anti‐NMDAR encephalitis
Clonally expanded plasma cells/blasts (cePc) were identified among intrathecal CD138+ cells by sequence analysis of Ig heavy‐chain amplicons obtained by single‐cell RT‐PCR. In Figure 2, an example of one of the patients with anti‐NMDAR encephalitis is shown: From this patient, a total of 120 single CD138+ cells were collected by FACS (Fig. 2A) and subjected to single‐cell PCR. Readable sequences were obtained from 81 Ig light‐chain amplicons (51 kappa chains, 30 lambda chains) and 37 IgG heavy‐chain amplicons. Unrooted phylogenetic trees constructed from IgG heavy (Fig. 2B), lambda (Fig. 2C), and kappa light‐chain sequences (Fig. 2D) are shown: Clonally related sequences aggregate together as overlapping “leaves” on the same “branch” of the phylogenetic tree. Sequences, which are very different from another or even belong to different germ line gene families, are separated by distance. Recombinant human monoclonal antibodies (rhuMab) were reconstructed from matching IgH and IgL variable gene regions of seven cePc (Fig. 2, circled). All identified cePc sequences were hypermutated, containing silent as well as replacement mutations indicating an ongoing antigen‐driven intrathecal immune response (Table 1).
Figure 2.
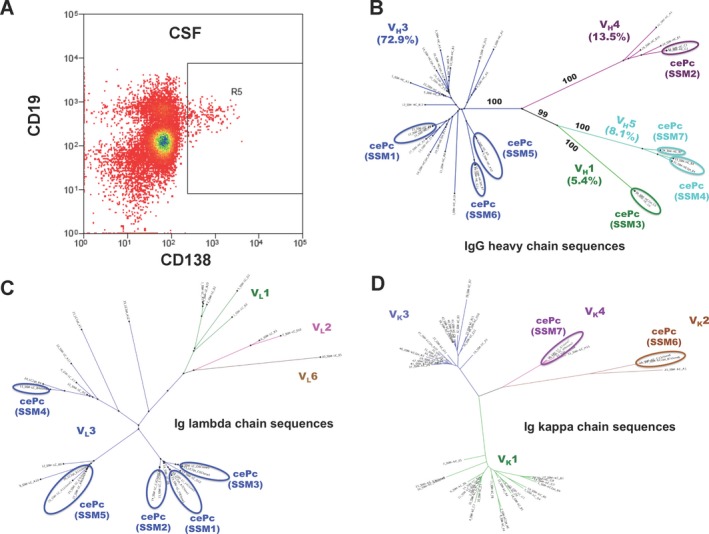
Presence of clonally expanded plasma cells with hypermutated antigen receptors indicates an intrathecal antigen‐driven immune response in patients with anti‐NMDAR encephalitis. Shown are unrooted phylogenetic trees of the IgG‐VH (B), V lambda (C), and V kappa (D) gene family repertoires of sequences obtained from CSF plasma cells of a patient with anti‐NMDAR encephalitis by single cell RT‐PCR. Each colored branch of the tree represents a single V family. The external node of a branch represents a “leaf” or a “sequence.” The branch length and distance between sequences correspond to sequence similarity/dissimilarity, shorter and closer branches relate to a greater sequence similarity. The bootstrap support values for each branch were calculated based on 100 resampling of the original dataset. Ovals indicate clonally expanded plasma cells (cePc) 1–7, from which recombinant monoclonal antibodies SSM 1–7 were derived. The gate (R5) in the FACS panel (A) shows the cell population used for single cell analysis.
Table 1.
Number of amino acid mutations in the sequences of reconstructed cePc‐derived rhuMAb compared with nearest germline gene sequences
| rhuMab | Germline V(D)J gene | Amino acid mutations in the IgG‐VH and matching VL genes of the cePc sequences from the corresponding germline genes | Total mutations | |||||
|---|---|---|---|---|---|---|---|---|
| FR1 | CDR1 | FR2 | CDR2 | FR3 | CDR3 | |||
|
SSM1.HC SSM1.λC |
IGHV3(D3)J6 IGLV3J2 |
1 – |
1 – |
1 – |
1 1 |
4 – |
– – |
8 1 |
|
SSM2.HC SSM2.λC |
IGHV4(D5)J2 IGLV3J2 |
– – |
1 – |
1 – |
1 1 |
4 – |
– – |
7 1 |
|
SSM3.HC SSM3.λC |
IGHV1(D6)J6 IGLV3J2 |
– – |
– – |
– – |
4 1 |
4 – |
– – |
8 1 |
|
SSM4.HC SSM4.λC |
IGHV5(D6)J5 IGLV3J1 |
3 1 |
3 1 |
1 1 |
2 1 |
4 – |
– 1 |
13 5 |
|
SSM5.HC SSM5.λC |
IGHV3(D6)J2 IGLV3J2 |
1 – |
– – |
– – |
– – |
– – |
– 1 |
1 1 |
|
SSM6.HC SSM6.kC |
IGHV3(D3)J4 IGKV2J4 |
1 1 |
1 3 |
2 1 |
1 1 |
9 1 |
1 1 |
15 8 |
|
SSM7.HC SSM7.kC |
IGHV5(D1)J4 IGKV4J2 |
– 1 |
3 4 |
2 – |
1 – |
– 1 |
– 1 |
6 7 |
(–) Either no mutation or silent mutation (not leading to an amino acid change).
A recombinant human monoclonal antibody (rhuMAb) produced from clonally expanded intrathecal plasma cells specifically recognizes the NMDAR
To elucidate whether cePc are the actual source of the intrathecal autoantibody response, we cloned paired immunoglobulin heavy‐ and light‐chain variable gene regions from cePc essentially as previously described.19, 21 PEI mediated cotransfection of IgG‐HC and matching Ig‐LC expression vectors into FreeStyle 293‐F cells were carried out to produce recombinant monoclonal antibodies in vitro. Secreted rhuMAb were purified from the supernatant of transfected cells by affinity chromatography. Successful antibody production was confirmed by SDS gel electrophoresis (data not shown).
From the seven rhuMab we cloned and expressed from CSF cePc of anti‐NMDAR encephalitis patient SSM, rhuMab SSM5 was identified to react specifically with NMDAR, whereas the remaining showed no CNS tissue reaction at all (data not shown). To further characterize the antigen specificity of rhuMab, we performed additional assays.
The pattern of brain immunostaining and main epitope target of rhuMab SSM5 are similar to antibodies in serum and CSF from anti‐NMDAR encephalitis patients
To characterize the tissue reactivity of rhuMab SSM5, immunohistochemistry on mildly fixed sagittal rat brain sections was performed, as previously described.14 Patient‐derived rhuMab SSM5 (Fig. 3A), but not the isotype control rhuMab 12D7 (Fig. 3B), produced an intense pattern of brain reactivity characteristic of NMDAR as previously reported using patients' sera or CSF,14 with a strong emphasis of the hippocampal region. Tissue reactivity of rhuMab SSM5 was clearly reduced by preincubation of tissue sections with sera from patients with anti‐NMDAR encephalitis (Fig. 3D and E), but not by preincubation with serum of a healthy blood donor (Fig. 3C and E), suggesting that antibodies contained in patients' sera and rhuMab SSM5 compete for similar target epitopes of the NMDAR.
Figure 3.
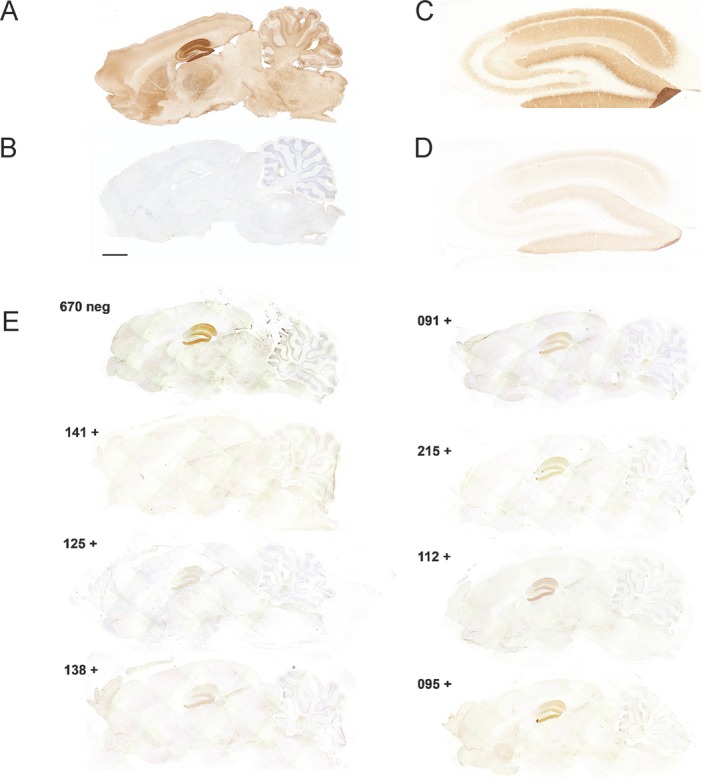
The pattern of brain tissue immunostaining of rhuMab SSM5 is characteristic of the NMDAR and blocked by serum antibodies from patients with anti‐NMDAR encephalitis. (A, B) Patient‐derived rhuMab reacts with brain tissue. Representative immunoreactivity of a patient‐derived rhuMab (SSM5) (A) and control rhuMab (12D7) (B) with sagittal sections of rat brain. SSM5 antibody, but not the control antibody shows a robust immunostaining of brain in a pattern characteristic of the NMDAR. Scale bar 2 mm. (C, D, E) Patient‐derived rhuMab competes for similar epitopes recognized by serum antibodies from patients with anti‐NMDAR encephalitis. Reactivity of biotinylated rhuMab SSM5 (diluted 1:20) with rat hippocampus preincubated with serum from a healthy subject (C) and serum from a patient with anti‐NMDAR encephalitis (diluted 1:2) (D). (E) Preincubation with sera of anti‐NMDAR encephalitis patients substantially blocks the reactivity of SSM5. In this experiment, the tissue sections were preincubated with sera from seven different patients (091, 095, 112, 125, 138, 141, 215) and one control without antibodies (670). There is competition of reactivity by each of the patients' sera, while control serum (top left) does not reduce rhuMab SSM5 reactivity (as expected). Case to case variation of competition suggests that anti‐NMDAR antibodies against epitopes recognized by rhuMab SSM5 are present in each of the patients but differ in concentration and/or affinity.
To further narrow down the epitope specificity of patient‐derived rhuMab SSM5, reactivity with HEK293 cells transfected with wild‐type GluN1/N2B or indicated GluN1 mutants was assessed by immunocytochemistry (Fig. 4A–C) as previously described.29 Patient‐derived rhuMab SSM5, but not isotype control rhuMab 12D7, specifically stained HEK293 cells cotransfected with wild‐type GluN1/GluN2b in a similar pattern as CSF from a patient with anti‐NMDAR encephalitis (green, Fig. 4A). Similar to patient's CSF, reactivity of rhuMab SSM5 with HEK293 transfectants was abrogated, when the main immunogenic hinge region of GluN1 was mutated replacing amino acid G369 by a nonpolar amino acid (mutant G369I) (Fig. 4B), while exchanging amino acid G369 for a polar residue (G369S) did not affect binding of rhuMab SSM5 or patient's CSF (Fig. 4C). Immunoprecipitation experiments with membrane fractions of brain tissue confirmed that rhuMab SSM5 specifically reacts with the 120 kDa GluN1subunit of the NMDAR (Fig. 4D).
Figure 4.
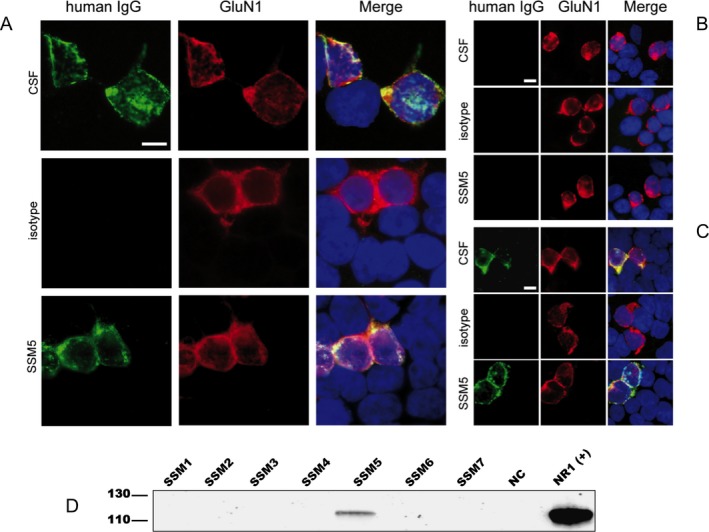
RhuMab SSM5 targets the same restricted epitope of the GluN1 subunit of the NMDAR as patient's CSF antibodies. (A–C) Patient‐derived rhuMab shows the same staining pattern as CSF from a patient with anti‐NMDAR encephalitis in immunohistochemistry with HEK cells expressing full‐length or mutant GluN1/GluN2B. Immunofluorescence with HEK293 cells expressing full‐length GluN1/GluN2B (A) or mutant GluN1 with full‐length GluN2B (B, C) and stained for human IgG (green) and with a commercial GluN1‐specific antibody (red); nuclear counterstaining with DAPI (blue). Cells were treated with human CSF (diluted 1:16), patient‐derived rhuMab (SSM5, 4.3 μg/mL) or control rhuMab (isotype, rhuMab 12D7, 4.3 μg/mL). The epitope‐disrupting G369I mutant (B) does not show staining by patients' CSF or rhuMab, while a mutant with less epitope disruption G369S (C) does not affect staining. Scale bar represents 10 μm. (D) Confirmation of antigen specificity of patient‐derived rhuMab by immunoprecipitation (IP). Proteins precipitated from brain membrane lysates were separated by PAGE electrophoresis and blotted onto PVDF membranes. Western blot detection was performed with a commercial GluN1‐specific antibody. A band of around 120 kDa corresponding to the GluN1 subunit of the NMDA receptor was identified using SSM5 rhuMab (5 μg, covalently coupled to Dynabeads) in the membrane fraction of brain tissue. Commercially available antibodies to the respective target antigen were used as positive control in IPs and in western blot detections. A rhuMab specific for an irrelevant target (12D7) was included as a negative control (NC) for IP experiments.
We therefore conclude that rhuMab SSM5 targets the same restricted epitope on the extracellular amino terminal domain of the GluN1 subunit of the NMDA receptor as has been described for disease‐relevant antibodies in serum and CSF of patients with anti‐NMDAR encephalitis.29
RhuMab SSM5 decreases the density of cell surface and synaptic NMDAR in cultured neurons
Hippocampal neurons were cultured for 24 h with patient‐derived rhuMab SSM5, isotype control rhuMab 12D7, or CSF of a patient with anti‐NMDAR encephalitis. Patient‐derived rhuMab (SSM5), but not the isotype control rhuMab (12D7), caused a significant decrease in cell surface NMDAR and synaptic NMDAR clusters, similar to that observed with CSF of a patient with anti‐NMDAR encephalitis (Fig. 5A, green, quantified in Fig. 5B). In contrast, PSD95, an excitatory postsynaptic protein, was not reduced (Fig. 5 A, red, quantified in Fig. 5B). Together, these results show that patient‐derived rhuMab SSM5 specifically reduces synaptic NMDAR clusters, as previously described for CSF of anti‐NMDAR encephalitis patients.9
Figure 5.
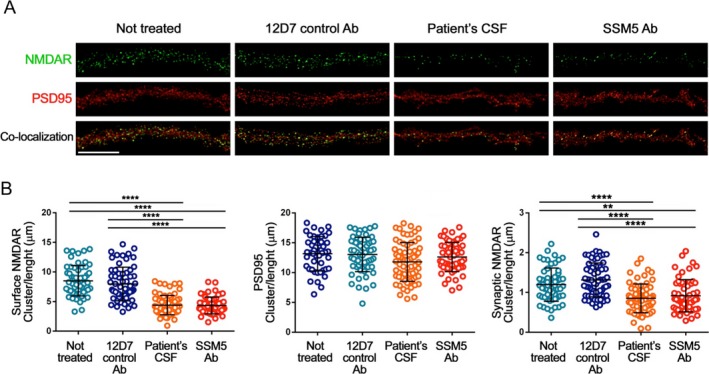
RhuMab SSM5 decreases the density of cell surface and synaptic NMDAR in cultured hippocampal neurons. (A) Representative dendrites from cultures of primary rat hippocampal neurons showing the density of NMDAR clusters (top panels), PSD95, an excitatory postsynaptic protein (middle panels), and synaptic NMDAR (colocalization of NMDAR with PSD95, lower panels) after 24 h treatment with control rhuMab 12D7 (4.5 μg/mL), CSF from a patient with anti‐NMDAR encephalitis (“patient's CSF,” 1:20 dilution), patient‐derived rhuMab SSM5 (4.5 μg/mL) or not treated; scale bar represents 10 mm. The quantification of these effects is shown in (B), demonstrating the density of surface NMDAR (left plot), PSD95 (middle plot) and synaptic NMDAR (right plot). Patient‐derived rhuMab SSM5, but not the control rhuMab 12D7 causes a significant reduction of surface and synaptic NMDAR clusters similarly to that of the CSF from a patient with anti‐NMDAR encephalitis. No effects occurred on PSD95. All graphs represent mean ± SD, n = 60 cells per condition. Significance of treatment effect was assessed by one‐way ANOVA (P < 0.0001 for NMDAR, synaptic NMDAR) with Bonferroni post hoc correction. **P < 0.001, ****P < 0.0001.
RhuMab SSM5 reduces NMDAR‐mediated currents in transfected Xenopus laevis oocytes
We next assessed the effects of patient‐derived rhuMab SSM5 on NMDAR function using two electrode voltage‐clamp experiments in X. laevis oocytes in vitro. Xenopus oocytes coexpressing GluN1/GluN2B were preincubated with patient‐derived rhuMab SSM5 in bath medium for 1 h prior to measurement (“antibody”), whereas GluN1/GluN2B expressing control oocytes were not preincubated (“control”) (Fig. 6A). Nontransfected oocytes were treated the same way, either preincubated with patient rhuMab (“uninjected, antibody”) or not preincubated (“uninjected”). Uninjected oocytes revealed no measurable currents. GluN1/GluN2B amplitudes in oocytes either without (“control”) or with (“antibody”) preincubation with autoantibody were measured and are shown normalized to control currents: RhuMab SSM5 significantly decreased NMDAR‐mediated currents by about 20% (Fig. 6B), supporting a functional impact of this patient‐derived rhuMab on NMDAR‐mediated currents.
Figure 6.

RhuMab SSM5 reduces NMDAR‐mediated currents in vitro and induces severe memory deficits in mice. (A, B) RhuMab SSM5 reduces NMDAR‐mediated currents in transfected Xenopus oocytes. (A) Representative current traces measured in Xenopus oocytes expressing GluN1‐1a plus GluN2B in response to superfusion with a solution containing 100 μmol/L glutamate plus 10 μmol/L glycine in Ba2+ Ringer. All currents were measured at −70 mV. GluN1‐1a/GluN2B expressing oocytes were preincubated with 4 μg/mL patient‐derived rhuMab in bath medium for 1 h prior to measurement (“antibody”), whereas GluN1‐1a/GluN2B expressing control oocytes were not preincubated with antibody (“control”). Control oocytes were treated the same way, either preincubated with patient rhuMab (“uninjected, antibody,” n = 5) or not preincubated (“uninjected,” n = 7). Uninjected oocytes revealed no measurable currents. (B) GluN1‐1a/GluN2B amplitudes in oocytes either without (“control”) or with (“antibody”) preincubation with autoantibody were measured and are shown normalized to control currents. Data are given as mean ± SEM. Statistical testing was performed by two‐tailed t‐test with an α error of 0.05. Significant difference is indicated by *P < 0.05; n indicates number of oocytes measured. (C–E) Cerebroventricular infusion of anti‐NMDAR rhuMab SSM5 causes severe reversible memory deficits. (C) Infusion of rhuMab SSM5 into the cerebroventricular system of mice for a total of 14 days causes deficits of memory. Novel object recognition index in mice treated with the patient‐derived rhuMab (SSM5, red circles) and the control rhuMab (12D7, green circles) (each at 90 μg/mL in saline solution). A high index indicates better object recognition memory. Note that mice infused with patient‐derived rhuMab showed a progressive decrease of memory that was maximal on Day 18. The total time of exploration of both objects was similar in both groups (not shown). Data are presented as mean ± SEM. Number of animals: SSM5: n = 7, 12D7: n = 7. Significance of treatment effect was assessed by two‐way ANOVA with an α error of 0.05 and post hoc testing with Bonferroni adjustment (asterisks). ***P < 0.001. (D, E) Animals infused with rhuMab SSM5 have a progressive increase of human IgG bound to hippocampus. (D) Immunostaining of human IgG in sagittal brain sections showing the hippocampus of representative animals infused with anti‐NMDAR monoclonal antibody (SSM5) (right panels) and control antibody (12D7) (left panels), sacrificed at the indicated experimental days. In animals infused with the SSM5 antibody, a gradual increase of IgG immunostaining until Day 18, followed by decrease of immunostaining is observed. Scale bar represents 200 μm. (E) Quantification of intensity of human IgG immunolabeling in hippocampus of mice infused with SSM5 antibody (dark gray columns) and control CSF (pale gray columns) sacrificed at the indicated time points. Mean intensity of IgG immunostaining in the group with the highest value (animals treated with anti‐NMDAR monoclonal antibody (SSM5) and sacrificed at Day 18) was defined as 100%. Data are presented as mean ± SEM. Number of animals: 5 animals per condition and time point. Significance of treatment effect was assessed by two‐way ANOVA with an α error of 0.05 (o) and post hoc testing with Bonferroni adjustment (*). °°°P < 0.001, **P < 0.01, ****P < 0.0001.
These results are in striking accordance with earlier results obtained by two electrode voltage‐clamp experiments in X. laevis oocytes showing a time‐dependent steady‐state reduction of the NMDAR‐mediated currents of about 30% within 16 min in oocytes exposed to dialyzed sera of patients with anti‐NMDAR encephalitis.33 The reason for the relatively small antibody‐mediated functional effects obtained in the X. laevis oocyte expression system compared to the strong effects in mice on NMDA receptor expression on the neuronal cell surface in vitro and memory impairment in vivo may be due to the necessity to record NMDAR‐mediated currents in Ca2+‐free media to block current inactivation in Xenopus oocytes.34 These recording conditions may influence conformational changes of the NMDA receptor induced by binding of the antibody or modulate antibody binding itself and thus diminish subsequent receptor internalization.
RhuMab SSM5 decreases the density of synaptic NMDAR and causes memory deficits in a mouse model of cerebroventricular antibody transfer
The in vitro results outlined above led us to determine the behavioral effects of patient‐derived rhuMab SSM5 in vivo: To this end, rhuMab SSM5 or isotype control rhuMab 12D7 was infused for 14 days into both ventricles of mice using osmotic mini‐pumps as previously described.11, 15 The most robust effect during the 14‐day infusion of patients' CSF15 was observed on the novel object recognition test, therefore we chose this test for assessing the effects of this patient‐derived rhuMab (Fig. 6C). Compared with animals infused with isotype control rhuMab 12D7 (green circles), those infused with patient‐derived rhuMab SSM5 (red circles) showed a progressive decrease of the object recognition index, indicative of a memory deficit.15 A high index indicates better object recognition memory and vice versa. The memory deficit became apparent on Day 10 and was maximal on Day 18 (4 days after the infusion of rhuMab had stopped, representing the cumulative effects of antibodies still circulating in CSF). On Day 25, the object recognition index had normalized and was similar to that of animals treated with isotype control rhuMab (Fig. 6C). These findings are similar to those reported with the model of infusion of patients' CSF NMDAR antibodies and could not be explained by other systemic effects (e.g., overall motor activity, weight loss) as described previously.15
In mice infused with rhuMab SSM5, impairment of memory was paralleled by a gradual increase of brain‐bound human IgG mainly in the hippocampus until Day 18, followed by a progressive decrease of immunostaining (Fig. 6D right panels, Fig. 6E dark gray columns), which was not observed in mice infused with control rhuMab 12D7 (Fig. 6D left panels, Fig. 6E pale gray columns).
Similar to studies in cultured hippocampal neurons mentioned above, the infusion of patient‐derived rhuMAb SSM5 (Fig. 7A upper panels, Fig. 7B dark gray), but not isotype control rhuMAb 12D7 (Fig. 7A lower panels, Fig. 7B light gray), caused a significant decrease of hippocampal cell surface NMDAR (Fig. 7A, red) and synaptic NMDAR clusters (defined as NMDAR clusters colocalizing with PSD95) in vivo, without affecting PSD95 (Fig. 7A, green) and without causing neuronal death (data not shown).
Figure 7.

RhuMab SSM5 decreases the density of synaptic NMDAR density in mice hippocampus after intraventricular infusion. (A, B) Anti‐NMDAR monoclonal antibody (SSM5) mediates a reduction of total and synaptic NMDAR in mice hippocampus. (A) 3D projection of the density of total clusters of NMDAR, PSD95, and synaptic clusters of NMDAR (defined as NMDAR clusters colocalizing with PSD95) in a hippocampal region from a representative animal of each experimental group. Merged images (merge: PSD95 [green]/NMDAR [red]) were postprocessed and used to calculate the density of clusters (density = spots/μm3). Scale bar represents 2 μm. (B) Quantification of the density of total NMDAR, total PSD95, and synaptic NMDAR clusters at Day 18 in a pooled analysis of hippocampal areas (CA1, CA2, CA3, and dentate gyrus) in animals treated with anti‐NMDAR monoclonal antibody (SSM5; dark gray) and control antibody (12D7; pale gray). Mean density of clusters in control antibody treated animals was defined as 100%. Data are presented as scatterplot plus mean ± SD. Number of animals: 5 animals per condition (18 hippocampal areas per animal = 90 hippocampal areas per condition). Significance of treatment effect was assessed by two‐tailed t‐test with an α error of 0.05. ****P < 0.0001.
These findings demonstrate that antibodies reconstructed from patient‐derived clonally expanded intrathecal plasma cells recapitulate key features of anti‐NMDAR encephalitis in vitro and in vivo.
Discussion
In this study, we show that a single recombinant human NMDAR‐specific monoclonal antibody, rebuilt from patients' clonally expanded CSF plasma cells by reverse genetics, is sufficient to reproduce epitope specificity, and key pathogenic features typical of anti‐NMDAR encephalitis in vitro and in vivo.
This disorder predominantly affects young women and children, with approximately 50% of women having an underlying ovarian teratoma that contains nervous tissue expressing NMDARs.2 It is believed that the ectopic expression of NMDARs contributes to triggering the immune response. The trigger of the disease in the rest of female patients as well as in young children and male patients is largely unknown, although in some cases other tumors (such as small‐cell lung cancer2) have been identified. In a small subset of patients the anti‐NMDAR immune response is triggered by herpes simplex encephalitis.4 Irrespective of the trigger of anti‐NMDAR encephalitis, data from previous studies suggested an enhancement of the immune response within the CNS, with potential antigen‐driven affinity maturation and clonal expansion of plasma cells.16 This hypothesis was suggested by three findings, (1) a frequent intrathecal synthesis of NMDAR antibodies even early in the disease course,1 (2) biopsy and autopsy material from patients showing brain inflammatory infiltrates containing plasma cells,6, 7 and (3) the better correlation of the course of the disease with CSF antibody titers than with serum antibody titers.5 All four treatment‐naive patients with recent onset anti‐NMDAR encephalitis in the current study showed intrathecal B‐cell and plasma cell accumulation and intrathecal production of NMDAR‐specific IgG. Moreover, using single‐cell RT‐PCR of FACS‐sorted CD138+ CSF plasma cells and recombinant antibody technology,19, 21 we identified expanded clones and were able to obtain seven different rhuMab (SSM1‐7) from intrathecal cePc of one of the anti‐NMDAR encephalitis patients. Ig sequences showed varying numbers of mutations from published germline sequences, indicating antigen‐driven expansion and ongoing somatic hypermutations. RhuMab SSM5 showed reactivity with NMDAR and was used for further studies, whereas the remaining showed no CNS tissue reaction at all.
We confirmed the antigen specificity of rhuMab SSM5 by immunohistochemistry of brain tissue sections and immunoprecipitation from brain membrane fractions.17 Moreover, this recombinant antibody competed with patients' CSF antibodies for the same restricted epitope on the extracellular amino terminal domain of the GluN1 subunit of the NMDA receptor,29 decreased the density of cell surface and synaptic NMDAR in cultured neurons, and significantly reduced NMDAR mediated currents in Xenopus oocytes expressing GluN1/GluN2B. Importantly, continuous infusion of rhuMab SSM5 into the cerebroventricular system of mice for 14 days resulted in progressive impairment of memory paralleled by accumulation of brain‐bound antibody and specific reduction of the density of synaptic NMDAR clusters, as previously reported in a similar model of infusion of patients' CSF.11, 15 Similar to this previously reported model,15 the impairment of memory was maximal on Day 18 and normalized by Day 25, consistent with a transitory, not structurally damaging effect of rhuMab SSM5. Taken together, using patient‐derived rhuMab SSM5 we were able to reproduce most relevant functional aspects previously demonstrated with sera or CSFs from patients with anti‐NMDAR encephalitis containing polyclonal antibody mixtures.
By reconstructing functional autoantibodies from CSF cePc in the form of rhuMab, we demonstrate the presence of a CNS‐specific antigen‐driven humoral immune response in the CSF compartment of patients with anti‐NMDAR encephalitis, consistent with studies showing that the sensitivity of NMDA receptor antibody testing is higher in CSF than in serum and CSF antibody levels correlate with the course of the disease.5 Whether autoreactive B‐cell affinity maturation indeed occurs inside the CNS compartment and some of the clonally related B cells migrate to the peripheral system to produce self‐reactive antibodies, or CNS‐specific B‐cell clones transmigrate the BBB and proliferate in the “B‐cell friendly” environment of the CNS, remains to be elucidated in further studies.
At this point we cannot answer, to which degree antibody‐mediated targeting of the NMDAR also accounts for the variety of other symptoms affecting approximately 75% of patients with anti‐NMDAR encephalitis, ranging from mood and psychiatric alterations to seizures and disorders of movements and consciousness.30, 31 It is important to keep in mind that in the human disease the antibodies are synthesized by plasma cells distributed throughout the brain and meninges suggesting a widespread presence of antibodies (along with inflammatory changes) in the brain parenchyma.13 In contrast, the passive transfer model used here favors the antibody binding in the hippocampus (due to passive diffusion and close proximity to the ventricular system and CSF) and at a lesser degree in other cortical regions, and lacks inflammatory infiltrates.11, 15 Future studies using other forms of antibody delivery, active immunization, or different mouse strains or additional rodent or non‐human primate species may answer whether “host” factors, or additional, so far unidentified intrathecal antibody specificities, contribute to the variety of symptoms observed in anti‐NMDAR encephalitis. However, a similar variety of symptoms is observed in models of NMDAR antagonists in which the spectrum of symptoms is dependent of the dose and intensity of effects on the NMDAR.32
Taken together, we unambiguously show that intrathecal accumulation of B cells and plasma cells corresponds to the clinical course in patients with anti‐NMDAR encephalitis. Moreover, presence of intrathecal cePc with hypermutated antigen receptors indicates an antigen‐driven intrathecal immune response in patients with anti‐NMDAR encephalitis. Consistently, a single recombinant human GluN1‐specific monoclonal antibody, rebuilt from intrathecal cePc by reverse genetics, is sufficient to reproduce NMDAR epitope specificity and key pathogenic features of the human disease in vitro and in vivo.
Having reconstructed pathogenic antineuronal antibody signatures in recombinant form permits further elucidation of disease mechanisms and development of targeted therapies in anti‐NMDAR encephalitis and will increase our understanding of the role of NMDAR in synaptic function, cognition and behavior in general.
Author Contribution
MM, SB, CB, and JS performed FACS sorting, single‐cell PCR, sequencing, cloning, expression, and purification and part of IHC characterization of rhuMabs under supervision of NG; FD and SB performed immunoprecipitation (IP) experiments under supervision of NK and NG; JP and FM performed immunohistochemistry with rat brain and immunocompetition assays, and studied the effects of rhuMab on primary cultures of hippocampal neurons and animal behavior under supervision of EMG, RM, and JD. KSG recruited and treated the patients under supervision of NM. KB performed the neuropsychological testing under the supervision of AJ. CCG analyzed flow cytometry data under the supervision of SGM and NM. NSS, GS, SK, NK, and NM performed electrophysiological analysis. KKF performed immunocytochemistry with transfected HEK cells under supervision of FL and KPW. AJB performed histopathological analysis of a former anti‐NMDAR encephalitis patient contributed by CEE. CW and CSN performed injection experiments of rhuMabs together with complement in rodent hippocampus for an earlier version of the manuscript. NG, JD, NM, HPH, SGM, and HW designed and supervised the project. NM and NG wrote the first draft of the manuscript. All authors contributed to and approved the final manuscript.
Conflict of Interest
All authors declare no relevant conflicts of interest.
Supporting information
Table S1. Clinical, magnetic resonance imaging, and electroencephalography findings in patients with anti‐NMDAR encephalitis.
Table S2. Routine CSF and PB analysis and antibody titers of patients with anti‐NMDAR encephalitis.
Table S3. Neuropsychological domains with respective tests applied during neuropsychological testing of patients with anti‐NMDAR encephalitis.
Table S4. Results of the different neuropsychological tests in patients with anti‐NMDAR encephalitis at initial assessment 1–16 weeks after admission.
Table S5. Results of the different neuropsychological tests in patients with anti‐NMDAR encephalitis at follow‐up assessment 8–48 months after initial assessment.
Table S6. Baseline characterization of the peripheral and intrathecal autoimmune response in anti‐NMDAR encephalitis using multi‐parameter flow cytometry before immunotherapy.
Table S7. Follow‐up characterization of the peripheral and intrathecal autoimmune response in anti‐NMDAR encephalitis using multiparameter flow cytometry at 5–7 months after initiation of immunotherapy.
Figure S1. Cerebral MRI in patients with anti‐NMDAR encephalitis.
Acknowledgments
We would like to thank Verena Schütte, Kerstin Gottschalk, and Schumina Säuberlich, Department of Neurology, WWU Münster, Germany, for excellent technical assistance. We are grateful for the complimentary initial screening of rhuMab specificity by Euroimmun, Lübeck, Germany. The expert technical help of Katharina Raba, FACS facility of the HHU Düsseldorf, Germany, and Jason Cline, Department of Neurology, HHU Düsseldorf, Germany is highly appreciated.
Funding Statement
This work was funded by German Research Foundation grants DFG, TR128, Project Z2 and DFG, INST 2105/27‐1; NIH grant RO1NS077851; Instituto Carlos III/FEDER grant FIS PI14/00203; CIBERER grant CB15/00010; Fundacio Cellex grant ; Walter und Ilse‐Rose‐Stiftung grant ; Forschungskommission of the Heinrich Heine University Düsseldorf, Germany grant ; Bundesministerium für Bildung und Forschung grant 031A232.
Contributor Information
Nico Melzer, Email: nico.melzer@ukmuenster.de.
Norbert Goebels, Email: norbert.goebels@uni-duesseldorf.de.
References
- 1. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long‐term outcome in patients with anti‐NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pruss H, Finke C, Holtje M, et al. N‐methyl‐D‐aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol 2012;72:902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armangue T, Leypoldt F, Malaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014;75:317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gresa‐Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow‐up of anti‐NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camdessanché J‐P, Streichenberger N, Cavillon G, et al. Brain immunohistopathological study in a patient with anti‐NMDAR encephalitis. Eur J Neurol 2011;18:929–931. [DOI] [PubMed] [Google Scholar]
- 7. Martinez‐Hernandez E, Horvath J, Shiloh‐Malawsky Y, et al. Analysis of complement and plasma cells in the brain of patients with anti‐NMDAR encephalitis. Neurology 2011;77:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moscato EH, Peng X, Jain A, et al. Acute mechanisms underlying antibody effects in anti‐N‐methyl‐D‐aspartate receptor encephalitis. Ann Neurol 2014;76:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti‐NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross‐talk between NMDA and Ephrin‐B2 receptors in anti‐NMDA encephalitis. Brain 2012;135(Pt 5):1606–1621. [DOI] [PubMed] [Google Scholar]
- 11. Planagumà J, Haselmann H, Mannara F, et al. Ephrin‐B2 prevents N‐methyl‐D‐aspartate receptor antibody effects on memory and neuroplasticity. Ann Neurol 2016;80:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vitureira N, Letellier M, Goda Y. Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr Opin Neurobiol 2012;22:516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody‐associated encephalitides: clues for pathogenesis. Brain 2012;135(Pt 5):1622–1638. [DOI] [PubMed] [Google Scholar]
- 14. Dalmau J, Tüzün E, H‐y W, et al. Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Planagumà J, Leypoldt F, Mannara F, et al. Human N‐methyl D‐aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015;138(Pt 1):94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dalmau J. NMDA receptor encephalitis and other antibody‐mediated disorders of the synapse: The 2016 Cotzias Lecture. Neurology 2016;87:2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kreye J, Wenke NK, Chayka M, et al. Human cerebrospinal fluid monoclonal N‐methyl‐D‐aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 2016;139(Pt 10):2641–2652. [DOI] [PubMed] [Google Scholar]
- 18. Obermeier B, Mentele R, Malotka J, et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med 2008;14:688–693. [DOI] [PubMed] [Google Scholar]
- 19. von Büdingen H‐C, Harrer MD, Kuenzle S, et al. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin‐specific antibodies. Eur J Immunol 2008;38:2014–2023. [DOI] [PubMed] [Google Scholar]
- 20. Golombeck KS, Bönte K, Mönig C, et al. Evidence of a pathogenic role for CD8(+) T cells in anti‐GABAB receptor limbic encephalitis. Neurol Neuroimmunol Neuroinflamm 2016;3:e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuenzle S, von Büdingen H‐C, Meier M, et al. Pathogen specificity and autoimmunity are distinct features of antigen‐driven immune responses in neuroborreliosis. Infect Immun 2007;75:3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tiller T, Meffre E, Yurasov S, et al. Efficient generation of monoclonal antibodies from single human B cells by single cell RT‐PCR and expression vector cloning. J Immunol Methods 2008;329(1–2):112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saitou N, Nei M. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987;4:406–425. [DOI] [PubMed] [Google Scholar]
- 24. Hillis DM, Bull JJ, White ME, et al. Experimental phylogenetics: generation of a known phylogeny. Science 1992;255:589–592. [DOI] [PubMed] [Google Scholar]
- 25. Raymond C, Tom R, Perret S, et al. A simplified polyethylenimine‐mediated transfection process for large‐scale and high‐throughput applications. Methods 2011;55:44–51. [DOI] [PubMed] [Google Scholar]
- 26. Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petit‐Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seebohm G, Chen J, Strutz N, et al. Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol Pharmacol 2003;64:70–77. [DOI] [PubMed] [Google Scholar]
- 29. Gleichman AJ, Spruce LA, Dalmau J, et al. Anti‐NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 2012;32:11082–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kayser MS, Kohler CG, Dalmau J. Psychiatric manifestations of paraneoplastic disorders. Am J Psychiatry 2010;167:1039–1050. [DOI] [PubMed] [Google Scholar]
- 31. Kayser MS, Dalmau J. Anti‐NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res 2016;176:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masdeu JC, Dalmau J, Berman KF. NMDA Receptor Internalization by Autoantibodies: a Reversible Mechanism Underlying Psychosis? Trends Neurosci 2016;39:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castillo‐Gomez E, Oliveira B, Tapken D, et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry, advance online publication, 9 August 2016; doi:10.1038/mp.2016.125 [DOI] [PubMed] [Google Scholar]
- 34. Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron 1999;22:645–648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical, magnetic resonance imaging, and electroencephalography findings in patients with anti‐NMDAR encephalitis.
Table S2. Routine CSF and PB analysis and antibody titers of patients with anti‐NMDAR encephalitis.
Table S3. Neuropsychological domains with respective tests applied during neuropsychological testing of patients with anti‐NMDAR encephalitis.
Table S4. Results of the different neuropsychological tests in patients with anti‐NMDAR encephalitis at initial assessment 1–16 weeks after admission.
Table S5. Results of the different neuropsychological tests in patients with anti‐NMDAR encephalitis at follow‐up assessment 8–48 months after initial assessment.
Table S6. Baseline characterization of the peripheral and intrathecal autoimmune response in anti‐NMDAR encephalitis using multi‐parameter flow cytometry before immunotherapy.
Table S7. Follow‐up characterization of the peripheral and intrathecal autoimmune response in anti‐NMDAR encephalitis using multiparameter flow cytometry at 5–7 months after initiation of immunotherapy.
Figure S1. Cerebral MRI in patients with anti‐NMDAR encephalitis.